

Abstract
Hydrogen peroxide (H2O2) is a central reactive oxygen species (ROS) that contributes to diseases from obesity to cancer to neurodegeneration but is also emerging as an important signaling molecule. We now report a versatile histochemical approach for detection of H2O2 that can be employed across a broad range of cell and tissue specimens in both healthy and disease states. We have developed a first-generation H2O2-responsive analogue named Peroxymycin-1, which is based on the classic cell-staining molecule puromycin and enables covalent staining of biological samples and retains its signal after fixation. H2O2-mediated boronate cleavage uncages the puromycin aminonucleoside, which leaves a permanent and dose-dependent mark on treated biological specimens that can be detected with high sensitivity and precision through a standard immunofluorescence assay. Peroxymycin-1 is selective and sensitive enough to image both exogenous and endogenous changes in cellular H2O2 levels and can be exploited to profile resting H2O2 levels across a panel of cell lines to distinguish metastatic, invasive cancer cells from less invasive cancer and nontumorigenic counterparts, based on correlations with ROS status. Moreover, we establish that Peroxymycin-1 is an effective histochemical probe for in vivo H2O2 analysis, as shown through identification of aberrant elevations in H2O2 levels in liver tissues in a murine model of nonalcoholic fatty liver disease, thus demonstrating the potential of this approach for studying disease states and progression associated with H2O2. This work provides design principles that should enable development of a broader range of histochemical probes for biological use that operate via activity-based sensing.
INTRODUCTION
Reactive oxygen species (ROS) are a family of redox-active small molecules that are broadly generated in living systems.1 While ROS have been long known to participate in immune responses,2 mounting data show that ROS can also serve as important signaling molecules in a diverse array of biological processes.3–7 In this context, a major ROS is hydrogen peroxide (H2O2), which can be produced by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) proteins8 in various cells and tissues upon stimulation with growth factors,9–11 cytokines,12 hormones,13 and neuro-transmitters.14 H2O2 can then activate many classes of downstream targets through reversible redox post-translational modifications,15–19 including phosphatases,20,21 kinases,22 transcription factors,23 and ion channels.24 On the other hand, however, aberrant production of H2O2 can result in oxidative stress, which can contribute to aging25 and development and progression of serious diseases, including cancer,25 obesity and diabetes,26,27 and neurodegenerative Alzheimer’s and Parkinson’s diseases.28,29 As such, the dual signal/stress nature of H2O2 provides motivation for developing new technologies to probe its contributions across a range of biological contexts.
In this regard, fluorescence imaging is a powerful approach for studying H2O2 in biological specimens owing to its high sensitivity, good spatial and temporal resolution, and non-invasive nature.30–50 Moreover, fluorescent probes that operate through activity-based sensing (ABS),31,51–53 such as through H2O2-mediated boronate cleavage,36,47 offer excellent selectivity toward H2O2 over other ROS36–65 and have been used to decipher principles of H2O2 signaling. For example, probes from our laboratory have been utilized to identify particular aquaporin subtypes as H2O2 channels,44 H2O2 sources and targets in stem cell maintainence and neurogenesis,45 H2O2/H2S crosstalk,46 and respiring mitochondria as primary source of H2O2 for brain cell signaling.41 Despite their utility, these probes are mostly limited to transient analysis of dissociated cells in culture and are not compatible with fixed samples that precludes assessment of a broader range of cell to tissue specimens. Indeed, immunostaining for the oxidized lipid product 4-hydroxy-2-nonenal (4-HNE) can be employed in fixed samples,66,67 but this method offers only an indirect measure of ROS levels.
Against this backdrop, we sought to develop a general ABS approach that would enable direct, selective, and sensitive histochemical analysis of H2O2 from cell to tissue samples. We turned our attention to puromycin, an aminonucleoside with an α-amino group that allows it to incorporate into nascent peptides at the ribosome,68 which leaves a permanent and dose-dependent mark on cells and tissues that can be readily detected by immunofluorescence using puromycin-specific antibodies.69–75 Because the α-amino group of puromycin is essential for incorporation in peptides, caging this moiety offers a general methodology for developing ABS probes for histochemical analysis in fixed biological specimens (Scheme 1a,b). We now report a puromycin-based method for histochemical detection of H2O2 that is generally applicable to both cell and tissue samples. Peroxymycin-1 utilizes the selective H2O2-mediated boronate oxidation reaction to generate puromycin and report H2O2 in a dose-dependent manner, whereas the negative control compound Ctrl-Peroxymycin-1, bearing the same boronate switch but with an extra carbon in the linker, cannot eliminate to the parent puromycin product (Scheme 1c). Peroxymycin-1 is capable of revealing changes in exogenous and endogenous H2O2 levels in cells and can be used to profile differences in basal H2O2 levels across breast cell lines from nontumorigenic cell models to nonmetastatic to metastatic cancer cell lines. Moreover, this probe enables H2O2 detection in fixed tissues after in vivo application, as demonstrated by the identification of liver-specific elevations in H2O2 in a diet-induced mouse model of nonalcoholic fatty liver disease (NAFLD). In addition to establishing the utility of Peroxymycin-1 as a versatile reagent for selective and sensitive detection of H2O2 in cells and tissues, this work provides a starting point for the general design of histochemical probes that operate through an ABS mechanism.
Scheme 1.
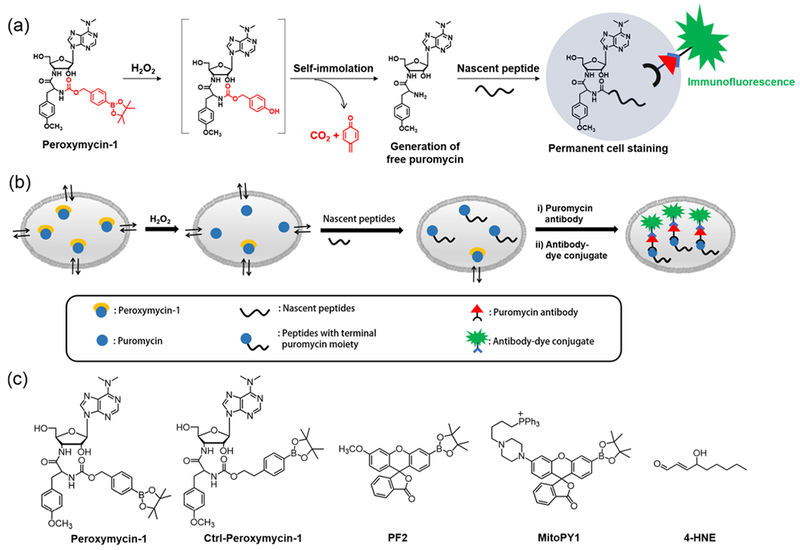
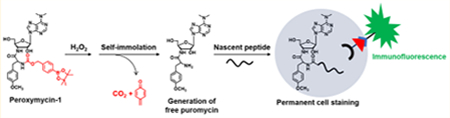
(a) Design and (b) Schematic Cartoon Showing the Working Principle of Peroxymycin-1 for Histochemical Detection of H2O2 in Various Biological Samples and (c) Chemical Structures of Probes Investigated in This Studya
aPeroxymycin-1 and Ctrl-Peroxymycin-1 are novel histochemical H2O2 probe and negative control compounds, respectively, developed in this study, whereas Peroxyfluor-2 (PF2) and Mitochondria Peroxy Yellow-1 (MitoPY1) are previously reported fluorescent probes for imaging H2O2 in cells, and 4-HNE is an oxidized lipid used to indirectly assess increased oxidative stress in fixed biological specimens.
RESULTS AND DISCUSSION
Design, Synthesis, and Characterization of Peroxymycin-1 and Ctrl-Peroxymycin-1.
Peroxymycin-1 was synthesized by conjugation of puromycin with 4-(hydroxymethyl)phenylboronic ester through a carbamate linkage. In the absence of H2O2, the caged probe lacking the free amino group cannot label newly synthesized cellular peptides and will be washed away after fixation. H2O2-mediated oxidation of phenylboronic ester into phenol moiety36–47,49,54–65 and subsequent self-immolation of the linker to release CO2 and quinone methide76–79 results in the formation of free puromycin (Scheme 2a), which can be incorporated into nascent peptides and enables permanent cell staining and detection by immunofluorescence. The negative control compound Ctrl-Peroxymycin-1 bears a p-(2-hydroxyethyl)-phenylboronic ester as the caging group. This boronate compound can be transformed by H2O2 into the corresponding phenol, but the ethyl linker is unable to undergo further selfimmolation and thus cannot form puromycin in a H2O2-mediated dose-dependent manner (Scheme 2b).
Scheme 2.
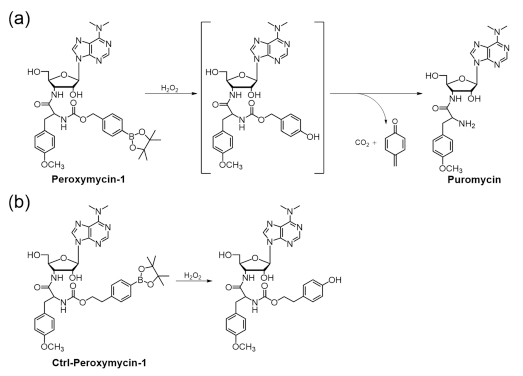
Reactions of Peroxymycin-1 and Ctrl-Peroxymycin-1 with H2O2a
aAs determined by LC–MS experiments.
The key step in the syntheses of these chemical probes is the preparation of 4-nitrophenyl carbonate intermediate (Scheme S1 in Supporting Information). Subsequent coupling reactions between such 4-nitrophenyl carbonate intermediates and puromycin dihydrochloride can yield either Peroxymycin-1 or its Ctrl-Peroxymycin-1 congener. In principle, this synthetic scheme can be generalized to wide variety of other responsive triggers that can cap the puromycin α-amino group, enabling the development of molecular probes for other analytes of biological interest. The probes were characterized by 1H NMR, 13C{1H} NMR, high-resolution mass spectrometry (HRMS), and liquid chromatography-coupled mass spectrometry (LC–MS).
