

ABSTRACT
Mammalian ULK1 (unc-51 like kinase 1) and ULK2, Caenorhabditis elegans UNC-51, and Drosophila melanogaster Atg1 are serine/threonine kinases that regulate flux through the autophagy pathway in response to various types of cellular stress. C. elegans UNC-51 and D. melanogaster Atg1 also promote axonal growth and defasciculation; disruption of these genes results in defective axon guidance in invertebrates. Although disrupting ULK1/2 function impairs normal neurite outgrowth in vitro, the role of ULK1 and ULK2 in the developing brain remains poorly characterized. Here, we show that ULK1 and ULK2 are required for proper projection of axons in the forebrain. Mice lacking Ulk1 and Ulk2 in their central nervous systems showed defects in axonal pathfinding and defasciculation affecting the corpus callosum, anterior commissure, corticothalamic axons and thalamocortical axons. These defects impaired the midline crossing of callosal axons and caused hypoplasia of the anterior commissure and disorganization of the somatosensory cortex. The axon guidance defects observed in ulk1/2 double-knockout mice and central nervous system-specific (Nes-Cre) Ulk1/2-conditional double-knockout mice were not recapitulated in mice lacking other autophagy genes (i.e., Atg7 or Rb1cc1 [RB1-inducible coiled-coil 1]). The brains of Ulk1/2-deficient mice did not show stem cell defects previously attributed to defective autophagy in ambra1 (autophagy/Beclin 1 regulator 1)- and Rb1cc1-deficient mice or accumulation of SQSTM1 (sequestosome 1)+ or ubiquitin+ deposits. Together, these data demonstrate that ULK1 and ULK2 regulate axon guidance during mammalian brain development via a noncanonical (i.e., autophagy-independent) pathway.
KEYWORDS: ATG1, ATG7, autophagy, axon guidance, neurodevelopment, RB1CC1, ULK1/2
Introduction
The serine/threonine-protein kinases ULK1 (unc-51 like kinase 1) and ULK2 are evolutionarily conserved serine/threonine kinase orthologs of the yeast autophagy related (Atg) family member Atg1, that have redundant roles in the regulation of autophagy.1 Autophagy targets long-lived proteins or organelles for degradation in lysosomes, and the products of this process are then recycled for other cellular pathways.2,3 The canonical ULK/Atg1 complex is composed of ULK1, ATG13, RB1CC1/FIP200/ATG17, and ATG101.4-6 It initiates autophagosome formation, at least in part by phosphorylating components of the autophagy-inducing class III phosphatidylinositol 3-kinase complex (e.g., PI3K3C/Vps34, PIK3R4/Vps15, BECN1/Vps30/ATG6, ATG14).7-9 ULK/Atg1 also promotes membrane recycling via ATG9.10-12 Consistent with the established role of ULK1/2 in autophagy, disrupting ULK1 expression in mice results in a defect in autophagy-mediated clearance of mitochondria during red blood cell maturation, and mice lacking both ULK1 and ULK2 expression die shortly after birth due to a defect in glycogen metabolism, which is similar to other autophagy-defective mice.13,14
Although best characterized for their roles in autophagy, mammalian Ulk1 and Ulk2 have first been cloned based on their high degree of homology to the Caenorhabditis elegans unc-51 gene.15,16 Disrupting unc-51 expression in C. elegans results in uncoordinated movement due to widespread abnormalities in axon guidance and fasciculation.17 Similar to unc-51 mutants, Drosophila melanogaster atg1 mutants exhibit abnormal axonal tracts in the ventral nerve cord, including premature truncation and defective midline crossing of longitudinal tracts at the embryonic stage and abnormal defasciculation of the mushroom body axons in larvae.18,19 The axon guidance and defasciculation defects in invertebrates are associated with defects in trafficking of key guidance molecules.20 Indeed, ULK/Atg1 has an evolutionarily conserved role in the endoplasmic reticulum (ER)-to-Golgi trafficking of specific cargo, such as SLC6A4/SERT (solute carrier family 6 [neurotransmitter transporter, serotonin], member 4), via phosphorylation of the COPII scaffold SEC16A (SEC16 homolog A, endoplasmic reticulum export factor). Disrupting unc-51 function in C. elegans leads to the accumulation of SLC6A4 in neuronal cell bodies instead of in axons.21 Results from primary mammalian neuronal cultures suggest that ULK1 and ULK2 also share redundant functions in neurite outgrowth. Dominant-negative disruption of ULK1 function in murine cerebellar granule neurons in culture inhibits neurite formation and extension.22 Knocking down the expression of Ulk1, Ulk2, or both genes leads to extensive neurite branching and stalled axonal growth in murine dorsal root ganglion neurons in culture, which is associated with impaired endocytosis of the nerve growth factor-NTRK1/TRKA receptor complex.23
Although the contribution of autophagy-related ULK/Atg1 function to neurite outgrowth has not been explored, several studies have suggested that neural stem and progenitor cells activate autophagy to meet high-energy demands during differentiation. Indeed, inhibition of autophagy by either pharmacological (e.g., 3-methyladenine) or genetic (e.g., Atg5 or ambra1 deficiency) approaches increases the proliferation of neural progenitor cells and impairs neuronal differentiation during embryonic development.24-26 Impairment of autophagy by deleting Rb1cc1 leads to the loss of stem cells and impairs differentiation in the postnatal brain but not in embryos.27 In addition to the neuronal stem cell defects in ambra1- and Rb1cc1-deficient mice, adult mice lacking central nervous system (CNS) expression of key components of the canonical autophagy pathway (e.g., Atg5, Atg7, and Rb1cc1) show widespread degeneration of neurons containing SQSTM1/p62+ and ubiquitin+ deposits,28-30 similar to those found in the CNS of patients with neurodegenerative disorders (e.g., amyotrophic lateral sclerosis and frontotemporal dementia).31
Although ULK1 and ULK2 clearly have the potential to influence various aspects of neurodevelopment, the role of Ulk1 and Ulk2 in the developing murine CNS remains poorly characterized. Here, we demonstrate that ULK1 and ULK2, though not required for constitutive autophagy in the CNS, play an important role in axon guidance during mammalian forebrain development.
