

Abstract
The use of advanced polarizable potentials in quantum mechanical/molecular mechanical (QM/MM) simulations has been shown to improve the overall accuracy of the calculation. We have developed a density-based potential called the Gaussian electrostatic model (GEM), which has been shown to provide very accurate environments for QM wave functions in QM/MM. In this contribution we present a new implementation of QM/GEM that extends our implementation to include all components (Coulomb, exchange—repulsion, polarization, and dispersion) for the total intermolecular interaction energy in QM/MM calculations, except for the charge-transfer term. The accuracy of the method is tested using a subset of water dimers from the water dimer potential energy surface reported by Babin et al. (J. Chem. Theory Comput. 2013 9, 5395—5403). Additionally, results of the new implementation are contrasted with results obtained with the classical AMOEBA potential. Our results indicate that GEM provides an accurate MM environment with average root-mean-square error <0.15 kcal/mol for every intermolecular interaction energy component compared with SAPT2+3/aug-cc-pVTZ reference calculations.
Graphical Abstract
The accurate and reliable study of large systems, such as biomolecules, remains a challenge for the quantum chemistry community. Hybrid QM/MM simulations have become one of the standard methods for the study of reaction mechanisms in biochemical and condensed-phase chemical systems.1–3 The accuracy of these simulations depends on two factors, the level of theory/basis set employed for the QM subsystem4–10 and the classical potential for the MM subsystem.11–14 The former has received a significant amount of attention, whereas the latter has lagged behind. Most classical force fields approximate the energy of the system by summing bonded and nonbonded interactions. Conventional non-polarizable potentials generally use atom-centered point charges to approximate the Coulomb interactions and a Lennard-Jones function for van der Waals (vdW) interactions. The use of point charges in close proximity to the QM wave function can result in the overpolarization of the QM subsystem.15,16 This problem can be addressed by using delocalized charges or by using molecular density-based potentials.11,17–20 Several methods that reproduce the electronic charge density have been proposed to improve the accuracy of electrostatic interactions.21–24 New force fields have been introduced to prevent the loss of anisotropy by employing distributed multipoles25–27 yet have failed to provide correct charge penetration effects at close range.28,29 However, this shortcoming can be avoided by employing damping functions or by a continuous description of the molecular charge density.11,13
The Gaussian electrostatic model (GEM), has been developed to provide an accurate potential based on molecular electronic densities.30–33 GEM uses the density fitting formalism to expand the molecular density with Hermite Gaussian functions. In this method, the molecular electronic density fitting is performed by the minimization of the error of the Coulomb self-interaction energy,30,34–37 where the approximate density is expanded with the use of auxiliary Hermite Gaussian basis functions (ABS). After the calculation of the Hermite coefficients with the density fitting procedure, distributed multipoles can also be obtained.31,38,39 The fitted densities are then employed to calculate each term in the QM energy decomposition analysis (EDA), that is, Coulomb, exchange, polarization, charge transfer, and dispersion.31 GEM has been shown to produce accurate energies and forces for a range of systems.11,31,32 The present method is similar in spirit to the polarizable density embedding (PDE).40,41 Recently, an approximate version of GEM, termed GEM*, has been developed to enable the use of the GEM densities in molecular dynamics (MD) simulations.42 This new force field has been implemented in a modified version of pmemd, pmemd.gem, in the AMBER suite of programs. The functional form for GEM* combines frozen core contributions (Coulomb and exchange—repulsion) from GEM, with the polarization, (modified) vdW, and bonded terms from AMOEBA. It has also been previously shown that the use of GEM in a QM/MM implementation can provide a significantly more accurate electrostatic environment.11 However, that work focused exclusively on the electrostatic interactions between the QM and MM subsystems.
In this work, we present the first implementation of GEM for QM/MM calculations, QM/GEM, which involves all of the components of the total intermolecular interaction energy, except for the charge-transfer term. To our knowledge, this is the first implementation of a QM/MM program where the MM environment is explicitly represented by molecular electron density used to calculate separate Coulomb, exchange, polarization, and dispersion contributions.