Reactivity of Peroxymycin-1 with Hydrogen Peroxide.
Peroxymycin-1 maintains good stability in aqueous solution buffered to physiological pH, showing only conversion of the pinacol boronate to the parent boronic acid with no release of puromycin in the absence of H2O2 (Figure S1). Treatment of Peroxymycin-1 with H2O2 produces puromycin, as shown by LC–MS analysis (Figure 1a,b), confirming the reaction depicted in Scheme 2a. The pseudo-first-order rate constant of the reaction at 25 °C was determined to be 1.0 × 10−3 s−1, with control experiments showing negligible puromycin generation from Peroxymycin-1 in the absence of H2O2 (Figure 1C)
Figure 1.
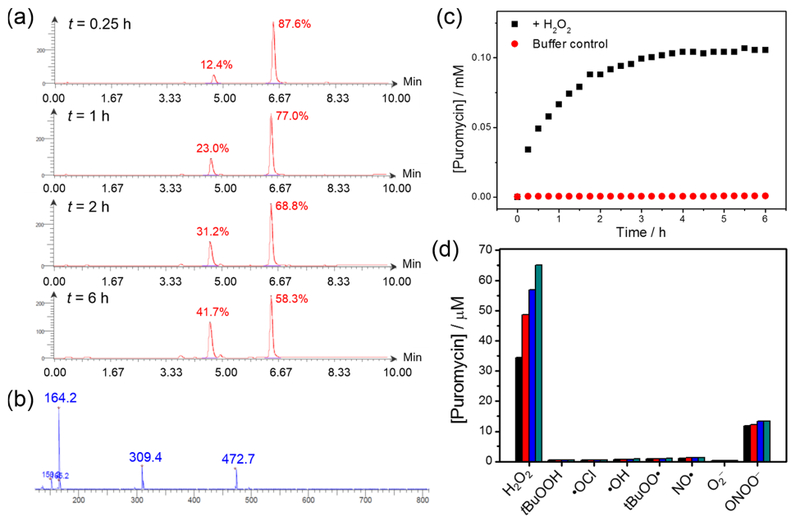
(a) LC chromatograms of the reaction between Peroxymycin-1 (0.3 mM) and H2O2 (0.1 mM) in phosphate buffer (20 mM, pH 7.4)/methanol solution mixture (2:1 v/v) with 3 vol % dimethyl sulfoxide (DMSO) at different time intervals. (b) MS of the peak with retention time 4.62 min, confirming formation of puromycin from the reaction between Peroxymycin-1 and H2O2. (c) Generation of puromycin from the solution of Peroxymycin-1, with and without H2O2, over time. (d) Selectivity of Peroxymycin-1 toward H2O2 over other reactive oxygen and nitrogen species. All species were at 100 μM except ONOO− was at 50 μM. Time points were taken at 15 min (black bars), 30 min (red bars), 45 min (blue bars), and 60 min (green bars).
In another control experiment, the reaction between Ctrl-Peroxymycin-1 and H2O2 was also studied by LC–MS and shows only the corresponding phenol as a boronate oxidation product without self-immolative cleavage to produce puromycin (Figure S2). These data further support the pathway depicted in Scheme 2, in which Peroxymycin-1 reacts with H2O2 through oxidation and subsequent self-immolation to yield free puromycin, rather than a mechanism involving direct cleavage of the carbamate linkage by H2O2. Finally, Peroxymycin-1 exhibits high selectivity for H2O2 over other ROS and reactive nitogen congeners as shown by LC–MS analyses (Figure 1d). Only peroxynitrite at 50 μM, which is far higher than estimated physiologically relevant concentrations,80 gives any response, and the production of puromycin is an order of magnitude less than for H2O2. As we will show, the use of an NO synthase inhibitor can help readily distinguish ROS versus reactive nitrogen species (RNS) boronate reactivity.
Histochemical Detection of Cellular H2O2 by Use of Peroxymycin-1.
With in vitro characterization results in hand, establishing the sensitivity and selectivity of Peroxymycin-1 for H2O2, we sought to apply this new chemical tool for histochemical H2O2 detection in cells. In initial experiments, HeLa cells were treated with either Peroxymycin-1 or puromycin at equivalent doses (1 μM) and monitored at different time intervals in order to assess cellular uptake and reaction of the probe with H2O2 relative to the time required for incorporating puromycin into cellular proteins (Figure 2, Figures S3 and S4). Puromycin serves as a positive control representing the fully deprotected product of Peroxymycin-1. Treated cells were then fixed and immunostained with primary α-puromycin antibody and secondary antibody-Alexa Fluor 488 and subsequently imaged by confocal fluorescence microscopy. Puromycin-treated cells revealed strong green fluorescence within 0.5 h with saturation at 1 h (Figure S4), suggesting fast uptake and incorporation of puromycin into cellular proteins. In contrast, Peroxymycin-1-treated cells show no significant green fluorescence at 0.5 h, but we were pleased to observe increases in fluorescence intensity at longer time points from 1 to 6 h (Figure 2a,c), presumably due to reaction of the probe with H2O2 followed by incorporation of the puromycin product into nascent cellular peptides. The statistically significant turnon of green fluorescence from cells treated with Peroxymycin-1 (Figure 2a) suggests that this reagent is sensitive enough to detect basal levels of H2O2 in cell culture. To compare the sensitivity of this new histochemical method to a fluorescent probe counterpart, we treated HeLa cells with Peroxyfluor-2 (PF2), a boronate probe with negligible background in the absence of H2O2 owing to its closed lactone structure that shows strong green fluorescence upon reaction with H2O2.43 HeLa cells treated with 10 μM PF2 for 0.5, 4, and 6 h and then washed display negligible fluorescence (Figure 2b), suggesting that this reagent is not sensitive enough to detect basal H2O2 levels in HeLa cells under these conditions. Because PF2 is not cell-trappable, we also employed a mitochondria-targeting H2O2 fluorescent probe, Mitochondria Peroxy Yellow-1 (MitoPY1),40 that is retained in cells. This probe was not effective for imaging basal H2O2 in HeLa cells under similar experimental conditions (e.g., same laser power and receiver gain but with excitation at 514 nm instead of 488 nm; Figure S5) but required higher laser power and receiver gain to generate a usable fluorescence signal over background (Figure S5c). As such, we suggest that the higher sensitivity of Peroxymycin-1 over PF2 and MitoPY1 comes from two main origins. First, covalent staining for the histochemical Peroxymycin-1 reagent can lead to a permanent mark that survives sample washing and fixation, as opposed to PF2 and other fluorescent dyes, where probe leakage can decrease signal-to-noise responses.81,82 Second, immunostaining offers advantages of low background noise owing to specific interactions from antibodies83 and higher signal due to the potential for multivalent binding of secondary antibody–dye conjugates onto the primary antibody.83 Taken together, the data show that Peroxymycin-1 exhibits excellent sensitivity for cellular detection of H2O2.
Figure 2.
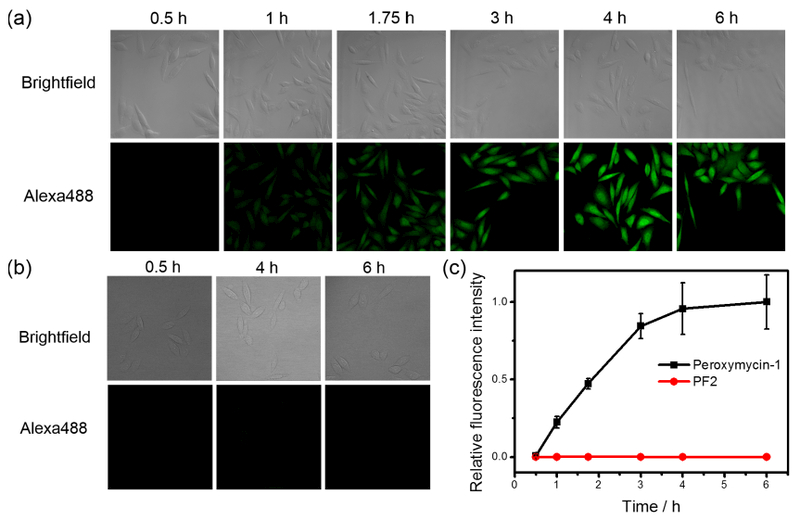
(a) Confocal fluorescence microscopic images of HeLa cells treated with Peroxymycin-1 (1 μM) for indicated time intervals. The cells were then washed, fixed, stained and imaged. (b) Confocal fluorescence microscopic images of HeLa cells treated with PF2 (10 μM) for 0.5, 4, and 6 h, respectively. The medium was then replaced by phosphate-buffered saline (PBS) solution and imaged. All images in panels a and b were recorded by use of the same imaging parameters with the Alexa488 channel. (c) Normalized cellular fluorescence intensities of the HeLa cells as determined by ImageJ, showing superior sensitivity for H2O2 detection for the new histochemical boronate H2O2 probe over a fluorescent probe counterpart. Error bars denote standard derivation (SD; n = 5).
Peroxymycin-1 Enables Histochemical Detection of Cellular H2O2 under Oxidative Stress or Physiological Stimulation Conditions.