Results
Ulk1/2 deficiency in the CNS results in abnormal axon guidance
Because of the previously described role of ULK/Atg1 orthologs in axon guidance and growth and that of ULK1/2 in neurite growth, we decided to examine the integrity of the major axon tracts formed by projection neurons in ulk1/2 double-knockout (DKO) mice. Previous reports indicate that interbreeding of ulk1- and ulk2-knockout (KO) mice on a mixed (C57BL/6J; 129/SV) background yields progeny with the expected Mendelian ratios at birth.14,32 We interbred C57BL/6J-enriched Ulk1+/−;ulk2−/− mice, and after genotyping 206 embryos at embryonic day (E) 18.5, we identified 63 (30.58%) Ulk1+/+;ulk2−/− embryos and 29 (14.08%) ulk1−/−;ulk2−/− embryos. These ratios suggest that the loss of both genes in a C57BL/6J-enriched genetic background increases early embryonic mortality. Nevertheless, we observed no significant differences in body weight (Fig. S1A) or gross brain morphology (Fig. S1B) in E18.5 ulk1/2-DKO embryos compared to littermate controls (Ulk1+/+;ulk2−/− or Ulk1+/−;ulk2−/−). Visual inspection of newborn pups and genotyping of pups on or after postnatal day (P) 1 confirmed the previously reported perinatal mortality of live-born ulk1/2-DKO pups.14,32
The axons of commissural projection neurons extend to the contralateral cortex, thereby connecting the 2 cortical hemispheres. The cell bodies of commissural projection neurons in rodents reside primarily in cortical layers II and III (80%), layer V (20%), and to a lesser extent in layer VI. Their axons traverse the telencephalic midline, giving rise to 3 commissural tracts: the corpus callosum (CC), the hippocampal commissure (HC), and the anterior commissure (AC). We used an antibody against the axonal marker cell adhesion molecule L1 like (CHL1) to examine the 3 commissural tracts in E18.5 embryos. Although the major axonal tracts formed properly in the ulk1- and ulk2-KO brains (Fig. 1A), the CC was thinner in the ulk1/2-DKO brains compared to that in littermate controls (Fig. 1A and B), and many of the callosal axons were overfasciculated (Fig. 1A10–10′). Although the AC in ulk1/2-DKO embryos was severely hypoplastic, the HC showed no overt abnormalities (Fig. 1A11–11′).
Figure 1.
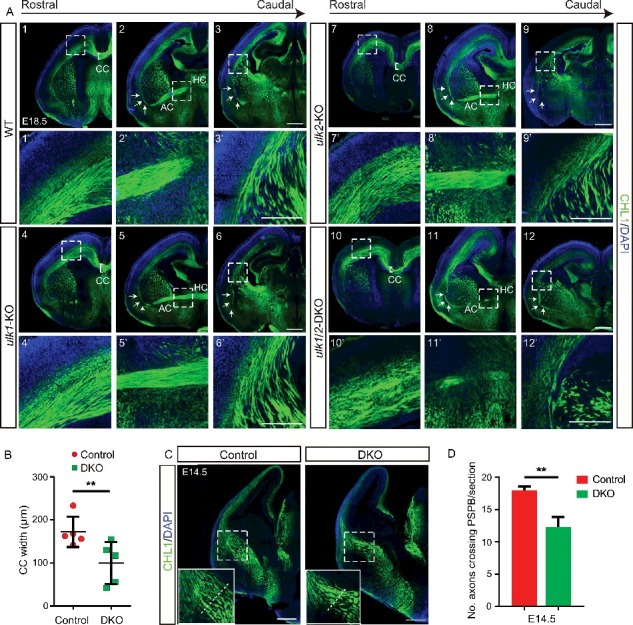
ULK1 and ULK2 are required for axonal guidance in the forebrain. (A) Brain sections of E18.5 WT (1, 2, 3), ulk1-KO (4, 5, 6), ulk2-KO (7, 8, 9), and ulk1/2-DKO (10, 11, 12) mice were stained with antibodies against the general axonal marker CHL1 (pseudocolored green). The callosal axons are heavily overfasciculated, and the corpus callosum is thinner in ulk1/2-DKO mice (10′). The anterior commissure fails to develop (11′). CTAs and TCAs progress abnormally as they cross the PSPB (12′). The ulk1/2-DKO axons are highly disorganized and extend aberrantly towards the external capsule (arrows in 2–3, 5–6, 8–9, and 11–12). The CTAs and TCAs are also overfasciculated. (B) Quantification of the dorsoventral width of the corpus callosum of control (n = 5) and ulk1/2-DKO mice (n = 5) at E18.5. (C-D) The axon pathfinding defects in ulk1/2-DKO mice are detected as early as E14.5 (dashed lines). Abbreviations: AC, anterior commissure; CC, corpus callosum; CTAs, corticothalamic axons; EC, external capsule; HC, hippocampal commissure; CHL1, cell adhesion molecule L1-like; PSPB, pallial-subpallial boundary; TCAs, thalamocortical axons. Scale bars: 500 µm in (A1-A12, and C); 250 µm in (A1′-A12′). **P <0.01 in (B and D).
We also examined the integrity of the corticothalamic axons (CTAs), which originate from neurons residing in layer VI of the cortex and project to the thalamus, and the thalamocortical axons (TCAs), which originate from neurons in various thalamic nuclei and project to corresponding cortical areas. The CTAs and TCAs of ulk1/2-DKO mice showed several abnormalities as they crossed the pallial-subpallial boundary (PSPB). Instead of forming a parallel fan-like structure, the ulk1/2-deficient axons were disorganized, overfasciculated, and traversed the PSPB more anteroventrally than axons in littermate controls (Figs. 1D12–12′ and S1C). The abnormal path of the CTAs and TCAs in ulk1/2-deficient animals was already evident at E14.5, with fewer axons crossing the PSPB (Fig. 1C and D), suggesting that ULK1 and ULK2 regulate an intermediate step in axon pathfinding (i.e., crossing the midline or specific boundaries).