The total energy of the system in QM/GEM calculations can be written as
| 1 |
The last term in eq 1 corresponds to the intermolecular interactions between the QM and MM subsystems, which can be separated as
| 2 |
where is the Coulomb interaction, corresponds to the exchange—repulsion between the GEM densities and the QM wave function, and and represent the polarization and dispersion between the two subsystems. In this implementation the exchange— repulsion component can be computed after a successful SCF cycle
| 3 |
where Kexch is the proportionality parameter, ρA(rA) denotes the electronic density of molecule A corresponding to the QM subsystem, and is the GEM density for molecule B in the MM subsystem. Alternatively, the exchange—repulsion term can be explicitly included in the effective Hamiltonian, Heff, similar to recent work by Giovannini et al.43 In our implementation, the modified external potential includes both the GEM Coulomb and exchange fields (see the Supporting Information)
| 4 |
| 5 |
This implementation of QM/GEM uses LICHEM12 to interface a modified version of Psi4 to calculate the required three center Coulomb and overlap integrals, with TINKER to calculate the polarization and dispersion terms (see the Supporting Information).44–46 In this initial implementation, the GEM densities for each MM subsystem are fitted individually (at the ωB97XD/aug-cc-pVTZ level) for each dimer instead of using the same fitted density for every MM subsystem as in our previous implementation.11 Consequently, two different scenarios are taken into account for each dimer within the subset, first monomer under the field of the second monomer and vice versa. These fitted densities are used for the evaluation of the Coulomb and overlap integrals needed for the calculation of and . The fitting is performed using either Cholesky decomposition (Chol) or Tychonov regularization (Tych).47 For the latter, a total molecular charge constraint (via Lagrange multipliers) is applied to ensure that the density integrates to the correct number of electrons.11 Furthermore, the accuracy of the fitting with the Tychonov regularization is tuned by optimizing the eigenvalue cutoff, λ.11 We have previously shown that the optimal fitting for Coulomb may not provide the best reproduction of the density for the exchange—repulsion interaction.11 Thus a case is also considered where sets of fitting coefficients are obtained for each term (λ1 for Coulomb and λ2 for exchange—repulsion).
The exchange—repulsion term is calculated by means of the Wheatley—Price overlap model via the two implemented methods described above (see eqs 3—5 and the Supporting Information),48,49 The proportionality parameter for the exchange term after SCF (eq 3), Kexch, is calculated via least-squares fitting and is fitted to reproduce the corresponding EDA components obtained from SAPT2+3 for a fitting set that correspond to the 10 water dimers, as reported by Tschumper et al. (see the Supporting Information)50. The proportionality parameter for the combined Coulomb and exchange— repulsion embedding (eqs 4 and 5) has been fitted by an iterative process. Here the exchange—repulsion embedding included in Ĥeff affects the QM density and thus results in a different response depending on the value of . Therefore, is fitted iteratively using least-squares fitting to the SAPT2+3 reference for the same 10 water dimer set (see the Supporting Information). The calculated proportionality parameters are similar to previously reported parameters for GEM* and GEM.11,42
The QM/MM polarization component is computed based on the AMOEBA model by approximating the QM wave function using the procedure employed in LICHEM including Tholé damping. This method has been show to account for >80% of the total QM/MM polarization component.12 The dispersion component is calculated by employing three terms of the dispersion expansion and fitting the required Cn (n = 6, 8, 10) coefficients to the same 10 dimer reference set as for the exchange—repulsion term. The results for the total intermo-lecular interactions calculated with QM/GEM are compared with the total intermolecular energy obtained from the SAPT2+3 reference and with QM/AMOEBA.12,27,51–53 Therefore, this implementation comprises direct embedding from the two first-order terms ( and ) and two responses from the MM environment ( and ) calculated after the SCF cycle. All QM/GEM calculations have been performed using the ωB97XD/aug-cc-pVTZ level for the QM subsystem.44,54–56 The GEM density has been fitted using two auxiliary basis sets.11 Results for the A2DG auxiliary basis set are discussed below (results with A2 are provided in the Supporting Information).
The capability of this initial QM/GEM implementation is assessed by comparing total and component-wise intermolec-ular interactions for a subset of the water dimer potential energy surface (PES) reported by Paesani and coworkers.57 A QM EDA has been performed on the full data set at the SAPT2+3/aug-cc-pVTZ level, as implemented in Psi4 (see the Supporting Information).44,54–56,58 The population of the Paesani data set has been reduced to the subset of water dimers that involve monomers with internal structures similar to the AMOEBA equilibrium geometry (see the Supporting Information).59 The distances and angles that are taken into account and their nomenclature are depicted in Figure S.2. The final subset corresponds to dimers with AMOEBA angle and bond energies ≤10 kcal/mol, H—O—H angles of 98.0 < θ < 118.0°, H—O bond distances of 0.87 < r < 1.07 Å, and O—O intermonomer distances >3.2 Å (see Figure S.3 and Table S.1). The resulting subset contains 4074 dimers, 2356(1718) of which have attractive(repulsive) total intermolecular interaction energy. The resulting subset of water dimers is further subdivided into 10 different clusters using the k-means clustering method, as implemented in the Scikit-learn60 package to facilitate the error analysis. The centroids for the clusters are defined by using the same geometrical criteria that are used to form the subset (see the Supporting Information). The nomenclature of the clusters, their population, and the intermolecular O—O distance of the centroids () are depicted in Table 1.