We next moved on to evaluate the ability of Peroxymycin-1 to respond to changes in H2O2 levels under oxidative stress conditions. HeLa cells were pretreated with H2O2 (50 μM) for 2 h and washed before they were incubated with Peroxymycin-1, Ctrl-Peroxymycin-1, or puromycin (1 μM) for 4 h. As anticipated, Peroxymycin-1-stained cells exposed to H2O2 show a clear, statistically significant increase in fluorescence intensity compared to Peroxymycin-1-stained control cells without H2O2 pretreatment (Figure 3). On the other hand, neither puromycin-stained cells nor Ctrl-Peroxymycin-1-stained cells show differences in fluorescence intensity when samples are exposed to H2O2 or mock vehicle (Figure 3). Notably, the lack of observed changes in fluorescence intensity with puromycin-stained cells upon H2O2 addition indicates that ROS treatment does not alter its cellular uptake and incorporation into proteins, whereas the lack of signal from Ctrl-Peroxymycin-1-stained cells shows that there is also no off-target release of puromycin from this reagent. As such, only Peroxymycin-1 is responsive to oxidative stress induced by H2O2 treatment owing to its ability to react with this ROS and undergo a self-immolative cleavage to release puromycin. Finally, cells coincubated with Peroxymycin-1 and H2O2 within 2 h can saturate to a comparable fluorescence intensity to cells stained with puromycin (Figure S6), indicating a relatively fast and clean H2O2-mediated conversion of Peroxymycin-1 into puromycin and subsequent incorporation of the aminonucleoside into cellular proteins.
Figure 3.
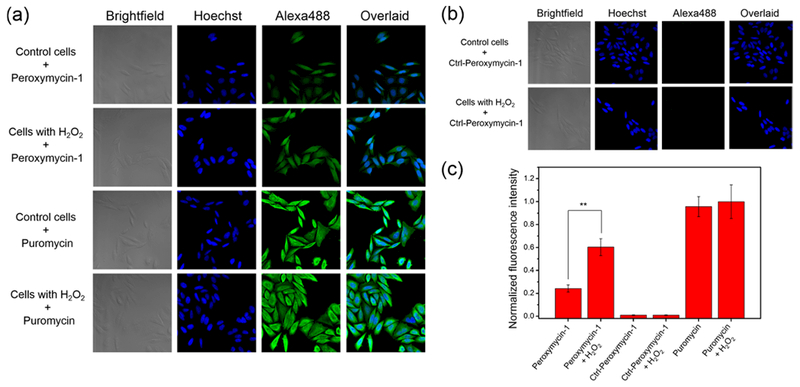
Confocal fluorescence microscopic images of HeLa cells, with or without pretreatment with H2O2 (50 μM) for 2 h, stained with (a) Peroxymycin-1 or puromycin (1 μM) or (b) Ctrl-Peroxymycin-1 (1 μM) for 4 h. The cells were then subsequently washed, fixed, stained, and imaged. All images were recorded by use of the same imaging parameters with the Alexa488 channel. (c) Cellular fluorescence intensities of HeLa cells as determined by ImageJ. Error bars denote SD (n = 5). **p < 0.01.
Next, we utilized Peroxymycin-1 for detection of endogenous H2O2 production through treatment of HeLa cells with paraquat, a small-molecule inducer of ROS and oxidative stress.40 HeLa cells coincubated with Peroxymycin-1 (1 μM) and paraquat (1 mM) showed patent increases in fluorescence intensity compared to Peroxymycin-1-stained control cells without paraquat treatment (Figure 4). In control experiments, we observed no significant changes in fluorescence between puromycin-stained cells with or without paraquat exposure, showing that Peroxymycin-1 responds to paraquat via ROS detection rather than changes in cellular uptake or puromycin incorporation (Figure 4). The data establish that Peroxymycin-1 is an effective chemical tool for detection of endogenous H2O2 production in cells.
Figure 4.
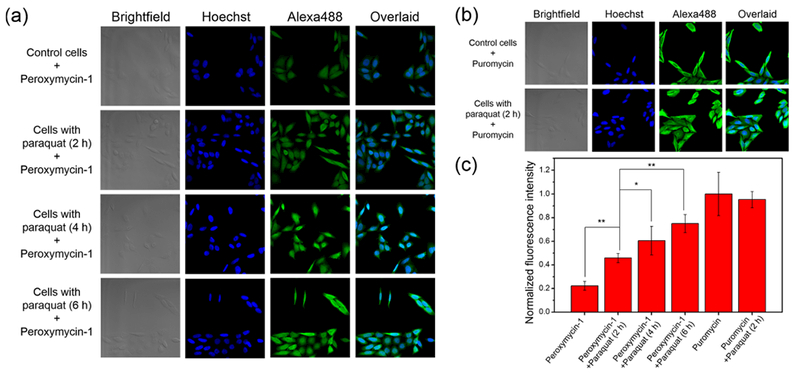
Confocal fluorescence microscopic images of HeLa cells stained with (a) Peroxymycin-1 (1 μM) or (b) puromycin (1 μM) for 6 h, with or without coincubation of paraquat (1 mM) for the indicated time intervals. The cells were subsequently washed, fixed, stained, and imaged. All images were recorded by use of the same imaging parameters with the Alexa488 channel. (c) Cellular fluorescence intensities of HeLa cells as determined by ImageJ. Error bars denote SD (n = 5). *p < 0.05 and **p < 0.01.
We then proceeded to apply Peroxymycin-1 to detect changes in cellular H2O2 levels upon physiological stimulation under signaling conditions. We turned our attention to A431 cells, which possess high expression of epidermal growth factor receptors (EGFR) and thus can respond to EGF stimulation for endogenous generation of H2O2 through a Nox/phosphoinositide 3-kinase (PI3K) pathway.9,10,38,43 A431 cells were stimulated with EGF (100 ng/mL) for 20, 40, or 60 min and then washed and incubated with Peroxymycin-1 (1 μM) for 4 h (Figure 5, Figure S7). EGF stimulation triggers a statistically significant increase in fluorescence intensity with increasing EGF incubation times. On the other hand, Ctrl-Peroxymycin-1-stained A431 cells, with or without EGF stimulation, revealed no observable differences in immunofluorescence, indicating that this probe does not give off-target production of puromycin (Figure 5b,c). As another control, puromycin-stained cells with or without EGF stimulation also do not show any significant changes in fluorescence intensity, again ruling out any alterations in rates of cellular uptake and puromycin incorporation with EGF exposure (Figure 5a,c). Indeed, the collective results establish that Peroxymycin-1 enables imaging of changes in intracellular H2O2 levels under both stress and signaling conditions.
Figure 5.
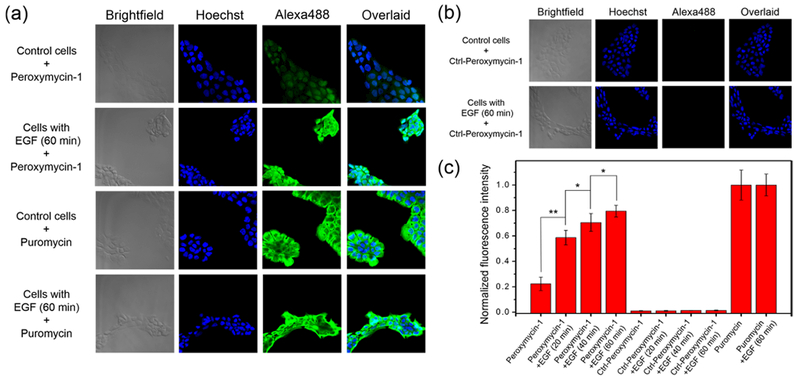
Confocal fluorescence microscopic images of A431 cells stained with (a) Peroxymycin-1 or puromycin (1 μM) or (b) Ctrl-Peroxymycin-1 (1 μM) for 4 h, with or without pretreatment with EGF (100 ng/mL) for 60 min. The cells were subsequently washed, fixed, stained, and imaged. All images were recorded by use of the same imaging parameters with the Alexa488 channel. (c) Cellular fluorescence intensity of A431 cells as determined by ImageJ. Error bars denote SD (n = 5). *p < 0.05 and **p < 0.01.
To further validate that the increases in cellular immunofluorescence observed with Peroxymycin-1 in this EGF/A431 cell model are due to H2O2 and not peroxynitrite or related RNS, we performed control experiments in which A431 cells were pretreated with a nitric oxide (NO) synthase inhibitor, l-NG-nitroarginine methyl ester (l-NAME; 100 μM; 25 min),38 along with EGF (100 ng/mL; 40 min) and then washed, incubated with Peroxymycin-1 (1 μM) for 4 h, and imaged (Figure 6). Inhibition of NO generation blocks formation of peroxynitrite and other RNS. Therefore, A431 cells pretreated with l-NAME and EGF should show elevated H2O2 levels but not peroxynitrite levels. Indeed, inhibition with l-NAME does not block EGF-stimulated increases in Peroxymycin-1 immunofluorescence (Figure 6), showing that the probe selectively detects elevations in H2O2 level without any peroxynitrite interference under these biological conditions.
Figure 6.
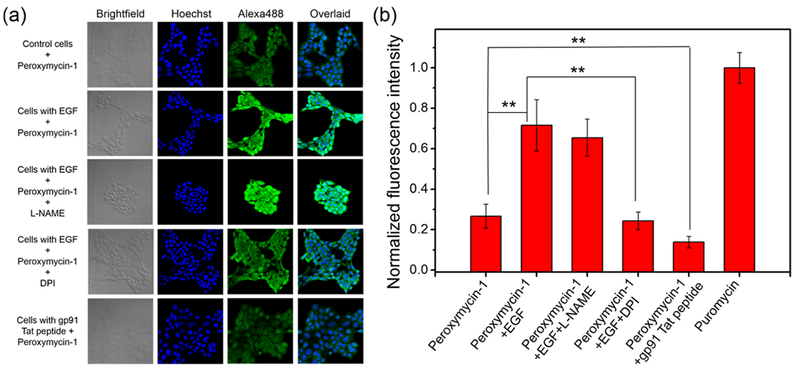
(a) Confocal fluorescence microscopic images of A431 cells with or without EGF stimulation (100 ng/mL) for 40 min, with or without various Nox or NO synthase inhibitors, followed by washing and incubation with Peroxymycin-1 (1 μM) for 4 h. The cells were subsequently washed, fixed, stained, and imaged. All images were recorded by use of the same imaging parameters with the Alexa488 channel. (b) Cellular fluorescence intensities of A431 cells as determined by ImageJ. Error bars denote SD (n = 5). **p < 0.01.