ULK1 and ULK2 regulate axon guidance via a noncanonical pathway
RB1CC1 and ATG13 are components of the canonical 3-MDa ULK/Atg1 kinase complex that regulates selective and nonselective autophagy in response to cellular stress.33,34 ULK/Atg1 also interacts with the COPII scaffold SEC16A to promote ER-to-Golgi trafficking of specific cargo in the absence of stress.21 Therefore, we immunoprecipitated endogenous ULK1 from extracts prepared from the cerebral cortex of P0 mice to determine which of these interactions may be relevant in the developing cortex. We observed interactions between ULK1 and SEC16A and ATG13 in mice lacking CNS expression of Rb1cc1 [Rb1cc1flox/flox;Nes/Nestin-Cre, hereafter referred to as Rb1cc1 conditional knockout (cKO) mice (Rb1cc1-cKO)], as well as in littermate controls, in which the interaction between ULK1 and RB1CC1 was also preserved (Fig. 2A). There were no obvious differences in the steady-state levels of components of the core autophagy machinery, including MAP1LC3B (hereafter referred to as LC3B), ATG13, RB1CC1, and ATG14, in the cortex from control and ulk1/2-DKO mice (Fig. 2B and C). We observed no significant increases in steady-state levels of the autophagy substrate SQSTM1 (Fig. 2B) or expression of Ulk/ATG1 paralogs (i.e., Ulk3, Ulk4, and Stk36) (Fig. S2A) in the brains of E18.5 ulk1/2-DKO animals compared to those in controls. Similarly, we did not detect an appreciable difference in steady-state levels of SQSTM1 in the cortex of ulk1−/−;Ulk2−/flox;Nes-Cre (hereafter referred to as Ulk1/2-cDKO mice) and controls at P0 (Fig. 2D and E) or the presence of SQSTM1+ and ubiquitin+ inclusions at P21 (Fig. 2F), despite the depletion of Ulk1 and Ulk2 in the brains of Ulk1/2-cDKO mice (Fig. S2B). By contrast, mice lacking CNS expression of either Atg7 (Atg7flox/flox;Nes-Cre, hereafter referred as Atg7-cKO) or Rb1cc1 showed dramatic increases in the steady-state level of SQSTM1 at P0 (Fig. 2D and E) and accumulation of SQSTM1+ and ubiquitin+ deposits at P21 (Fig. 2F). Finally, we observed no significant difference in the number of double-membrane bound vesicles in the callosal axons of control or Ulk1/2-cDKO mice (Fig. S3A and B). Together, these results indicate that unlike RB1CC1 and ATG7, the autophagy-inducing kinases, ULK1 and ULK2, are not required for the constitutive autophagy-mediated turnover of ubiquitinated proteins in the CNS.
Figure 2.

ULK1 and ULK2 are not required for autophagy-mediated turnover of ubiquitinated protein in the cerebral cortex. (A) Immunoprecipitation of ULK1 by using extracts prepared from the cortex of P0 mice and performed in the presence or absence of epitope-specific blocking peptides. The interaction between ULK1, ATG13, and RB1CC1 was detected in extracts prepared from P0 cortex, whereas that between ULK1 and SEC16A was detected in extracts prepared from P0 cortex in the presence or absence of RB1CC1. (B) Extracts prepared from the cortex of E18.5 control and ulk1/2-DKO mice were immunoblotted with antibodies against autophagy proteins, including SQSTM1, LC3B, ATG13, RB1CC1, and ATG14. (C) Quantification of the SQSTM1 level in the E18.5 cortex of control (n = 3) and ulk1/2-DKO mice (n = 3). (D and E) Immunoblot and immunostaining analyses of the cortex from control, Ulk1/2-cDKO, Atg7-cKO, and Rb1cc1-cKO mice at P0 by using antibodies against SQSTM1. (F) SQSTM1 (pseudocolored red) and ubiquitin (pseudocolored green) staining of the cortex from control, Ulk1/2-cDKO, Atg7-cKO, and Rb1cc1-cKO mice at P21. Sections were counterstained with DAPI (pseudocolored blue). Abbreviations: BP, blocking peptides; IP, immunoprecipitation; ns, not significant. Scale bars: 50 µm.
We next determined whether the effects of Ulk1/2 deficiency on the formation of axonal tracts in the CNS were recapitulated by loss of Ulk1/2, Rb1cc1 or Atg7 by using cKO mice. Ulk1/2-cDKO mice show progressive hippocampal degeneration, beginning around 8 wk of age.21 Although some of the Ulk1/2-cDKO mice survive as long as 4 mo, 40% die within the first 24 h after birth.21 Therefore, we examined CHL1 staining of brain sections from P1 Ulk1/2-cDKO mice and littermate controls. The staining revealed dysgenesis of the CC (including the presence of Probst bundles), hypoplastic AC, and disruption of CTAs and TCAs in the Ulk1/2-cDKO mice (Fig. 3), similar to that observed in the ulk1/2-DKO mice (Fig. 1A). Mice lacking CNS expression of Rb1cc1 or Atg7 showed no defects in commissural axon tracts or disruption of CTAs and TCAs (Fig. 3).
Figure 3.

RB1CC1 and ATG7 are not required for axon guidance in the forebrain. CHL1 (green) staining of serial sections of P1 Ulk1/2-cDKO brains reveals abnormally fasciculated axons, including the CTAs and TCAs (inset), corpus callosum dysgenesis, and anterior commissure hypoplasia. These axon guidance abnormalities are similar to those identified in the ulk1/2-DKO mice and are not observed in autophagy-deficient Atg7-cKO or Rb1cc1-cKO mice. Abbreviations: AC, anterior commissure; CC, corpus callosum; EC, external capsule; HC, hippocampal commisure; IC, internal capsule; CHL1, cell adhesion molecule L1-like; Pb, Probst bundle. Scale bars: 500 µm.
RB1CC1 is essential for maintaining the neuronal stem cell pools in postnatal brains,27 and the dentate gyrus is 1 of only 2 brain regions with continuous neurogenesis in adult animals. Therefore, we examined the dentate gyrus of Ulk1/2-cDKO and Rb1cc1-cKO mice in more detail. We observed no significant differences in the numbers of SOX2+ neuronal stem cells in the subgranular zone of the dentate gyrus, MKI67/KI67+ mitotic neurons, or mature neurons (RBFOX3/NEUN+) in the granular zone of the dentate gyrus in Ulk1/2-cDKO compared to littermate controls (Fig. 4A and B). In contrast and as expected based on previous studies,27 the numbers of neuronal stem cells, mitotic neurons, and mature neurons were all significantly (P < 0.05) decreased in the Rb1cc1-cKO dentate gyrus at P21 (Fig. S4). Therefore, consistent with the notion that ULKs are dispensable for autophagy in the CNS of unstressed animals, they also are not required for the RB1CC1- and autophagy-mediated postnatal regulation of neural stem cell function. Together with the observation that the axon guidance defects in the Ulk1/2-deficient mice are not present in Atg7- or Rb1cc1-deficient mice, these data suggest that ULK1 and ULK2 regulate the development of the mouse forebrain via a noncanonical pathway that is distinct from its role in autophagy.
Figure 4.

ULK1 and ULK2 are not required for maintaining the neural stem cells in the hippocampus. (A) Immunostaining of markers for neuronal stem cells (SOX2; green), mitotic cells (MKI67/KI67; red), and mature neurons (RBFOX3/NEUN; blue) in the dentate gyrus of adult control and Ulk1/2-cDKO mice. (B) The mean number ± SEM of the 3 cell types in the dentate gyrus showed no statistical significance between controls (n = 3) and Ulk1/2-cDKO (n = 3) mice. Abbreviations: GZ, granule zone; ns, not significant; SGZ, subgranular zone.