Table 1.
Clusters Obtained with the k-Means Clustering Approach, Their Populations and the O—O Distances of the Centroids
| cluster | population | (Å) |
|---|---|---|
| 0 | 347 | 5.332 |
| 1 | 464 | 4.882 |
| 2 | 894 | 4.381 |
| 3 | 321 | 5.402 |
| 4 | 378 | 5.312 |
| 5 | 323 | 5.383 |
| 6 | 447 | 5.429 |
| 7 | 267 | 5.351 |
| 8 | 264 | 5.353 |
| 9 | 369 | 5.423 |
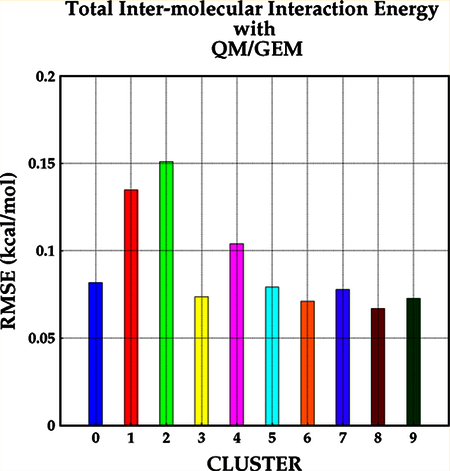
Figure 1 shows the calculated maximum unsigned error (MAX) and root-mean-square error (RMSE) obtained with Tychonov regularization using λ1 = 0.00003 for Coulomb interactions and λ2 = 0.001 for exchange—repulsion interactions. The component-by-component comparison is presented for the Coulomb () and exchange—repulsion () terms. The errors for the polarization and dispersion terms () are presented together to enable their comparison with the induction and dispersion terms from the SAPT2+3 reference calculations (see the Supporting Information for detailed decomposition results). The error analysis for results obtained via Cholesky decomposition and Tychonov regularization using only λ1 and exchange—repulsion terms computed after SCF cycle are provided in the Supporting Information. The calculated RMSE (MAX) for the Coulomb term is below 0.05 kcal/mol (0.2 kcal/mol), with the exception of MAX = 0.3 kcal/mol for cluster 2, compared with the Cholesky decomposition, where three clusters are observed to have MAX > 0.2 kcal/mol (1, 2, and 4). About 92% of all 4074 water dimers exhibit unsigned error (UE) for ≤ 0.05 kcal/mol when the fitting is performed with the Tychonov regularization compared with 87% of dimers fitted using the Cholesky decomposition.
Figure 1.
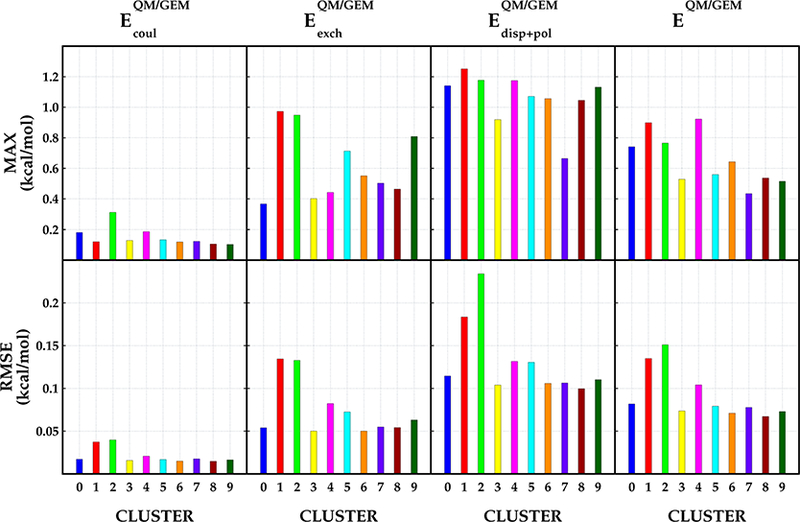
Errors per cluster with respect to SAPT. The errors corresponding to the Coulomb interaction energy are depicted on the first column, and the errors corresponding to the exchange interaction energy are given on the second column. The computed errors for the sum of dispersion and polarization energies are depicted on the third, while the error for total energy is given on the fourth column.