To identify the molecular source of H2O2 production upon EGF stimulation, A431 cells were treated with EGF (100 ng/mL; 40 min) in the presence of the broad-spectrum Nox inhibitor diphenyleneiodonium (DPI; 5 μM).38 As expected, Peroxymycin-1-stained, EGF-stimulated A431 cells show a statistically significant decrease in immunofluorescent signal with DPI incubation (Figure 6), establishing that Nox is responsible for the physiological H2O2 burst in this cell model.
Finally, with the high selectivity and sensitivity of H2O2 established for Peroxymycin-1 for use in histochemical cellular assays, we sought to utilize this reagent for imaging basal H2O2 production derived from Nox. To this end, A431 cells were pretreated with gp91 Tat peptide (100 μM; 30 min), an isoform-specific inhibitor of Nox2 that can suppress H2O2 production,38 and then stained with Peroxymycin-1 (1 μM; 4 h). Indeed, gp91 Tat peptide-treated cells exhibit markedly lower levels of Peroxymycin-1 immunofluorescence compared to controls without inhibitor treatment (Figure 6). For comparison, the fluorescent H2O2 probes PF2 and MitoPY1 failed to show statistically significant decreases in signal in response to Nox2 inhibition by the gp91 Tat peptide (Figures S8 and S9), again illustrating that this histochemical approach affords improved sensitivity through enabling permanent staining of the cells in response to H2O2 with low background noise and improved multivalent signal through immunostaining.
Peroxymycin-1 Enables H2O2 Profiling across Multiple Cell Types.
The high sensitivity and selectivity of the histochemical approach to H2O2 detection enabled by Peroxymycin-1 can be exploited to profile levels of this ROS across a panel of cell types. In this context, elevated levels of ROS in cancer can contribute to cell growth, proliferation, and migration in this disease,84 and the ability to assess relative ROS levels across related but distinct types of cells can help characterize and correlate this biochemical marker with key oncogenic properties such as metastatic potential. To achieve this goal, we sought to profile H2O2 levels across a panel of related cancer and nontumorigenic breast cell lines. These lines include highly metastatic breast cancer (MDA-MB-231), less-invasive breast cancer (MDA-MB-468 and MCF-7), non-tumorigenic breast epithelial cell (MCF-10A), invasive breast cancer (HS 578T), and normal breast cell (HS 578Bst) models. Samples of each of these cell lines were labeled with Peroxymycin-1 (1 μM) for 4 h, immunostained, and imaged (Figure 7, Figures S10 and S11). Interestingly, we observed a correlation between H2O2 levels and metastatic potential, as the highly metastatic MDA-MB-231 cell lines showed higher H2O2-dependent fluorescence intensities compared to less invasive MDA-MB-468 and MCF-7 and nontumorigenic MCF-10A cells (p < 0.01; Figure S10a). Moreover, for HS 578T and HS 578Bst cells, which are derived from primary invasive ductal carcinoma and normal adjacent tissues, respectively, the former cell line exhibited a higher fluorescence intensity (Figure S10a). To normalize for potential differences in cell size and puromycin incorporation rate across this panel of cell lines, we conducted control experiments with puromycin for the same time interval (Figures S10 and S11) and used these data as an internal standard for the Peroxymycin-1-stained cells. Indeed, these normalized fluorescence intensities follow the order of metastatic/invasive cancer cells > cancer cells > nontumorigenic cells (Figure 7b), suggesting that metastatic/invasive cancer cells show higher H2O2 levels than less invasive cancer cells and nontumorigenic cells. These imaging results are consistent with chemoproteomics experiments showing higher levels of protein sulfenic acids, which are major initial oxidation products of proteins by H2O2, in metastatic/invasive breast cancer cells relative to healthy control cells,85 and highlight the potential of this histochemical approach to rapidly assess and compare multiple types of biological specimens in the context of ROS status.
Figure 7.
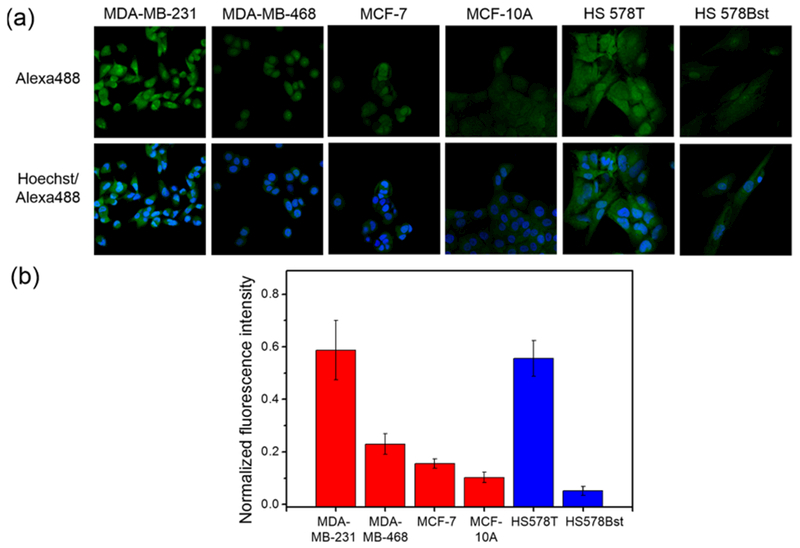
(a) Confocal fluorescence microscopic images of highly metastatic breast cancer cells (MDA-MB-231), breast cancer cells (MDA-MB-468 and MCF-7), nontumorigenic breast epithelial cells (MCF-10A), invasive breast cancer cells (HS 578T), and normal breast cells (HS 578Bst) assayed with Peroxymycin-1 (1 μM) for 4 h. The cells were subsequently washed, fixed, stained, and imaged. All images were recorded by use of the same imaging parameters. (b) Comparative fluorescence intensities from cells treated with Peroxymycin-1 normalized by fluorescence intensities from cells treated with puromycin as an internal standard (n = 5).
Peroxymycin-1 as a Histochemical Probe for Detection of H2O2 in Tissues: Identification of Elevations in Liver Peroxide Levels in a Diet-Induced Model of Nonalcoholic Fatty Liver Disease.
Technologies to reliably and directly assess ROS levels within intact tissues, and in particular the levels of specific individual ROS molecules, remain insufficiently developed. Indeed, one of the most common traditional histochemical assays that is employed to probe ROS and inflammation in tissue biopsies is staining for 4-hydroxy-2-noneal (4-HNE),68,69 which is an oxidized lipid metabolite that shows no selectivity toward a particular ROS and thus offers limited and indirect information on overall redox status. As such, we sought to apply our Peroxymycin-1 approach and validate its utility for histochemical H2O2 detection from tissue specimens derived from in vivo animal models, particularly during disease development and progression. For the present study, we chose to apply this reagent to a mouse model of nonalcoholic fatty liver disease (NAFLD), as it is the most common liver disease in developed countries and is strongly associated with obesity, diabetes, liver cirrhosis, and cancer.86–88 In addition, lipid oxidation products such as 4-HNE have been found in fatty liver,87,88 providing motivation to develop a complementary method to directly and selectively monitor potential changes in H2O2 status during NAFLD onset and progression. To establish a diet-induced NAFLD model with hepatic steatosis, 8-week-old male C57BL/6 mice were fed either normal chow (NC) or a high-fat diet (HFD; 60% of kilocalories from fat; Research Diets D12492) for 20 weeks.89,90 As expected, a significant increase in body weight of mice fed with HFD, as compared to those fed with NC (Figure S12), was observed, along with ectopic lipid deposition in livers of HFD mice (Figure S13). Peroxymycin-1 (10 mg/kg) was then introduced to either NC or HFD mice through intraperitoneal injection. The mice were euthanized 4 h after injection and their liver tissues were then harvested. Dot blots of homogenized liver tissue lysates (2 μg) from HFD mice injected with Peroxymycin-1 revealed a higher fluorescence ratio for anti-puromycin to anti-lamin B1 staining (the latter was employed for staining nuclear envelope and hence for normalization of total protein spotted) compared to what was observed for NC mice (Figure 8a), suggesting elevated H2O2 levels in liver tissues of HFD mice compared to NC controls. With these results in hand, we moved on to utilize Peroxymycin-1 for histochemical imaging of H2O2 in fixed tissue. Liver tissues were fixed, sectioned, immunostained, and imaged by confocal fluorescence microscopy. The images obtained show higher fluorescence intensities in the HFD tissue sections compared to NC controls, validating the elevation of H2O2 in the NAFLD disease model induced by HFD feeding (Figure 8b). Negative control experiments without staining primary α-puromycin antibody (Figure 8b) confirm that immunofluorescence from the tissue samples originates from specific interactions between primary and secondary antibodies. Complementary positive control experiments using 4-HNE staining are also consistent with higher levels of global oxidation in liver tissues from HFD mice (Figure 8c). Taken together, these results identify that HFD feeding can trigger an increase in H2O2 in the liver and establish Peroxymycin-1 as a unique chemical tool for directly assessing H2O2 pools with high selectivity and sensitivity in a broad range of cell and tissue specimens.
Figure 8.