ULK1 and ULK2 regulate the midline crossing of callosal axons
We next sought to further characterize the neurodevelopmental defects in ulk1/2-deficient mice to gain insight into the role of mammalian orthologs of yeast ATG1 in the CNS. To exclude the possibility that the abnormalities in the callosal axons, CTAs, and TCAs in ulk1/2-deficient mice were caused by impaired production of cortical-projection neurons, we examined the cortical layer structure of E18.5 ulk1/2-DKO brains. Nissl staining and the expression of the callosal neuron marker special AT-rich sequence binding protein 2 (SATB2), layer V marker BCL11B/CTIP2 (B cell leukemia/lymphoma 11B), and layer VI marker TBR1 (T-box brain gene 1) were all normal in the ulk1/2-DKO cortex (Fig. 5A and B). Thus, ULK1 and ULK2 appeared to be dispensable for specification of projection neurons in the embryonic cortex. The ulk1/2-DKO embryos did not show an increase in apoptosis (Fig. 5C and D) or neural tube closure defects (data not shown), both of which are seen in ambra1-deficient animals.24
Figure 5.
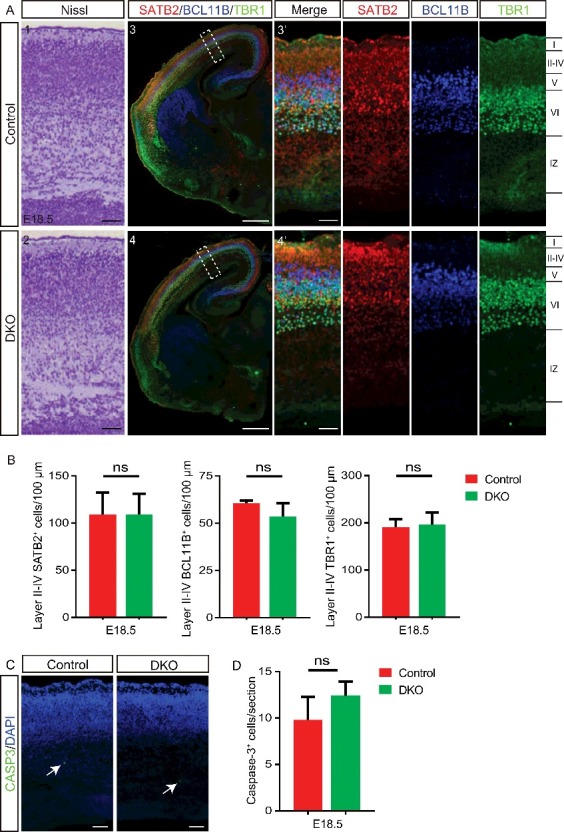
The cortical layers of the ulk1/2-DKO brain are grossly normal. (A) Nissl staining (A1-A2) and expression of the callosal neuron marker SATB2 (red), layer V marker BCL11B/CTIP2 (blue), and layer VI marker TBR1 (green) in control and ulk1/2-DKO cortices at E18.5 (A3-A4), and (B). The quantifications of SATB2+, BCL11B/CTIP2+, and TBR1+ cells in layers II-IV, V, and VI, respectively, are not statistically significant (ns), indicating that Ulk1/2 are not required for neurogenesis. (C and D) Staining of cleaved CASP3 (green) in E18.5 control and ulk1/2-DKO cortices (C) and the number of cleaved-CASP3+ cells per section (D) was quantified (n = 3 for control and ulk1/2-DKO mice). Arrows in (C) point to the sparse apoptosis present in both the control and ulk1/2-DKO cortices. Scale bars: 50 µm in (A1-A2, A3′-A4′), and C; 500 µm in (A3-A4).
To trace the projection of callosal axons across the midline, we embedded crystals of the fluorescent carbocyanine dye 1,1´-dioctadecyl-3,3,38,38, tetramethylindocarbocyanine perchlorate (Dil) into the parietal cortex of control and ulk1/2-DKO embryos at E18.5. Compared to that of controls, the fluorescence intensity of callosal axons reaching the contralateral cortex of ulk1/2-DKO brains was significantly (P < 0.01) reduced (Fig. 6A and B). Luxol Fast Blue staining for myelinated axons showed significantly reduced CC thickness in Ulk1/2-cDKO brains at 2 months, indicating persistent deformity of the CC in Ulk1/2-deficient mice (Fig. 6C and D).
Figure 6.

ULK1 and ULK2 regulate development of the corpus callosum. (A and B) Dil was placed in the parietal cortex of E18.5 ulk1/2-DKO (n = 2) and control (n = 2) mice to trace the midline crossing of callosal axons. Representative sections (A) and quantification (B) highlight the dramatic reduction in fluorescence intensity in the contralateral cortex (rostral and caudal levels) of ulk1/2-DKO mice (asterisks in A). (C) Luxol Fast Blue (LFB) and Nissl staining reveals persistent anomalies of the corpus callosum in the adult Ulk1/2-cDKO mice. (D) The dorsoventral width of the corpus callosum of adult control (n = 3) and Ulk1/2-cDKO mice (n = 3) was quantified, and the average width of each mutant was normalized to that of its corresponding control. There is a significant reduction in the width of the corpus callosum in Ulk1/2-cDKO mice relative to that in controls. (E) Representative brain sections from E18.5 ulk1/2-DKO mice, P0 Ulk1/2-cDKO mice, and age-matched controls. The midline glial structure developed properly. (F) CALR (calreticulin) immunostaining (green) of the glutamatergic guidepost neurons in control and ulk1/2-DKO brains at E15.5 and E18.5 showed that the production of those neurons is unaltered in the Ulk1/2-deficient mice. Abbreviations: CC, corpus callosum; GW, glial wedge; IGG, indusium griseum glia, MZG, midline zipper glia; Pb, Probst bundle. Scale bars: 500 µm in (A1-A4, and C1-C2); 100 µm in (A1′-A4′, C1′-C2′, and E-F). **P <0.01 in (B and D).
Abnormal CC development can result from cell-autonomous defects or various noncell autonomous abnormalities, such as environmental axon guidance cues from midline glia and guidepost neurons. Having ruled-out obvious defects in cortical neurogenesis (Fig. 5), we examined the midline glial structures and guidepost neuron population. Glial cells stained with antibodies directed against GFAP (glial fibrillary acidic protein) were properly formed in the midline glial structures, including the glial wedge, indusium griseum glia, and midline zipper glia (Fig. 6E). The guidepost neurons marked by CALR/calreticulin appeared normal in the mutants at E15.5; thus, their abnormal distribution along the midline at E18.5 was most likely a consequence of improper positioning of the axons crossing the midline rather than the cause (Fig. 6F).