As expected, the exchange—repulsion results are found to be more accurate with the Tychonov regularization using λ1 and λ2, with calculated MAX(RMSE) for all dimers below 1.0(0.15) kcal/mol (Figure 1). Conversely, when only one set of Hermite coefficients (optimized for Coulomb) or when the Cholesky decomposition is used for exchange—repulsion, the MAX- (RMSE) is observed to increase above 1.0(0.2) kcal/mol for almost half of the clusters (Figures S.4 and S.5). About 86% of the water dimers have UE ≤ 0.1 kcal/mol using the Tychonov method with individual fitting for Coulomb and exchange— repulsion (λ1 and λ2), while 83 and 82% of the water dimers have UE ≤ 0.1 kcal/mol without individual fitting using the Tychonov regularization or Cholesky decomposition, respectively. Computing the exchange—repulsion term after SCF cycle results in higher errors, where ~80% of the water dimers have UE ≤ 0.1 kcal/mol for both Tychonov method without individual fitting and Cholesky decomposition (see the Supporting Information).
The errors for the polarization+dispersion term are larger than for the frozen core (Coulomb and exchange—repulsion) terms for all of the fitting schemes, with RMSE values below 0.15 kcal/mol for most clusters and MAX > 1.0 kcal/mol for almost every cluster, regardless of the fitting method. The decrease in accuracy for these two components is mainly due to the approximations used for the calculation of these two terms and the lack of the charge transfer term. Nevertheless, ~78% of the water dimers have UE ≤ 0.1 kcal/mol irrespective of the fitting method.
For the total intermolecular interaction, the RMSE(MAX) for almost all clusters is observed to be <0.1(0.8) kcal/mol except for clusters 1 and 2 (1 and 4) (RMSE ≤ 0.15; MAX < 1.0 kcal/mol) with Tychonov regularization using λ1 and λ2. Similar errors are observed with the other two fitting schemes (Cholesky and Tychonov with only one λ), although the maximum and RMS errors are increased to 1.2 kcal/mol for three clusters.
Figure 2 shows the MAX and RMSE values for the dimers subset, calculated at the same QM level of theory (ωB97XD/ aug-cc-pVTZ) using the AMOEBA potential to represent the MM environment. The overall RMSE with AMOEBA is similar to those observed with GEM. Both methods result in UE ≤ 0.4 kcal/mol for ~99% of the water dimers. However, three dimers have UE > 1.0 kcal/mol with AMOEBA compared with GEM. This is mostly due to the penetration error because the high UE dimers in AMOEBA, which have intermolecular distances <2.0 Å, are well within the region of intermolecular density overlap. In the case of GEM, only one dimer has 1.0 kcal/mol > UE > 0.9 kcal/mol. This dimer has intramolecular angles of 98.9 and 105.3°, indicating that the error is likely arising due to the intramolecular strain of the monomers. It should be noted that in our current implementation the computational cost of the QM/GEM calculation is roughly five times more expensive than for QM/AMOEBA. This is due to the fact that the density of the molecule in the MM subsystem is fitted explicitly each time (as described above). However, as we have previously reported, prefitted densities can be employed,32 which will significantly improve the computational efficiency in subsequent implementations.
Figure 2.
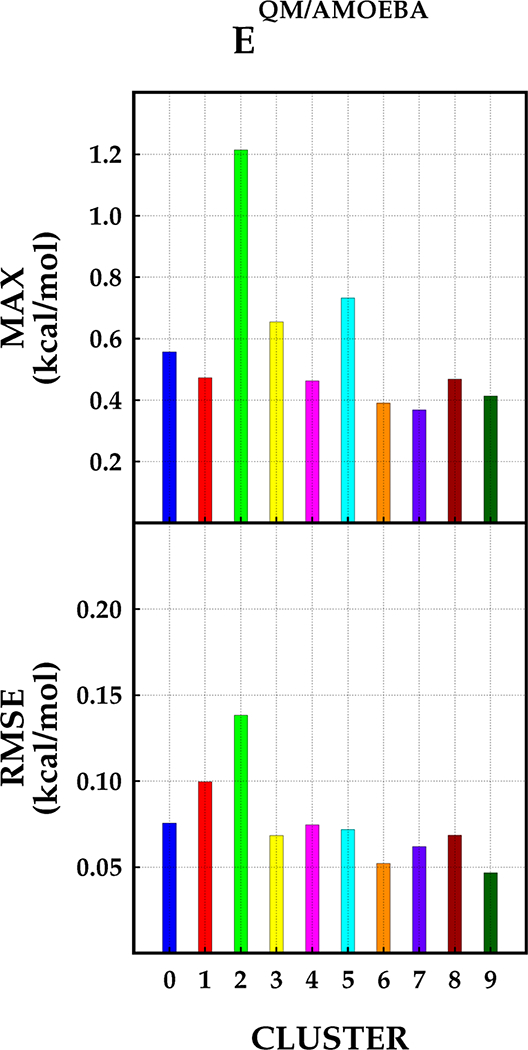
Total intermolecular interaction energy errors per cluster for QM/AMOEBA with respect to SAPT.