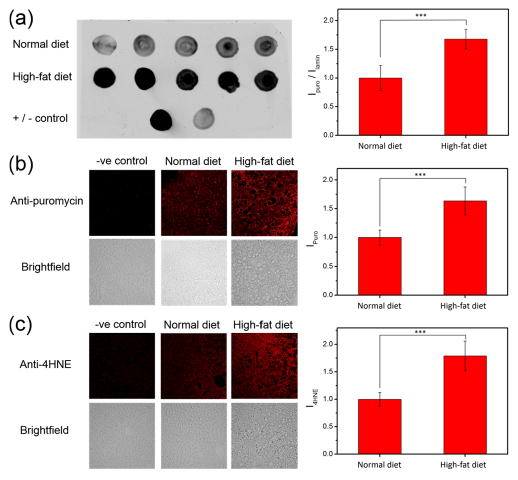
(a) Dot blots of liver tissue lysates from individual normal-chow diet and high-fat diet mice injected with Peroxymycin-1 (10 mg/kg). (b) Immunofluorescence from liver tissues of Peroxymycin-1-treated mice stained with anti-puromycin and anti-mouse-Alexa647 as primary and secondary antibody, respectively. (c) Immunofluorescence from liver tissues of Peroxymycin-1-treated mice stained with anti-4-HNE and anti-goat-Alexa647 as primary and secondary antibody, respectively. Error bars denote SD (n = 5). ***p < 0.001.
CONCLUDING REMARKS
We have presented the design, synthesis, and evaluation of Peroxymycin-1, a first-generation histochemical probe that enables detection of H2O2 across a broad range of cell and tissue specimens. Peroxymycin-1 is prepared by caging the α-amino group of puromycin with H2O2-responsive aryl boronate through a self-immolative carbamate linkage. This caging reaction is highly versatile, as demonstrated by the simple preparation of negative control compound, Ctrl-Peroxymycin-1, by a similar synthetic scheme.
We have highlighted the utility of this reagent by monitoring changes in H2O2 in cell models under oxidative stress or physiological redox signaling conditions, assessing basal levels of H2O2 across a panel of healthy and cancer cell lines where increases in H2O2 correlate with greater metastatic potential and identifying elevations in liver H2O2 levels in a diet-induced mouse model of NAFLD. The permanent and dose-dependent staining properties of Peroxymycin-1 are robust to sample washing and fixation, and additional features of antibody specificity and multivalent binding provide a platform with improved sensitivity relative to standard fluorescent probes. Also, incorporation of immunostaining into the detection platform offers a broader selection of potential H2O2 readouts. By simply changing the dye on secondary antibody, the H2O2 levels can be visualized at different excitation and emission wavelengths and as such should facilitate multianalyte detection using multicolor fluorescence imaging. Moreover, the readouts are not limited to fluorescence and in principle can be extended to different detection modalities such as chemiluminescence or bioluminescence. On balance, one potential drawback to note is that because immunostaining requires fixation of biological specimens, real-time monitoring of H2O2 levels is thus limited. Together with the incorporation time of puromycin into nascent peptides, Peroxymycin-1 may show lower temporal resolution in imaging H2O2 fluxes as compared to fluorescent H2O2 probes.
Nevertheless, with its permanent staining feature, excellent signal-to-noise ratio, and high selectivity and sensitivity, Peroxymycin-1 enables histochemical analysis of H2O2 in a broad range of cell and tissue specimens. Traditional methods for measuring ROS levels in fixed tissues utilize staining of cellular oxidation products such as 4-NHE and nitrotyrosine,66,67 which are inherently indirect assays for ROS, and differences in the expression levels of cellular proteins/lipids across different specimens can lead to variations in determination of ROS levels. More importantly, these assays show no specificity toward a particular ROS. On the other hand, small-molecule-based ROS fluorescent probes like 2′,7′-dichlorodihydrofluorescein (H2DCF) have been utilized for assessing ROS levels in tissues,91 but potential artifacts including low photostability, lack of selectivity toward a particular ROS, propensity for oxidation by cytochrome c and heme peroxidases, and artificial generation of ROS make the use of this probe challenging without proper controls.92 As a result, the direct and specific reaction with H2O2 and the robust performance on H2O2 imaging in a variety of biological specimens suggest that Peroxymycin-1 has value as a first-generation H2O2-specific histochemical probe.
In a broader chemical context, the present work represents a highly versatile and general strategy for developing fixable molecular probes for sensitive histochemical detection of target analytes in biological samples, particularly in fixed specimens. Indeed, puromycin is a promising general scaffold for developing histochemical reagents for a diverse array of biologically relevant analytes by introduction of other responsive triggers, akin to privileged dyes like fluorescein, boron-dipyrromethene (BODIPY), cyanine, and their analogues for studying cellular communications and signaling mediated by the target analytes.
EXPERIMENTAL SECTION
Materials and Reagents.
Puromycin dihydrochloride was purchased from Cayman Chemical. 4-Bromophenylethyl alcohol and 4-(hydroxymethyl)phenylboronic acid pinacol ester were purchased from AK Scientific. 4-Nitrophenyl chloroformate, potassium acetate, N,N-diisopropylethylamine, and 4-(dimethylamino)pyridine were purchased from Sigma–Aldrich. Bis(pinacolato)diboron was purchased from Oakwood Chemical. [1,1′-Bis(diphenylphosphino)-ferrocene]dichloropalladium(II) was purchased from Matrix Scientific. For the materials and reagents for in vitro and biological assays, α-puromycin antibody [3RH11] was purchased from Kerafast. AlexaFluor 488 anti-mouse immunoglobulin G (IgG), AlexaFluor 647 anti-mouse IgG, AlexaFluor 647 anti-goat IgG and Hoechst 33342 were purchased from Invitrogen. Hydrogen peroxide solution (3 wt % in water), potassium superoxide, tert-butyl hydroperoxide (70 wt % in water), paraquat dichloride hydrate, sodium sulfide nonahydrate, glutathione, cysteine, and α-lipoic acid were purchased from Sigma–Aldrich. Sodium hypochlorite was purchased from Fluka. Iron(II) sulfate heptahydrate was purchased from Alfa Aesar. PROLI NONOate was purchased from Cayman Chemical. Dulbecco’s modified Eagle’s medium (DMEM; high glucose and no phenol red), GlutaMAX, fetal bovine serum (FBS), and phosphate-buffered saline (PBS) were purchased from Thermo Fisher Scientific. 4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)phenylethanol93 and 4-nitrophenyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl carbonate94 were synthesized according to literature methods. All other reagents were of analytical grade and were used without further purification. Milli-Q water was used in all experiments unless otherwise stated.
Physical Measurements and Instrumentation.
1H NMR and 13C{1H} spectra were collected at 25 °C on Bruker AVB-400, AVQ-400, and AV-300 spectrometers at the College of Chemistry NMR Facility at the University of California, Berkeley. All chemical shifts are reported in the standard δ notation of parts per million relative to residual solvent peak as an internal reference. Splitting patterns are indicated as follows: br, broad; s, singlet; d, doublet; t, triplet; m, multiplet; dd, doublet of doublets. Low-resolution electrospray mass spectra were recorded on a liquid chromatograph–mass spectrometer (Agilent Technology 6130, quadrupole LC/MS). High-resolution mass spectra were collected at the College of Chemistry Mass Spectrometry Facility at the University of California, Berkeley. Reaction kinetics of the probes with species of interest and other substrates were investigated by LC–MS (Agilent Technology 6130, quadrupole LC/MS) coupled with photodiode array for detection (λ = 275 nm). Confocal microscopic images were recorded on a Zeiss laser scanning microscope 710 with a 40× water-immersion objective lens using Zen 2009 software (Carl Zeiss).
Syntheses.
4-Nitrophenyl [4-(4,4,5,5-Tetramethyl-1,3,2-dioxa-borolan-2-yl)phenyl]ethyl Carbonate.
4-Nitrophenyl chloroformate (189.7 mg, 0.94 mmol) was dissolved in dry dichloromethane (20 mL). 4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)phenylethanol93 (212.3 mg, 0.86 mmol) was added to the solution, followed by dropwise addition of 4-(dimethylamino)pyridine in dichloromethane (1.1 mmol, 2 mL) at 0 °C. The reaction mixture was allowed to reach room temperature and stirred for 2 h. The reaction was quenched by saturated aqueous NaHCO3 solution, and extracted with dichloro-methane. The organic layer was further washed by saturated aqueous NaHCO3 solution two times and once by saturated aqueous NaCl solution, dried over MgSO4, and filtered. Volatile organic solvent was evaporated under reduced pressure, and the crude product was purified by column chromatography on silica gel with hexane/ethyl acetate (4:1 v/v) as eluent, yielding the desired product as a white solid (255.9 mg, 72%). 1H NMR (CDCl3, 300 MHz) δ 8.22 (2H, d, J = 7.8 Hz), 7.79 (2H, d, J = 7.8 Hz), 7.28 (4H, m), 4.48 (2H, t, J = 6.9 Hz), 3.08 (2H, t, J = 6.6 Hz), 1.34 (12H, s). 13C{1H} NMR (CDCl3, 75 MHz) δ 155.5, 152.3, 145.3, 140.0, 135.2, 128.4, 125.2, 121.8, 83.8, 69.4, 35.1, 24.9. LRMS (ESI) m/z [M + Na+]+ calcd for C21H24BNO7N, 436.15; found, 436.1.
Peroxymycin-1.