ULK1/2 deficiency impairs the organization of the somatosensory cortex
To characterize the axon guidance defects in ulk1/2-deficient CTAs and TCAs in more detail, we first embedded Dil crystals into the presumptive somatosensory cortex of E18.5 embryos to label CTAs and trace their projections. CTAs crossing the PSPB in control brains were highly organized, but those in ulk1/2-DKO brains tended to traverse the striatum aberrantly (Figure 7A1–1′ and 3–3′). Despite the disorganization and overfasciculation of the axons, the CTAs still found and targeted their final destination in the dorsal thalamus (dTh), as evidenced by the Dil signal in the dTh of ulk1/2-DKO brains (Fig. 7A2–4). We also immunostained the brains of Ulk1/2-cDKO mice and controls at P21 for SLC17A6/VGLUT2 (solute carrier family 17 [sodium-dependent inorganic phosphate cotransporter], member 6) and found no obvious differences in the intensity or distribution of this presynaptic marker in the dTh (Fig. 7B). COX/cytochrome oxidase staining highlighted the intact array of barreloids in the thalamus (Fig. 7B). Therefore, despite the abnormal fasciculation and path taken by the CTAs, we did not detect gross abnormalities in postnatal circuitry in the dTh.
Figure 7.

ULK1 and ULK2 are required for proper organization of the somatosensory cortex. (A) Representative images of fluorescence signal from Dil crystals embedded in the somatosensory cortex reveals the disorganized CTA projections crossing the PSPB in E18.5 ulk1/2-DKO brains. These Ulk1/2-deficient axons, despite their disorganization and abnormal fasciculation, ultimately reach their targeted destination and innervate the dTh (A2 and A4). (B) Normal SLC17A6 staining (red) and cytochrome oxidase staining in the dTh in the ulk1/2-DKO brains at P21 suggests proper cortical innervation of that structure. (C) Dil tracing shows the severely disorganized TCA projections in the ulk1/2-DKO brains. (D) Representative images of CALR (calreticulin) staining (green) reveal a dramatic reduction in the number of TCA projections reaching the cortex in E18.5 ulk1/2-DKO brains. (E) Representative images of sections immunostained using antibodies against the cortical barrel markers SLC6A4 (green) and SLC17A6 (red). Arrows indicate barrels. (F) The number of barrel units per section was quantified from P7 control (n = 3) and Ulk1/2-cDKO (n = 3) brains. **P <0.01. (G) Cytochrome oxidase staining reveals the disorganization of the somatosensory barrel cortex in P7 Ulk1/2-cDKO brains. Abbreviations: A, anterior; COX, cytochrome oxidase; dTh, dorsal thalamus; M, medial; VPL, ventral posterolateral nucleus; VPM, ventral posteromedial nucleus. Scale bars: 100 µm in (A1′-A3′, B, C2′-C4′, D1′-D2′, E, and G); 500 µm in (A1-A4, C1-C4, and D1-D2).
TCAs project to the cortex through the intermediate zone during embryonic development and migrate radially to layer IV at birth.35 The extensive TCA arbors segregate into clusters surrounded by rings of layer IV neurons' somas, whose dendrites are oriented to synapse with TCA afferents. Thalamic innervations are essential to remodel cortical neurons postnatally, and this organization gives rise to the barrels in the somatosensory cortex, which relay information from a single whisker.35 To trace TCA projections, we embedded Dil crystals into the dTh of E18.5 brains. Similar to the CTAs, the TCAs of ulk1/2-DKO brains were disorganized and overfasciculated as they crossed the PSPB (Fig. 7C). In E18.5 ulk1/2-DKO mice, the decreased levels of cortical staining using an antibody against the TCA marker CALR/calreticulin suggested that fewer TCAs reached the cortex after crossing the PSPB than did in controls (Fig. 7D). We assessed the organization of the TCA projections postnatally in the brains of P7 mice by immunolabeling them for the cortical barrel markers SLC6A4 and SLC17A6/VGLUT2. The Ulk1/2-cDKO brains had fewer intact barrels (Fig. 7E and F). The somatosensory map in the Ulk1/2-cDKO mice, as revealed by COX staining, was highly disorganized compared to that in the controls (Fig. 7G).
CNTN2 (contactin 2) is mislocalized in corticothalamic axons
The defasciculation defects resulting from Ulk1/2 deficiency were similar to those observed in unc-51- or atg1-mutant invertebrates. In unc-51-mutant C. elegans, the axon guidance receptor UNC-5 is mislocalized to the cell soma and fails to mediate repulsion of axons away from the UNC-6/netrin-rich ventral midline.36 In Drosophila, ATG1 regulates axonal localization of Fas2 (Fasciclin 2), an IgG-family cell adhesion molecule that is important for axon guidance and fasciculation in the mushroom body. Therefore, we hypothesized that ULK1 and ULK2 may also regulate cellular localization of specific axon guidance receptors in projection neurons. Although most of the axon guidance receptors that we examined, including NCAM1 (neural cell adhesion molecule 1), PSA-NCAM1, DCC, and ROBO1 were clearly present in the cortex and striatum of ulk1/2-DKO and littermate controls (Fig. 8A and data not shown), the CTA marker CNTN2/TAG-1 (contactin 2) was notably absent from the striatum (i.e., distal terminus of the CTAs) in ulk1/2-DKO embryos (Fig. 8B). The expression of CNTN2 in the dorsal cortex (i.e., proximal region of the CTAs) was unaltered (Fig. 8B). Immunoblot analyses performed on dissected regions of the cortex (i.e., proximal CTAs) and striatum (i.e., distal CTAs) from E18.5 brains revealed a significant reduction (P <0.05) in the level of CNTN2 in the distal CTAs of the ulk1/2-DKO animals (Fig. 8C to E). Combined with the results of the tracing experiments, which indicated that the CTAs are able to reach their final destination in the dTh of ulk1/2-DKO brains (Fig. 7A2 to 4), these results suggest that ULK1 and ULK2 regulate the trafficking of specific axon guidance molecules, such as the CTA marker CNTN2, in developing mouse brains similar to the function of ULK/Atg1 orthologs in invertebrates.
Figure 8.

Abnormal axonal fasciculation in the Ulk1/2-deficient animals is associated with mislocalization of CNTN2 in the projection neurons. (A) Neuronal cell adhesion molecule distribution was unaltered in the ulk1/2-DKO brains compared to that in the controls. (B) The intensity of CNTN2 immunostaining (red) was dramatically decreased in distal CTAs of the ulk1/2-DKO brain at E18.5. All of the sections were counterstained with CHL1 cell adhesion molecule (green). (C) Western blot analyses of the extracts prepared from the cortex (proximal CTAs) and striatum (distal CTAs) confirmed normal expression of neuronal cell adhesion molecule but significantly decreased CNTN2 levels in the striatum. (D and E) Quantification of the NCAM1 and CNTN2 levels in control and ulk1/2-DKO cortex and striatum. Abbreviations: Cnt, controls; NCAM1, neuronal cell adhesion molecule 1; ns, not significant.*P < 0.05. Scale bars: 200 µm.