Overall, it is observed that the use of AMOEBA provides an accurate representation for the total intermolecular interaction. However, the comparison of the Coulomb intermolecular interaction reveals that the term-by-term interactions have reduced accuracy (see the Supporting Information). Thus the accuracy of the total intermolecular interaction with AMOEBA is mostly due to error cancellation between the Coulomb, polarization and buffered Halgren terms. Similar results for AMOEBA as an embedding environment in close proximity of a QM wave function have been previously reported.19
In summary, we have presented an initial implementation of QM/GEM involving four energy components for the GEM subsystem. The results indicate that GEM provides a very accurate environment in a QM/MM setting with the additional advantage of a physically intuitive decomposition of the intermolecular interaction terms. The average RMSE is <0.2 kcal/mol for every term as well as for the total energy, with MAX below 1.0 kcal/mol for second-order and total interactions and 0.2 kcal/mol for the frozen-core terms. The introduction of a total molecular charge constraint to ensure that the density integrates to the correct number of electrons, coupled to the individual optimization of eigenvalue cutoffs for Coulomb and exchange—repulsion, is shown to improve the accuracy of the GEM environment. It is noteworthy that even with a small fitting set consisting only 10 water dimers, the accuracy of the total intermolecular interaction energy with GEM is very similar to AMOEBA, yet unlike AMOEBA, GEM does not result in MAXs over 1.0 kcal/mol. Because all of the energy components are computed individually with GEM, further improvements can be achieved by the introduction of an explicit charge-transfer term, improvement of the auxiliary basis set, or a better description for the dispersion energy term. This proof of principle constitutes an incentive toward the future application of this technology for full hybrid polarizable QM/ MD using PDE.61
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grant R01GM108583 to G.A.C. We thank the UNT Chemistry Department for the use of the HPC Cluster CRUNTCH3 and NSF grant no. CHE-1531468.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.8b01412.
Scheme for subset selection, clustering details, fitting details for exchange and dispersion parameters, QM/ GEM errors for subset, QM/AMOEBA errors for subset, QM/GEM results separated by monomers included in the QM subsystem, and SAPT2+3/aug-cc-pVTZ results for full Paesani PES (PDF)
Term-by-term and total intermolecular interaction energy components (XLSX)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Senn H; Thiel W QM/MM Methods for Biological Systems In Atomistic Approaches in Modern Biology; Topics in Current Chemistry 268; Springer: Berlin, 2007; pp 173–290. [Google Scholar]
- (2).Senn HM; Thiel W QM/MM Methods for Biomolecular Systems. Angew. Chem., Int. Ed 2009, 48, 1198–1229. [DOI] [PubMed] [Google Scholar]
- (3).van der Kamp MW; Mulholland AJ Combined Quantum Mechanics/Molecular Mechanics (QM/MM) Methods in Computational Enzymology. Biochemistry 2013, 52, 2708–2728. [DOI] [PubMed] [Google Scholar]
- (4).Claeyssens F; Ranaghan KE; Manby FR; Harvey JN; Mulholland AJ Multiple high-level QM/MM reaction paths demonstrate transition-state stabilization in chorismate mutase: correlation of barrier height with transition-state stabilization. Chem. Commun 2005, 5068–5070. [DOI] [PubMed] [Google Scholar]
- (5).Woods CJ; Manby FR; Mulholland AJ An efficient method for the calculation of quantum mechanics/molecular mechanics free energies. J. Chem. Phys 2008, 128, 014109. [DOI] [PubMed] [Google Scholar]
- (6).van der Kamp MW; Zurek J; Manby FR; Harvey JN; Mulholland AJ Testing High-Level QM/MM Methods for Modeling Enzyme Reactions: Acetyl-CoA Deprotonation in Citrate Synthase. J. Phys. Chem. B 2010, 114, 11303–11314. [DOI] [PubMed] [Google Scholar]