Puromycin dihydrochloride (20 mg, 36.7 μmol) and 4-nitrophenyl [4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-benzyl carbonate94 (13.3 mg, 33.3 μmol) were dissolved in dry dimethylformamide (4 mL). 4-(Dimethylamino)pyridine in dimethyl-formamide (5 μmol; 1 mL) and N,N-diisopropylethylamine (23.3 μL; 134 μmol) were then added to the reaction mixture, and the solution was stirred at room temperature for 1 h. The reaction was then quenched by saturated aqueous NaHCO3 solution and extracted with ethyl acetate. The organic layer was further washed by saturated aqueous NaHCO3 solution two times and once by saturated aqueous NaCl solution, dried over MgSO4, and filtered. Volatile organic solvent was evaporated under reduced pressure, and the crude product was purified by column chromatography on silica gel with dichloromethane/methanol (9:1 v/v) as eluent, yielding the desired product as a white solid (16.6 mg, 68%). 1H NMR (CDCl3, 400 MHz): δ 8.05 (1H, s), 7.93 (1H, s), 7.77 (2H, d, J = 7.6 Hz), 7.28 (2H, d, J = 7.6 Hz), 7.12 (2H, d, J = 8.0 Hz), 6.84 (2H, d, J = 8.0 Hz), 6.59 (1H, br s), 5.63 (1H, d, J = 6.4 Hz), 5.48 (1H, d, J = 4.4 Hz), 5.02–5.11 (3H, m), 4.74 (1H, m), 4.43 (1H, m), 4.37 (1H, m), 4.02 (1H, s), 3.88 (1H, d, J = 12.8 Hz), 3.76 (3H, s), 3.69 (1H, d, J = 12.8 Hz), 3.10–3.67 (6H, br s), 3.00–3.10 (1H, m), 2.90–3.00 (1H, m), 1.33 (12H, s). 13C{1H} NMR (CDCl3, 100 MHz) δ 172.1, 158.7, 156.2, 154.7, 151.3, 148.4, 139.1, 137.7, 135.0, 130.3, 128.4, 127.0, 121.0, 114.2, 90.9, 84.9, 83.9, 72.7, 67.0, 62.4, 56.6, 55.3, 51.7, 38.3, 24.9. HRMS (ESI) m/z [M + H+]+ calcd for C36H47BN7O9, 732.3528; found, 732.3530.
Ctrl-Peroxymycin-1.
The procedure was similar to that for Peroxymycin-1 except that 4-nitrophenyl [4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]ethyl carbonate (13.7 mg, 33.3 μmol) was used instead of [4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-benzyl carbonate, yielding the desired product as a white solid (15.4 mg, 62%). 1H NMR (CDCl3, 400 MHz) δ 8.05 (1H, s), 7.92 (1H, s), 7.72 (2H, d, J = 7.6 Hz), 7.18 (2H, d, J= 7.6 Hz), 7.12 (2H, d, J = 7.2 Hz), 6.85 (2H, d, J = 8.4 Hz), 6.59 (1H, br s), 5.51 (1H, d, J = 7.2 Hz), 5.47 (1H, s), 5.10 (1H, br s), 4.76 (1H, s), 4.30–4.45 (2H, m), 4.24 (2H, t, J = 6.4 Hz), 4.05 (1H, s), 3.89 (1H, d, J = 12.0 Hz), 3.75 (3H, s), 3.67 (1H, d, J = 12.0 Hz), 3.10–3.67 (6H, br s), 3.00–3.10 (1H, m), 2.85–2.95 (3H, m), 1.31 (12H, s). 13C{1H} NMR (CDCl3, 100 MHz) δ 172.1, 158.7, 156.2, 154.8, 151.0, 148.0, 141.1, 137.7, 135.0, 130.3, 128.4, 121.1, 114.3, 91.0, 84.9, 83.8, 72.9, 65.7, 62.3, 56.5, 55.3, 51.5, 38.2, 35.6, 24.9. HRMS (ESI) m/z [M + H+]+ calcd for C37H49BN7O9, 746.3685; found, 746.3685.
LC–MS Analysis of ROS Reactivity.
Peroxymycin-1 and Ctrl-Peroxymycin-1 (0.3 mM), respectively, were dissolved in phosphate buffer (20 mM, pH 7.4)/methanol solution mixture (2:1 v/v) with 3 vol % DMSO and allowed to stand at room temperature. At predetermined time intervals, aliquot of the solution mixture were taken for LC–MS analysis. Separation was achieved by gradient elution from 5% to 100% MeOH in water (constant 0.1 vol % formic acid) over 8 min, isocratic elution with 100% MeOH from 8 to 12 min, and returned to initial conditions and equilibrated for 3 min. The LC chromatograms were recorded by monitoring absorption at 275 nm. For reactions with H2O2, probes (0.3 mM) were dissolved in phosphate buffer (20 mM, pH 7.4)/methanol solution mixture (2:1 v/v) with 3 vol % DMSO. H2O2 was then added to the solution mixture (final concentration = 0.1 mM), and the reaction mixture was allowed to stand at room temperature. At predetermined time intervals, aliquots of the solution mixture were taken for LC–MS analysis as previously described. Other ROS were generated as described previously93 and then added to the solution of Peroxymycin-1 with final concentration of 0.1 mM (except peroxynitrite at 0.05 mM).80 An aliquot of the solution was then taken for LC–MS analysis as described above.
Cell Culture.
Cells were grown in the UC Berkeley Tissue Culture Facility. Human cervical epithelial carcinoma (HeLa), human epidermoid carcinoma (A-431), and human breast carcinoma (MDA-MB-231 and MDA-MB-468) were maintained in DMEM medium (high glucose) supplemented with GlutaMAX and 10 vol % FBS. Human breast carcinoma (MCF-7) was maintained in DMEM medium (high glucose) supplemented with GlutaMAX, 10 vol % FBS, 1 vol % nonessential amino acids, and 1% sodium pyruvate. Human breast carcinoma (HS 578T) was maintained in DMEM medium (high glucose) supplemented with GlutaMAX, 10 vol % FBS, 1% sodium pyruvate and insulin (10 μg/mL). Human nontumorigenic breast epithelial cells (MCF-10A) were maintained in DMEM/F12 [with L-glutamate, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and phenol red] supplemented with 5 vol % horse serum, EGF (20 ng/mL), hydrocortisone (0.5 mg/mL), cholera toxin (100 ng/mL), insulin (10 μg/mL), and 1 vol % penicillin/streptomycin. Human normal breast epithelial cells (HS 578Bst) were maintained in ATCC Hybri-Care Medium (46-X) supplemented with sodium bicarbonate (1.5 g/mL), EGF (30 ng/mL), and 10 vol % FBS. All cells were incubated in 5% CO2 humidified air and were subcultured when 80% confluence was reached. For cell imaging experiments, the cells were plated on 8-well Lab Tek borosilicate chambered coverglass slides (Nunc) or 24-well plates with 12 mm coverslips and allowed to grow to ca. 60% confluency before cell imaging experiments were performed.
Immunohistochemical Staining Protocol for Cells.
Cells were incubated with the probe compound (1 μM) for indicated time intervals. The cells were then washed three times with PBS solution and fixed by 4% paraformaldehyde at room temperature for 10 min. The cells were washed with PBS solution and PBS solution + 0.1 vol % Triton X-100, followed by incubation with α-puromycin antibody [3RH11] (Kerafast; 1:500 v/v) in PBS solution containing 10 vol % FBS and 0.1 vol % Triton X-100 at 37 °C for 30 min. Subsequently, the cells were washed with PBS solution and PBS solution + 0.1 vol %-Triton X-100 and stained by α-mouse secondary antibody-Alexa Fluor 488 (1:100 v/v) in PBS solution containing 10 vol % FBS and 0.1 vol % Triton X-100 at 37 °C for 30 min. The cells were then washed with PBS solution and stained by Hoechst 33342 at room temperature for 10 min. After that, the cells were washed three times with PBS solution and imaged by confocal fluorescence microscopy.
Stimulation and Inhibition of H2O2 and/or NO Production in A431 Cells.
EGF stimulation was carried out by pretreatment of A431 cells with EGF (100 ng/mL) for 20, 40, and 60 min; samples were then washed with PBS and incubated with Peroxymycin-1 (1 μM) for 4 h. For the study with l-NAME, A431 cells were pretreated with l-NAME (100 μM) for 25 min and then washed with PBS and incubated with EGF (100 ng/mL) for 40 min, followed by washing and incubation with Peroxymycin-1 (1 μM) for 4 h. For the DPI experiment, cells were pretreated with a mixture of DPI (5 μM) and EGF (100 ng/mL) for 40 min, followed by incubation with a mixture of DPI (5 μM) and Peroxymycin-1 (1 μM) for 4 h. For inhibition of H2O2 production by gp91 Tat peptide, cells were pretreated with gp91 Tat peptide (100 μM) for 30 min and then incubated with a mixture of gp91 Tat peptide (100 μM) and Peroxymycin-1 (1 μM) for 4 h.
Confocal Fluorescence Microscopy Imaging.
Confocal fluorescence cell imaging was performed with a Zeiss laser scanning microscope 710 with a 40× water-immersion objective lens using Zen 2009 software (Carl Zeiss). Hoechst 33342 was excited with a 405 nm diode laser, and emission was collected on a META detector between 450 and 500 nm. Alexa Fluor 488 was excited at 488 nm with an Ar laser, and emission was collected on a META detector between 500 and 625 nm. Alexa Fluor 647 was excited with a 633 nm HeNe laser, and emission was collected on a META detector between 638 and 759 nm. PF2 was excited at 488 nm with an Ar laser, and emission was collected on a META detector between 493 and 630 nm. MitoPY1 was excited at 514 nm, and emission was collected on a META detector between 525 and 640 nm. PBS solution was used as the imaging buffer for all confocal experiments. Image analysis was performed by use of ImageJ. A region of interest (ROI) was created, and cellular fluorescence intensity was measured. The reported cellular fluorescence intensity was determined by averaging the measured intensity of five different ROIs from at least three different images in triplicate experiments. Statistical analyses were performed with a two-tailed Student’s t-test (MS Excel).
Animal Work.
Eight-week-old male C57BL/6 mice (Jackson Laboratory) were fed with either standard chow or high-fat diet (60% of kilocalories from fat; Research Diets D12492) for 20 weeks. Body weight of the mice was measured weekly. All animal studies were approved by and performed according to the guidelines of the Animal Care and Use Committee of the University of California, Berkeley.
Histochemical Detection of H2O2 in Liver Tissues of Mice Fed with Normal Chow or High-Fat Diet.