Discussion
Here we provide the first description of neurodevelopmental defects associated with Ulk1/2 deficiency in mammals. Using germline KO and CNS-specific cKO mouse models, we demonstrate that the loss of Ulk1 and Ulk2 expression in the CNS leads to pathfinding defects affecting callosal axons, AC axons, CTAs, and TCAs via a noncanonical (i.e., autophagy-independent) pathway.
The autophagy-inducing activity of ULK/Atg1 depends on its stable interaction with ATG13 and RB1CC14,5 and can be regulated by AMBRA1-mediated ubiquitination of ULK1.37 Although a kinase complex composed of ULK1, ATG13, and RB1CC1 was detected in the cortex of wild-type postnatal mice, Ulk1/2 deficiency in the CNS did not result in histopathological findings identified in other autophagy-deficient mice. For example, ulk1/2-DKO and Ulk1/2-cDKO mice did not show the excessive neuronal cell death characteristic of ambra1-KO embryos, the accumulation of SQSTM1+ and ubiquitin+ deposits, the loss of cortical neurons typically seen in postnatal Atg7-cKO or Rb1cc1-cKO mice, or the loss of neuronal stem cells observed in Rb1cc1-deficient mice. Together, these observations indicate that ULK1 and ULK2 are dispensable for not only constitutive autophagy-mediated turnover of SQSTM1 and ubiquitinated proteins in the CNS but also autophagy-mediated neuronal stem cell maintenance.
Although these observations are somewhat surprising, given the well-established roles of ULK1/2 in selective and nonselective autophagy induced by various metabolic stresses,1,38 recent studies have indicated that certain forms of autophagy (including starvation-induced autophagy) can proceed independently of ULK/Atg1, albeit not as efficiently as in the presence of ULK/Atg1.32,39,40 Such a pathway may be sufficient to promote the turnover of SQSTM1 and ubiquitinated proteins in the CNS and maintain cellular homeostasis in developing Ulk1/2-deficient mouse brains. Although we found no evidence of increased RNA expression of Ulk3, Ulk4, or Stk36 in the brains of ulk1/2-deficient mice, one of these ULK/Atg1 orthologs or an as-yet unidentified kinase may compensate for the loss of ULK1/2 in the CNS.
Just as histopathological features associated with autophagy defects were not identified in Ulk1/2-deficient mice, the neurodevelopmental abnormalities that we found in Ulk1/2-deficient mice were not recapitulated in mice lacking the expression of Rb1cc1 or Atg7 in the CNS. Similar results were reported in C. elegans, where the axon guidance and defasciculation defects observed in unc-51 mutants were not recapitulated in epg-1 (Atg13) or epg-9 (Atg101) mutants.41,42 Together, these findings suggest that ULK/Atg1 regulates axon guidance via a noncanonical pathway, which does not require the formation of the canonical autophagy-inducing ULK1/2-ATG13-RB1CC1/ATG17 complex. We recently demonstrated that ULK/Atg1, by interacting with and phosphorylating the COPII scaffold SEC16A, regulates the assembly of ER-exit sites containing the cargo adapter SEC24C and the trafficking of associated cargoes.21 The interaction between ULK1 and SEC16A occurs in the absence of ATG13 or RB1CC1, and ULK1/2-dependent ER-to-Golgi trafficking can proceed in Atg13-depleted cells, despite the disruption of the ULK1/2-ATG13-RB1CC1/ATG17 complex.21 This noncanonical function of ULK1/2 in ER-to-Golgi trafficking may contribute to axon growth and guidance.
Among the cargoes whose ER-to-Golgi trafficking is regulated by ULK1/2 is the serotonin transporter SLC6A4.21 SLC6A4 is transiently expressed in TCAs, where it helps regulate serotonin levels.40 The appropriate level of serotonin at synaptic junctions is essential for the normal organization and development of the thalamocortical afferent circuits, and impaired uptake of serotonin due to genetic or pharmacological disruption of SLC6A4 function in TCAs impairs barrel cortex formation.43,44 The disorganization of the somatosensory cortex in Ulk1/2-cDKO mice is similar to that resulting from genetic or pharmacological disruption of SLC6A4 activity in developing mouse brains.45 Additional studies are required to test the hypothesis that defective ER-to-Golgi trafficking of SLC6A4 and/or other secretory cargo in TCAs contributes to the disorganization of the somatosensory cortex in Ulk1/2-cDKO mice.
ULK/Atg1 family members regulate additional aspects of membrane and protein trafficking, which may contribute to ULK1/2-dependent axonal guidance. For example, the NCAM-like axon guidance molecule Fas2, which regulates the fasciculation of mushroom body axons, is mislocalized in Atg1-mutant D. melanogaster due to a defect in the kinesin-mediated vesicle transport pathway.19 ATG1/UNC-51-mediated phosphorylation of the kinesin adaptor UNC-76 increases its affinity for SYT-1/synaptotagmin-1, a major transmembrane protein of synaptic vesicles, and promotes the transport of synaptic vesicles.18 In C. elegans, the interaction between UNC-51 and VAB-8, a kinesin-like molecule that controls the cell surface expression of SAX-3/ROBO1 in touch neuron growth cones, is essential for anteroposterior axon growth.46,47 Together, these findings suggest that ULK/Atg1 has a conserved role in regulating kinesin-mediated vesicular transport that facilitates the trafficking of axon guidance molecules. In C. elegans, UNC-51 also binds to and phosphorylates UNC-14, a RUN-domain protein that acts as an effector of RAP and RAB members of the RAS oncogene family of GTPases and regulates membrane trafficking;46,48 disrupting this interaction leads to aberrant localization of the UNC-6/netrin receptor UNC-5 to the cell soma (rather than axons) and a failure to repulse axons away from the UNC-6/netrin-rich ventral midline.36 In mammals, ULK1 modulates RAS and RAB5-mediated endocytosis through its interactions with the synaptic GTPase-activation protein, SYNGAP1, and the synaptic scaffold protein, SDCBP/Syntenin, which recruits RAB5 and ULK1 to a subset of synaptic vesicles.49
ULK1 and ULK2 have also been implicated in endocytosis and trafficking of the NTRK1 receptor complex, processes that involve modulation of RAB5 activity.23,50 Thus, the restricted localization of the CTA marker CNTN2 to the proximal region of the CTAs may result from a defect in ULK/Atg1-dependent protein or vesicle trafficking. Although CNTN2 promotes axon growth and fasciculation in vitro and in vivo through homophilic or heterophilic interactions,51,52 disruption of CNTN2 function alone is insufficient to explain the aberrant fasciculation and axon guidance defects in the ulk1/2-deficient mice because cntn2-deficient mice do not show overt phenotypes affecting the CC, or defasciculation defect.53 Mislocalized CNTN2 may function in a dominant manner due to its ability to interact with other axon guidance molecules.54,55 CNTN2 is likely to be one of several molecules involved in axon growth and guidance whose function is altered in ulk1/2-deficient brains.