- (7).Claeyssens F; Ranaghan KE; Lawan N; Macrae SJ; Manby FR; Harvey JN; Mulholland AJ Analysis of chorismate mutase catalysis by QM/MM modelling of enzyme-catalysed and uncatalysed reactions. Org. Biomol. Chem 2011, 9, 1578–1590. [DOI] [PubMed] [Google Scholar]
- (8).van der Kamp MW; Mulholland AJ Combined Quantum Mechanics/Molecular Mechanics (QM/MM) Methods in Computational Enzymology. Biochemistry 2013, 52, 2708–2728. [DOI] [PubMed] [Google Scholar]
- (9).Bennie SJ; van der Kamp MW; Pennifold RCR; Stella M; Manby FR; Mulholland AJ A Projector-Embedding Approach for Multiscale Coupled-Cluster Calculations Applied to Citrate Synthase. J. Chem. Theory Comput 2016, 12, 2689–2697. [DOI] [PubMed] [Google Scholar]
- (10).Lonsdale R; Fort RM; Rydberg P; Harvey JN; Mulholland AJ Quantum Mechanics/Molecular Mechanics Modeling of Drug Metabolism: Mexiletine N-Hydroxylation by Cytochrome P450 1A2. Chem. Res. Toxicol. 2016, 29, 963–971. [DOI] [PubMed] [Google Scholar]
- (11).Cisneros GA; Piquemal J-P; Darden TA Quantum Mechanics/Molecular Mechanics Electrostatic Embedding with Continuous and Discrete Functions. J. Phys. Chem. B 2006, 110, 13682–13684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kratz EG; Walker AR; Lagardere L; Lipparini F; Piquemal J-P; Cisneros GA LICHEM: AQM/MM program for simulations with multipolar and polarizable force fields. J. Comput. Chem 2016, 37, 1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Gurunathan PK; Acharya A; Ghosh D; Kosenkov D; Kaliman I; Shao Y; Krylov AI; Slipchenko LV Extension of the Effective Fragment Potential Method to Macromolecules. J. Phys. Chem. B 2016, 120, 6562–6574. [DOI] [PubMed] [Google Scholar]
- (14).Andreussi O; Prandi IG; Campetella M; Prampolini G; Mennucci B Classical Force Fields Tailored for QM Applications: Is it Really a Feasible Strategy? J. Chem. Theory Comput 2017, 13, 4636–4648. [DOI] [PubMed] [Google Scholar]
- (15).König PH; Hoffmann M; Frauenheim T; Cui Q A Critical Evaluation of Different QM/MM Frontier Treatments with SCC- DFTB as the QM Method. J. Phys. Chem. B 2005, 109, 9082–9095. [DOI] [PubMed] [Google Scholar]
- (16).Vreven T; Byun KS; Komaromi I; Dapprich S; Montgomery JA; Morokuma K; Frisch MJ Combining Quantum Mechanics Methods with Molecular Mechanics Methods in ONIOM. J. Chem. Theory Comput 2006, 2, 815–826. [DOI] [PubMed] [Google Scholar]
- (17).Das D; Eurenius KP; Billings EM; Sherwood P; Chatfield DC; Hodošček M; Brooks BR Optimization of quantum mechanical molecular mechanical partitioning schemes: Gaussian delocalization of molecular mechanical charges and the double link atom method. J. Chem. Phys 2002, 117, 10534–10547. [Google Scholar]
- (18).Valderrama E; Wheatley RJ An environmental pseudopotential approach to molecular interactions: Implementation in MOLPRO. J. Comput. Chem 2003, 24, 2075–2082. [DOI] [PubMed] [Google Scholar]
- (19).Mao Y; Shao Y; Dziedzic J; Skylaris C-K; Head-Gordon T; Head-Gordon M Performance of the AMOEBA Water Model in the Vicinity of QM Solutes: A Diagnosis Using Energy Decomposition Analysis. J. Chem. Theory Comput 2017, 13, 1963–1979. [DOI] [PubMed] [Google Scholar]
- (20).Sousa SF; Ribeiro AJM; Neves RPP; Brás NF; Cerqueira NMFSA; Fernandes PA; Ramos MJ Application of quantum mechanics/molecular mechanics methods in the study of enzymatic reaction mechanisms. Wiley Interdisc. Rev: Comp. Mol. Sci 2017, 7, e1281. [Google Scholar]
- (21).Gavezzotti A Calculation of Intermolecular Interaction Energies by Direct Numerical Integration over Electron Densities. I. Electrostatic and Polarization Energies in Molecular Crystals. J. Phys. Chem. B 2002, 106, 4145–4154. [Google Scholar]
- (22).Volkov A; Coppens P Calculation of electrostatic interaction energies in molecular dimers from atomic multipole moments obtained by different methods of electron density partitioning. J. Comput. Chem 2004, 25, 921–934. [DOI] [PubMed] [Google Scholar]