Normal chow (NC) and high-fat diet (HFD) mice, respectively, were injected intraperitoneally with 10 mg/kg peroxymycin in PBS/PET [60% poly(ethylene glycol) 400, 30% ethanol, and 10% Tween-80] solution mixture (1:1 v/v). After 4 h, mice were euthanized and liver tissue was harvested. For dot blots, 10 mg of liver tissue was homogenized in RIPA buffer. Cleared lysates were quantified and spotted on a nitrocellulose membrane at protein content of 2 μg. After blocking, the membrane was incubated with anti-puromycin and anti-lamin B (Abcam 16048) primary antibodies (1:1000 v/v) at 4 °C overnight. Then the dot blot was incubated with dye-conjugated secondary antibodies (Invitrogen, 1:10 000 v/v) for 1 h at room temperature and visualized on a Licor Odyssey imaging system. ImageJ was used for blot quantification.
For confocal fluorescence imaging of liver tissue sections, liver tissues were fixed by 4% paraformaldehyde at 4 °C overnight. Then the tissues were moved into 30% sucrose solution for cryoprotection at 4 °C overnight. The tissues were then embedded in tissue freezing medium (TFM), and 30 μm sections were taken on a cryostat and subsequently adhered to slides. After being washed with PBS, the sections were incubated in PBS containing 0.5% bovine serum albumin (BSA) and 0.5% Triton X-100 at room temperature for 20 min, followed by incubation with anti-mouse IgG Fab (1:10 v/v) in PBS containing 0.5% BSA and 0.5% Triton X-100 at room temperature for 2 h. After that, the sections were washed with PBS containing 0.5% BSA and 0.5% Triton X-100 for 5 min at room temperature three times and then incubated with donkey serum (10 vol %) in PBS containing 0.5% BSA and 0.5% Triton X-100 at room temperature for 2 h, followed by incubation with mouse α-puromycin antibody (1:100 v/v) in PBS containing 0.5% BSA and 0.5% Triton X-100 at room temperature for 1 h. Then the sections were washed with PBS containing 0.5% BSA and 0.5% Triton X-100 at room temperature for 5 min three times and incubated with anti-mouse-Alexa647 antibody (1:250, v/v) in PBS containing 0.5% BSA and 0.5% Triton X-100 at room temperature for 1 h. The sections were washed with PBS containing 0.5% BSA and 0.5% Triton X-100 at room temperature for 10 min three times, mounted with coverslips, and imaged by confocal fluorescence microscopy.
For 4-HNE staining, after fixation, cryoprotection, and sectioning, the sections were washed with PBS, incubated in PBS containing 0.5% BSA and 0.5% Triton X-100 at room temperature for 20 min, and then incubated with donkey serum (10 vol %) in PBS containing 0.5% BSA and 0.5% Triton X-100 at room temperature for 2 h. After that, the sections were incubated with goat anti-4HNE antibody (1:100 v/v) in PBS containing 0.5% BSA and 0.5% Triton X-100 at room temperature for 1 h, washed with PBS containing 0.5% BSA and 0.5% Triton X-100 at room temperature for 5 min three times, and incubated with anti-goat-Alexa647 antibody (1:250 v/v) in PBS containing 0.5% BSA and 0.5% Triton X-100 at room temperature for 1 h. The sections were washed with PBS containing 0.5% BSA and 0.5% Triton X-100 at room temperature for 10 min three times, mounted with coverslips, and imaged by confocal fluorescence microscopy.
Supplementary Material
ACKNOWLEDGMENTS
We thank the NIH (GM 79465 and ES 004705) for support of this work. C.Y.-S.C thanks the Croucher Foundation for a postdoctoral fellowship. K.S. thanks the IVRI fund. C.J.C. is an Investigator of the Howard Hughes Medical Institute. We thank Alison Killilea and Carissa Tasto (UC Berkeley Tissue Culture Facility) for expert technical assistance.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b02279.
One scheme outlining synthesis of Peroxymycin-1 and Ctrl-Peroxymycin-1; 13 figures showing stability of Peroxymycin-1, LC–MS analysis of reaction mixture, confocal fluorescence microscopic images of HeLa and A431 cells stained with Peroxymycin-1, puromycin, Ctrl-Peroxymycin-1, PF2, or MitoPY1 and of different breast cells stained with Peroxymycin-1 or puromycin, and body weight and liver tissues from mice fed with NC or HFD (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Winterbourn CC Nat. Chem. Biol 2008, 4, 278. [DOI] [PubMed] [Google Scholar]
- (2).Rada B; Leto N Contrib. Microbiol 2008, 15, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Rhee SG Science 2006, 312, 1882. [DOI] [PubMed] [Google Scholar]
- (4).Stone JR; Yang S Antioxid. Redox Signaling 2006, 8, 243. [DOI] [PubMed] [Google Scholar]
- (5).D’Autréaux B; Toledano MB Nat. Rev. Mol. Cell Biol 2007, 8, 813. [DOI] [PubMed] [Google Scholar]
- (6).Dickinson BC; Chang CJ Nat. Chem. Biol 2011, 7, 504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Murphy MP; Holmgren A; Larsson NG; Halliwell B; Chang CJ; Kalyanaraman B; Rhee SG; Thornalley PJ; Partridge L; Gems D; Nyström T; Belousov V; Schumacker PT; Winterbourn CC Cell Metab. 2011, 13, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lambeth JD Nat. Rev. Immunol 2004, 4, 181. [DOI] [PubMed] [Google Scholar]
- (9).Sundaresan M; Yu Z-X; Ferrans VJ; Irani K; Finkel T Science 1995, 270, 296. [DOI] [PubMed] [Google Scholar]
- (10).Bae YS; Kang SW; Seo MS; Baines IC; Tekle E; Chock PB; Rhee SG J. Biol. Chem. 1997, 272, 217. [PubMed] [Google Scholar]
- (11).Dickinson BC; Peltier J; Stone D; Schaffer DV; Chang CJ Nat. Chem. Biol 2011, 7, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ohba M; Shibanuma M; Kuroki T; Nose KJ Cell Biol 1994, 126, 1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kimura T; Okajima F; Sho K; Kobayashi I; Kondo Y Endocrinology 1995, 136, 116. [DOI] [PubMed] [Google Scholar]
- (14).Mukhin YV; Garnovskaya MN; Collinsworth G; Grewal JS; Pendergrass D; Nagai T; Pinckney S; Greene EL; Raymond JR Biochem. J 2000, 347, 61. [PMC free article] [PubMed] [Google Scholar]
- (15).Gopalakrishna R; Jaken S Free Radical Biol. Med 2000, 28, 1349. [DOI] [PubMed] [Google Scholar]
- (16).Varadarajan S; Yatin S; Aksenova M; Butterfield DA J. Struct. Biol 2000, 130, 184. [DOI] [PubMed] [Google Scholar]
- (17).Jacob C; Giles GI; Giles NM; Sies H Angew. Chem., Int. Ed 2003, 42, 4742. [DOI] [PubMed] [Google Scholar]
- (18).Paulsen CE; Carroll KS Chem. Rev. 2013, 113, 4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Brewer TF; Garcia FJ; Onak CS; Carroll KS; Chang CJ Annu. Rev. Biochem. 2015, 84, 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lee SR; Kwon KS; Kim SR; Rhee SG J. Biol. Chem 1998, 273, 15366. [DOI] [PubMed] [Google Scholar]
- (21).Salmeen A; Andersen JN; Myers MP; Meng TC; Hinks JA; Tonks NK; Barford D Nature 2003, 423, 769. [DOI] [PubMed] [Google Scholar]
- (22).Guyton KZ; Liu Y; Gorospe M; Xu Q; Holbrook NJ J. Biol. Chem 1996, 271, 4138. [DOI] [PubMed] [Google Scholar]
- (23).Schmidt KN; Amstad P; Cerutti P; Baeuerle PA Chem. Biol 1995, 2, 13. [DOI] [PubMed] [Google Scholar]
- (24).Avshalumov MV; Rice ME Proc. Natl. Acad. Sci. U. S. A 2003, 100, 11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Finkel T; Serrano M; Blasco MA Nature 2007, 448, 767. [DOI] [PubMed] [Google Scholar]
- (26).Baynes JW Diabetes 1991, 40, 405. [DOI] [PubMed] [Google Scholar]
- (27).Inoguchi T; Li P; Umeda F; Yu HY; Kakimoto M; Imamura M; Aoki T; Etoh T; Hashimoto T; Naruse M; Sano H; Utsumi H; Nawata H Diabetes 2000, 49, 1939. [DOI] [PubMed] [Google Scholar]
- (28).Andersen JK Nat. Rev. Neurosci 2004, 10, S18. [Google Scholar]
- (29).Park L; Zhou P; Pitstick R; Capone C; Anrather J; Norris EH; Younkin L; Younkin S; Carlson G; McEwen BS; Iadecola C Proc. Natl. Acad. Sci U. S. A 2008, 105, 1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Giepmans BNG; Adams SR; Ellisman MH; Tsien RY Science 2006, 312, 217. [DOI] [PubMed] [Google Scholar]
- (31).Chan J; Dodani SC; Chang CJ Nat. Chem 2012, 4, 973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kaur A; Haghighatbin MA; Hogan CF; New EJ Chem. Commun 2015, 51, 10510. [DOI] [PubMed] [Google Scholar]