The observation that the hippocampal commissure develops normally, while the CC, AC, TCAs, and CTAs show pathfinding defects in the ulk1/2-DKO and Ulk1/2-cDKO mice indicates that ULK1 and ULK2 may regulate axonal guidance in a cell type-specific manner. There are several possible explanations for this phenomenon. First, the spatiotemporal expression pattern of Ulk1 and Ulk2 in the developing mouse brain may contribute to the cell type-specific differences in axon guidance. For example, based on data from Genepaint (www.genepaint.org), Ulk1 and Ulk2 are expressed at higher levels in the cortical plate (postmitotic neurons) than in the ventricular/subventricular zone (neural stem cells) at E14.5. The expression of Ulk1 and Ulk2 is also higher in the cortex than in the striatum and thalamus. Second, cell type- and/or developmental stage-specific differences in ULK1/2-interacting partners may influence their ability to regulate ULK/Atg1-dependent membrane and/or protein trafficking in different populations of neurons. Additional studies are required to more precisely define and elucidate the noncanonical roles of ULK1 and ULK2 that contribute to abnormal axon guidance in the brains of developing ulk1/2-deficient animals.
Materials and methods
Animals
All animal experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of St. Jude Children's Research Hospital (St. Jude). ulk1−/− mice and ulk2−/− mice have been described previously.13,14,32 Ulk1+/− and Ulk2+/− mice were backcrossed onto C57BL/6J background for 3 to 6 generations prior to breeding with each other. Resulting C57BL/6J-enriched Ulk1+/−;ulk2−/− mice were interbred to generate ulk1−/−;ulk2−/−double-knockout (ulk1/2-DKO) mice. The Nes-Cre line was obtained from the Jackson Laboratory (003771). Strategies to target Ulk2 locus and generate Ulk2flox mice have been described previously.21,32 Ulk1+/−;Ulk2f/f females were crossed with Ulk1+/−;ulk2−/−;Nes-Cre males to generate Ulk1/2Nes conditional DKO (Ulk1/2-cDKO) mice. Controls included those mice expressing Ulk1 but not Ulk2 in the relevant brain regions (Ulk1+/−;Ulk2−/flox;Nes-Cre and Ulk1+/+;Ulk2−/flox;Nes-Cre). The mice were maintained in a mixed (C57BL/6J and 129SV/J) background. Atg7f/f females (a generous gift from Dr. Masaaki Komatsu, Tokyo Metropolitan Institute of Medical Science) and Rb1cc1f/f females were crossed with Nes-Cre males to generate Atg7Nes-cKO (Atg7-cKO) and Rb1cc1Nes-cKO (Rb1cc1-cKO) mice.
Immunostaining and histology
Embryonic and newborn mouse brains were dissected in phosphate-buffered saline (PBS; Invitrogen, 14190–250) and fixed overnight in 4% paraformaldehyde (PFA; Santa Cruz Biotechnology, sc-281692) in PBS at 4°C. Mice older than P1 were transcardially perfused with 4% PFA and fixed overnight. The brains were then cryoprotected using 30% sucrose (Sigma Aldrich, S7903) in PBS overnight at 4°C and embedded in OCT (Invitrogen, 15–183–13) for cryosectioning. Frozen sections were washed with 0.2% Triton X-100 (Sigma Aldrich, X100) in tris-buffered saline with Tween 20 (TBST; Invitrogen, BP-2471–1) and incubated in blocking solution (i.e., 5% normal goat serum [Sigma Aldrich, G9023] or normal donkey serum [Sigma Aldrich, D9663] in TBST) for 1 h at room temperature. Sections were incubated with primary antibodies diluted in the blocking solution overnight at 4°C, washed with TBST, and incubated with Alexa Fluor-conjugated secondary antibodies (Invitrogen, A-11008, A21422, A21426, A21434, A21244) diluted at 1:1000 in the blocking solution for 2 h at room temperature. Sections were mounted in ProLong Gold antifade reagent with DAPI (Invitrogen, P-36931). The following primary antibodies were used: rat anti-CHL1 (Millipore, MAB5272), mouse anti-NEFM (Developmental Studies Hybridoma Bank, 2H3), mouse anti-CNTN2 (Developmental Studies Hybridoma Bank, 4G7), mouse anti-NCAM1 (Developmental Studies Hybridoma Bank, 5B8), mouse anti-MAP2 (Sigma Aldrich, M1406), mouse anti-SQSTM1 (Abnova, H00008878-M01), rabbit anti-ubiquitin (DAKO, Z0458), goat anti-SOX2 (Santa Cruz Biotechnology, sc-17320), rabbit anti- MKI67/KI67+ (Cell Signaling Technology, 9129), mouse anti-SATB2 (Abcam, ab51502), rat anti-BCL11B/CTIP2 (Abcam, ab18465), rabbit anti-TBR1 (Abcam, ab31940), rabbit anti-cleaved CASP3/Caspase-3 (Cell Signaling Technology, 9661) rabbit anti-GFAP (DAKO, Z033429), rabbit anti-CALR/calreticulin (Millipore, AB5054), rabbit anti-SLC6A4/5-HTT (Millipore, PC177L), guinea pig anti-SLC17A6/VGLUT2 (Millipore, AB2251), and guinea pig anti-RBFOX3/NEUN (Synaptic System, 266004).
For histological analysis, formalin-fixed brains were embedded in paraffin (StatLab, PPSEM), and 4-μm sagittal sections were cut, stained with Luxol Fast Blue (Poly Scientific, s235‐1), and counterstained with Nissl (Poly Scientific, s2548). To measure the number of axons crossing the PSPB, E14.5 brain sections from control (n = 3) and ulk1/2-DKO mice (n = 3) were stained with CHL1. A straight line was drawn along the PSPB, and the number of fluorescence peaks along the line were counted as the number of axons. For each animal, at least 3 sequential coronal sections collected at 100-μm intervals along the rostrocaudal axis were quantified. To quantify the dorsoventral CC width, low-magnification images were taken with a Marianas 2 microscope (3i, Denver, CO, USA). Using SlideBook 6.0 software, the CC width was measured in 5 sections collected at 100-µm intervals along the rostrocaudal axis in each animal. The numbers of SATB2+, BCL11B/CTIP2+, and TBR1+ neurons in an approximately 200-μm-wide columnar area, extending from the white matter to the pial surface of the somatosensory cortex, were manually counted using Imaris software. Statistical significance was determined using 2-tailed unpaired Student t tests.