- (23).Paricaud P; Předota M; Chialvo AA; Cummings PT From dimer to condensed phases at extreme conditions: Accurate predictions of the properties of water by a Gaussian charge polarizable model. J. Chem. Phys 2005, 122, 244511. [DOI] [PubMed] [Google Scholar]
- (24).Eckhardt CJ; Gavezzotti A Computer Simulations and Analysis of Structural and Energetic Features of Some Crystalline Energetic Materials. J. Phys. Chem. B 2007, 111, 3430–3437. [DOI] [PubMed] [Google Scholar]
- (25).Hermida-Ramón JM; Brdarski S; Karlstrom G; Berg U Inter- and intramolecular potential for the N-formylglycinamide-water system. A comparison between theoretical modeling and empirical force fields. J. Comput. Chem 2003, 24, 161–176. [DOI] [PubMed] [Google Scholar]
- (26).Gresh N; Cisneros GA; Darden TA; Piquemal J-P Anisotropic, Polarizable Molecular Mechanics Studies of Inter- and Intramolecular Interactions and Ligand-Macromolecule Complexes. A Bottom-Up Strategy. J. Chem. Theory Comput 2007, 3, 1960–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ponder JW; Wu C; Ren P; Pande VS; Chodera JD; Schnieders MJ; Haque I; Mobley DL; Lambrecht DS; DiStasio RA; et al. Current Status of the AMOEBA Polarizable Force Field. J. Phys. Chem. B 2010, 114, 2549–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Stone AJ The Theory of Intermolecular Forces; Oxford University: Oxford, U.K., 2000. [Google Scholar]
- (29).Freitag MA; Gordon MS; Jensen JH; Stevens WJ Evaluation of charge penetration between distributed multipolar expansions. J. Chem. Phys 2000, 112, 7300–7306. [Google Scholar]
- (30).Cisneros GA; Piquemal J-P; Darden TA Intermolecular electrostatic energies using density fitting. J. Chem. Phys 2005, 123, 044109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Piquemal J-P; Cisneros GA; Reinhardt P; Gresh N; Darden TA Towards a force field based on density fitting. J. Chem. Phys 2006, 124, 104101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Cisneros GA; Piquemal J-P; Darden TA Generalization of the Gaussian electrostatic model: Extension to arbitrary angular momentum, distributed multipoles, and speedup with reciprocal space methods. J. Chem. Phys 2006, 125, 184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Piquemal J-P; Cisneros GA In Many-Body Effects and Electrostatics in Multi-Scale Computations of Biomolecules; Cui Q, Meuwly M, Ren P, Eds.; PanStanford Publishing: Singapore, 2014; pp 269–300. [Google Scholar]
- (34).Dunlap BI; Connolly JWD; Sabin JR On first-row diatomic molecules and local density models. J. Chem. Phys 1979, 71, 4993–4999. [Google Scholar]
- (35).Koster AM Efficient recursive computation of molecular integrals for density functional methods. J. Chem. Phys 1996, 104, 4114–4124. [Google Scholar]
- (36).Elking DM; Cisneros GA; Piquemal J-P; Darden TA; Pedersen LG Gaussian Multipole Model (GMM). J. Chem. Theory Comput 2010, 6, 190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Cisneros GA; Elking DM; Piquemal J-P; Darden TA J. Phys. Chem. A 2007, 111, 12049–12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Cisneros GA Application of Gaussian Electrostatic Model (GEM) Distributed Multipoles in the Force Field. J. Chem. Theory Comput 2012, 8, 5072–5080. [DOI] [PubMed] [Google Scholar]
- (39).Torabifard H; Starovoytov ON; Ren P; Cisneros GA Development of an AMOEBA water model using GEM distributed multipoles. Theor. Chem. Acc 2015, 134, 101. [Google Scholar]
- (40).Olsen JMH; Steinmann C; Ruud K; Kongsted J Polarizable Density Embedding: A New QM/QM/MM-Based Computational Strategy. J. Phys. Chem. A 2015, 119, 5344–5355. [DOI] [PubMed] [Google Scholar]
- (41).Reinholdt P; Kongsted J; Olsen JMH Polarizable Density Embedding: A Solution to the Electron Spill-Out Problem in Multiscale Modeling. J. Phys. Chem. Lett 2017, 8, 5949–5958.+ [DOI] [PubMed] [Google Scholar]