- (33).Kaur A; Kolanowski JL; New EJ Angew. Chem. Int. Ed 2016, 55, 1602. [DOI] [PubMed] [Google Scholar]
- (34).Kolanowski JL; Liu F; New EJ Chem. Soc. Rev 2018, 47, 195. [DOI] [PubMed] [Google Scholar]
- (35).Tang Y; Lee D; Wang J; Li G; Yu J; Lin W; Yoon J Chem. Soc. Rev 2015, 44, 5003. [DOI] [PubMed] [Google Scholar]
- (36).Chang MCY; Pralle A; Isacoff EY; Chang CJ J. Am. Chem. Soc 2004, 126, 15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Miller EW; Albers AE; Pralle A; Isacoff EY; Chang CJ J. Am. Chem. Soc 2005, 127, 16652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Miller EW; Tulyathan O; Isacoff EY; Chang CJ Nat. Chem. Biol 2007, 3, 263. [DOI] [PubMed] [Google Scholar]
- (39).Srikun D; Miller EW; Domaille DW; Chang CJ J. Am. Chem. Soc 2008, 130, 4596. [DOI] [PubMed] [Google Scholar]
- (40).Dickinson BC; Chang CJ J. Am. Chem. Soc 2008, 130, 9638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Bao L; Avshalumov MV; Patel JC; Lee CR; Miller EW; Chang CJ; Rice MEJ J. Neurosci 2009, 29, 9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Srikun D; Albers AE; Nam CI; Iavarone AT; Chang CJ J. Am. Chem. Soc 2010, 132, 4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Dickinson BC; Huynh C; Chang CJ J. Am. Chem. Soc 2010, 132, 5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Miller EW; Dickinson BC; Chang CJ Proc. Natl. Acad. Sci. U. S.A 2010, 107, 15681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Dickinson BC; Peltier J; Stone D; Schaffer DV; Chang CJ Nat. Chem. Biol 2011, 7, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lin VS; Lippert AR; Chang CJ Proc. Natl. Acad. Sci. U. S. A 2013, 110, 7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lippert AR; Van de Bittner GC; Chang CJ Acc. Chem. Res 2011, 44, 793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Onoda M; Uchiyama S; Endo A; Tokuyama H; Santa T; Imai K Org. Lett 2003, 5, 1459. [DOI] [PubMed] [Google Scholar]
- (49).Lo LC; Chu CY Chem. Commun 2003, 2728. [DOI] [PubMed] [Google Scholar]
- (50).Maeda H; Fukuyasu Y; Yoshida S; Fukuda M; Saeki K; Matsuno H; Yamauchi Y; Yoshida K; Hirata K; Miyamoto K Angew. Chem., Int. Ed 2004, 43, 2389. [DOI] [PubMed] [Google Scholar]
- (51).Lee MH; Kim JS; Sessler JL Chem. Soc. Rev 2015, 44, 4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Chen X; Tian X; Shin I; Yoon J Chem. Soc. Rev 2011, 40, 4783. [DOI] [PubMed] [Google Scholar]
- (53).Yang Y; Zhao Q; Feng W; Li F Chem. Rev 2013, 113, 192. [DOI] [PubMed] [Google Scholar]
- (54).Cochemé HM; Quin C; McQuaker SJ; Cabreiro F; Logan A; Prime TA; Abakumova I; Patel JV; Fearnley IM; James AM; Porteous CM; Smith RA; Saeed S; Carre JE; Singer M; Gems D; Hartley RC; Partridge L; Murphy MP Cell Metab 2011, 13, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Smith RA; Hartley RC; Murphy MP Antioxid. Redox Signaling 2011, 15, 3021. [DOI] [PubMed] [Google Scholar]
- (56).Cochemé HM; Logan A; Prime TA; Abakumova I; Quin C; McQuaker SJ; Patel JV; Fearnley IM; James AM; Porteous CM; Smith RA; Hartley RC; Partridge L; Murphy MP Nat. Protoc 2012, 7, 946. [DOI] [PubMed] [Google Scholar]
- (57).Wen Y; Liu K; Yang H; Li Y; Lan H; Liu Y; Zhang X; Yi T Anal. Chem 2014, 86, 9970. [DOI] [PubMed] [Google Scholar]
- (58).Wen Y; Liu K; Yang H; Liu Y; Chen L; Liu Z; Huang C; Yi T Anal. Chem 2015, 87, 10579. [DOI] [PubMed] [Google Scholar]
- (59).Zhang W; Liu W; Li P; Huang F; Wang H; Tang B Anal. Chem 2015, 87, 9825. [DOI] [PubMed] [Google Scholar]
- (60).Xu J; Zhang Y; Yu H; Gao X; Shao S Anal. Chem 2016, 88, 1455. [DOI] [PubMed] [Google Scholar]
- (61).Liu J; Ren J; Bao X; Gao W; Wu C; Zhao Y Anal. Chem 2016, 88, 5865. [DOI] [PubMed] [Google Scholar]
- (62).Yuan L; Lin W; Xie Y; Chen B; Zhu SJ Am. Chem. Soc 2012, 134, 1305. [DOI] [PubMed] [Google Scholar]
- (63).Masanta G; Heo CH; Lim CS; Bae SK; Cho BR; Kim HM Chem. Commun 2012, 48, 3518. [DOI] [PubMed] [Google Scholar]
- (64).Kumar M; Kumar N; Bhalla V; Sharma PR; Qurishi Y Chem. Commun 2012, 48, 4719. [DOI] [PubMed] [Google Scholar]
- (65).Yi L; Wei L; Wang R; Zhang C; Zhang J; Tan T; Xi Z Chem. - Eur. J 2015, 21, 15167. [DOI] [PubMed] [Google Scholar]
- (66).Tuder RM; Zhen L; Cho CY; Taraseviciene-Stewart L; Kasahara Y; Salvemini D; Voelkel NF; Flores SC Am. J. Respir. Cell Mol. Biol 2003, 29, 88. [DOI] [PubMed] [Google Scholar]
- (67).McCormack AL; Atienza JG; Johnston LC; Andersen JK; Vu S; Di Monte DA J. Neurochem 2005, 93, 1030. [DOI] [PubMed] [Google Scholar]
- (68).Starck SR; Green HM; Alberola-Ila J; Roberts RW Chem. Biol 2004, 11, 999. [DOI] [PubMed] [Google Scholar]
- (69).Liu J; Xu Y; Stoleru D; Salic A Proc. Natl. Acad. Sci. U. S. A 2012, 109, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Fontaine SD; DiPasquale AG; Renslo AR Org. Lett 2014, 16, 5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Fontaine SD; Spangler B; Gut J; Lauterwasser EMW; Rosenthal PJ; Renslo AR ChemMedChem 2015, 10, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Buhr F; Kohl-Landgraf J; tom Dieck S; Hanus C; Chatterjee D; Hegelein A; Schuman EM; Wachtveitl J; Schwalbe H Angew. Chem., Int. Ed 2015, 54, 3717. [DOI] [PubMed] [Google Scholar]
- (73).Spangler B; Morgan CW; Fontaine SD; Vander Wal MV; Chang CJ; Wells JA; Renslo AR Nat. Chem. Biol 2016, 12, 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Ge J; Zhang CW; Ng XW; Peng B; Pan S; Du S; Wang D ; Li L; Lim KL; Wohland T; Yao SQ Angew. Chem., Int. Ed 2016, 55, 4933. [DOI] [PubMed] [Google Scholar]
- (75).Du S; Wang D; Lee J-S; Peng B; Ge J; Yao SQ Chem. Commun 2017, 53, 8443. [DOI] [PubMed] [Google Scholar]
- (76).Amir RJ; Pessah N; Shamis M; Shabat D Angew. Chem., Int. Ed 2003, 42, 4494. [DOI] [PubMed] [Google Scholar]
- (77).Sagi A; Weinstain R; Karton N; Shabat DJ Am. Chem. Soc 2008, 130, 5434. [DOI] [PubMed] [Google Scholar]
- (78).Karton-Lifshin N; Segal E; Omer L; Portnoy M; Satchi-Fainaro R; Shabat DJ Am. Chem. Soc 2011, 133, 10960. [DOI] [PubMed] [Google Scholar]
- (79).Alouane A; Labruère R; Le Saux T; Schmidt F; Jullien L Angew. Chem. Int. Ed 2015, 54, 7492. [DOI] [PubMed] [Google Scholar]
- (80).Szabo C; Ischiropoulos H; Radi R Nat. Rev. Drug Discovery 2007, 6, 662. [DOI] [PubMed] [Google Scholar]
- (81).Clark HA; Kopelman R; Tjalkens R; Philbert MA Anal. Chem 1999, 71, 4837. [DOI] [PubMed] [Google Scholar]
- (82).Cobbold PH; Rink TJ Biochem. J 1987, 248, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Fritschy JM; Härtig W Immunofluorescence In eLS (formerly the Encyclopedia of Life Sciences); John Wiley & Sons Ltd.: Chichester, U.K., 2001; DOI: 10.1038/npg.els.0001174. [DOI] [Google Scholar]
- (84).Hamanaka RB; Chandel NS Trends Biochem. Sci 2010, 35, 505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Seo YH; Carroll KS Proc. Natl. Acad. Sci. U. S. A 2009, 106, 16163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Uppal V; Mansoor S; Furuya KN Curr. Gastroenterol. Rep 2016, 18, 24. [DOI] [PubMed] [Google Scholar]
- (87).Hardy T; Oakley F; Anstee QM; Day CP Annu. Rev. Pathol.: Mech. Dis 2016, 11, 451. [DOI] [PubMed] [Google Scholar]
- (88).Browning JD; Horton JD J. Clin. Invest 2004, 114, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Wang CY; Liao JK Methods Mol. Biol 2012, 821, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Sato A; Kawano H; Notsu T; Ohta M; Nakakuki M; Mizuguchi K; Itoh M; Suganami T; Ogawa Y Diabetes 2010, 59, 2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).McLellan ME; Kajdasz ST; Hyman BT; Bacskai BJ J. Neurosci 2003, 23, 2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Wang X; Fang H; Huang Z; Shang W; Hou T; Cheng A; Cheng HJ Mol. Med 2013, 91, 917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Carroll V; Michel BW; Blecha J; VanBrocklin H; Keshari K; Wilson D; Chang CJ J. Am. Chem. Soc 2014, 136, 14742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Jourden JLM; Cohen SM Angew. Chem. Int. Ed 2010, 49, 6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.