- (42).Duke RE; Starovoytov ON; Piquemal J-P; Cisneros GA GEM*: A Molecular Electronic Density-Based Force Field for Molecular Dynamics Simulations. J. Chem. Theory Comput 2014, 10, 1361–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Giovannini T; Lafiosca P; Cappelli C A General Route to Include Pauli Repulsion and Quantum Dispersion Effects in QM/MM Approaches. J. Chem. Theory Comput 2017, 13, 4854–4870. [DOI] [PubMed] [Google Scholar]
- (44).Parrish RM; Burns LA; Smith DGA; Simmonett AC; DePrince AE; Hohenstein EG; Bozkaya U; Sokolov AY; Di Remigio R; Richard RM; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput 2017, 13, 3185–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ponder JW; Ren P; Piquemal J-P TINKER: Software Tools for Molecular Design, v. 8.1; Washington University: St. Louis, MO, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lagardere L; Jolly L-H; Lipparini F; Aviat F; Stamm B; Jing ZF; Harger M; Torabifard H; Cisneros GA; Schnieders MJ; et al. Tinker-HP: a massively parallel molecular dynamics package for multiscale simulations of large complex systems with advanced point dipole polarizable force fields 2018, 9, 956–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Press W; Teukolsky SA; Vettering WT; Flannery BP Numerical Recipes in FORTRAN: The Art of Scientific Computing, 2nd ed.;Cambridge University Press: Cambridge, U.K., 1992. [Google Scholar]
- (48).Wheatley RJ; Price SL An overlap model for estimating the anisotropy of repulsion. Mol. Phys 1990, 69, 507–533. [Google Scholar]
- (49).Mitchell JBO; Price SL A Systematic Nonempirical Method of Deriving Model Intermolecular Potentials for Organic Molecules: Application To Amides. J. Phys. Chem. A 2000, 104, 10958–10971. [Google Scholar]
- (50).Tschumper GS; Leininger ML; Hoffman BC; Valeev EF; Schaefer HF III; Quack M Anchoring the water dimer potential energy surface with explicitly correlated computations and focal point analyses. J. Chem. Phys 2002, 116, 690–701. [Google Scholar]
- (51).Ren P; Ponder JW Consistent treatment of inter- and intramolecular polarization in molecular mechanics calculations. J. Comput. Chem 2002, 23, 1497–1506. [DOI] [PubMed] [Google Scholar]
- (52).Ren P; Ponder JW Polarizable Atomic Multipole Water Model for Molecular Mechanics Simulation. J. Phys. Chem. B 2003, 107, 5933–5947. [Google Scholar]
- (53).Grossfield A; Ren P; Ponder JW Ion Solvation Thermodynamics from Simulation with a Polarizable Force Field. J. Am. Chem. Soc 2003, 125, 15671–15682. [DOI] [PubMed] [Google Scholar]
- (54).Chai J-D; Head-Gordon M Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- (55).Dunning T Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys 1989, 90, 1007–1023. [Google Scholar]
- (56).Kendall RA; Dunning TH Jr.; Harrison RJ Electron affinities of the first?row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys 1992, 96, 6796–6806. [Google Scholar]
- (57).Babin V; Leforestier C; Paesani F Development of a ʼʼFirst Principles” Water Potential with Flexible Monomers: Dimer Potential Energy Surface, VRT Spectrum, and Second Virial Coefficient. J. Chem. Theory Comput 2013, 9, 5395–5403. [DOI] [PubMed] [Google Scholar]
- (58).Jeziorski B; Moszynski R; Szalewicz K Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev 1994, 94, 1887–1930. [Google Scholar]
- (59).Laury ML; Wang L-P; Pande VS; Head-Gordon T; Ponder JW Revised Parameters for the AMOEBA Polarizable Atomic Multipole Water Model. J. Phys. Chem. B 2015, 119, 9423–9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Pedregosa F; Varoquaux G; Gramfort A; Michel V; Thirion B; Grisel O; Blondel M; Prettenhofer P; Weiss R; Dubourg V; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res 2011, 12, 2825–2830. [Google Scholar]
- (61).Loco D; Lagardere L; Caprasecca S; Lipparini F; Mennucci B; Piquemal J-P Hybrid QM/MM Molecular Dynamics with AMOEBA Polarizable Embedding. J. Chem. Theory Comput 2017, 13, 4025–4033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.