Immunoblot analyses
Tissues were lysed in Triton-based cell lysis buffer (40 mM HEPES, 120 mM NaCl, 1 mM EDTA, 1.5 mM Na3VO4, 50 mM NaF, 10 mM β-glycerophosphate, 20 mM MoO4, 0.5% Triton X-100, protease inhibitor [Roche, 1836170001], phosphatase inhibitor [Sigma Aldrich, P5726]). Proteins in cleared lysates were electrophoretically separated on 4% to 12% Bis-Tris gels (Life Technologies, NP0335BOX). Proteins were then transferred to PVDF membranes. After incubation with a 5% skim-milk block, blots were probed with antibodies directed against the following targets: ULK1 (Sigma Aldrich, A7481), SQSTM1 (Sigma Aldrich, P0067), LC3B (Novus Biologicals, NB100–2220), ATG13 (Sigma Aldrich, SAB4200100), RB1CC1 (Cell Signaling Technology, 12436), ATG14 (MBL, PD026), CNTN2 (R&D, AF4439), NCAM1 (Developmental Studies Hybridoma Bank, 5B8), and GAPDH (Sigma Aldrich, G9545). Membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies (GE Healthcare, RPN4301, NA931), and bands were detected using chemiluminescence-detection kits (Amersham, RPN2232).
Quantitative real-time PCR
Total RNA was extracted from mouse cortex using the RNeasy Mini Kit (Qiagen 74104), according to the manufacturer's instructions. The reverse-transcription reaction was carried out using the SuperScript III first-strand synthesis kit (Life Technologies, 18080051) per the manufacturer's instructions. TaqMan gene-expression assays containing FAM-labeled primer/probe sets specific for Ulk1 (Mm-00437238_m1), Ulk2 (Mm-00497023_m1), Ulk3 (Mm01151463_m1), Ulk4 (Mm01349664_m1), Stk36 (Mm00558201_m1), and Rn18s (Hs99999901_s1) were obtained from Applied Biosystems. The real-time PCR reactions were performed in a total-reaction volume of 25 µL by using FastStart TaqMan Probe Master reagent (Roche, 04673409001), and results were analyzed using the ABI 7900 real-time PCR-detection system (Applied Biosystems, 4351405). Relative expression was normalized to Rn18s RNA and calibrated to the respective controls.
Electron microscopy
Quantification of autophagosome-like structures in callosal axons of 2-mo-old mice by transmission electron microscopy was performed as described previously.56 Briefly, mice were anesthetized with CO2, perfused transcardially with 10 mL 0.1 M phosphate buffer (pH 7.4) followed by 10 mL 2.5% glutaraldehyde: 2% PFA in 0.1 M CaCO4. The brains from 2 Ulk1/2-cDKO and 2 littermate controls were removed and fixed in the same fixative and postfixed in 2% osmium tetroxide in 0.1 M sodium cacodylate buffer with 0.3% potassium ferrocyanide for 2 h. Vibratome sections (100 µm) of the brain were cut sagittally and collected in cold PBS, rinsed in phosphate buffer, dehydrated through a series of graded ethanol-to-propylene oxide solutions, infiltrated and embedded in epoxy resin (Electron Microscopy Sciences, EMbed 812), and polymerized at 70°C overnight. Semithin (0.5-µm) sections were stained with toluidine blue for light microscope examination. Ultrathin (80-nm) sections were cut and imaged using an FEI Tecnai F20 TEM FEG electron microscope (Oregon, OH, USA) with an AT XR41 camera (Advanced Microscopy Techniques, Woburn, MA, USA). Fifty-five random images (1980 µm2 total) were collected from the CC region of each animal. The double-membrane bound structures were identified by 2 specialists from the electron microscopy facility of St. Jude Children's Research Hospital, who were blinded to the genotypes. The numbers of autophagosome-like structures per field are presented as mean ± SEM.
Axonal tracing
E18.5 brains were fixed in 4% PFA overnight at 4°C. Small crystals of 1,1′-dioctadecyl-3,3,38,38, tetramethylindocarbocyanine perchlorate (DiI; Molecular Probes, D-3911) were inserted into the parietal cortex, somatosensory cortex, or dTh to trace different axonal tracks. The tissues were stored in 4% PFA at 37°C for 3 to 4 wk and then cut on a VT1000S vibratome (Leica, Buffalo Grove, IL, USA) into 100-µm sections. Sections were counterstained with Hoechst 33258 (Molecular Probes, H3569) and mounted with 50% glycerol. To quantify the callosal projection in the contralateral cortex, the Dil fluorescence intensity was measured within an area drawn in cortical layers II to IV. For each animal, at least 3 sections were quantified, and the measures from the ulk1/2-DKO (n = 2) brain were normalized to the controls (n = 2). Values are shown as mean ± SEM.
Immunoprecipitation
Endogenous ULK1 was extracted from the cortex of P0 WT mice by using a Triton-lysis buffer. The lysates were incubated with anti-ULK1 antibody (Santa Cruz Biotechnology, sc10900) overnight at 4°C and precipitated with Protein G agarose beads (Thermo Scientific, 20399). Eluate was electrophoretically separated on 4% to 12% Bis-Tris gels (Life Technologies, NP0335BOX). Proteins were then transferred to PVDF membranes. Blots were probed with antibodies directed against the following targets: ULK1, ATG13, RB1CC1, and GAPDH.
Cytochrome oxidase staining
Brains were removed and fixed overnight in 4% PFA at 4°C. Barrel cortices were flattened on superfrost plus slides and cut tangentially (parallel to layer IV) into 100-µm sections on a vibratome. Free-floating sections were incubated with CO-reaction solution, which included 0.5 mg/mL DAB, 0.5 mg/mL cytochrome C (Sigma Aldrich, C2506), and 40 mg/mL sucrose in PBS at 37°C until staining appeared. Sections were washed 3 times in PBS, mounted with Aqua Polymount (Polysciences, 18606), and imaged using Eclipse 80i bright-field light microscopy (Nikon, Minato, Tokyo, Japan).
Supplementary Material
Funding Statement
This work was supported by grants from the National Heart, Lung, and Blood Institute (R01 HL114697 to M.K.), and ALSAC.
Abbreviations
- AC
anterior commissure;
- AMBRA1
autophagy/Beclin 1 regulator 1;
- Atg
autophagy related;
- BCL11B/CTIP2
B-cell leukemia/lymphoma 11B
- CC
corpus callosum;
- CHL1
cell adhesion molecule L1-like;
- cKO
conditional knockout;
- CNS
central nervous system;
- CNTN2
contactin 2;
- CTA
corticothalamic axon;
- Dil
1,1´-dioctadecyl-3,3,38,38, tetramethylindocarbocyanine perchlorate;
- DKO
double knockout;
- dTh
dorsal thalamus;
- E
embryonic day;
- ER
endoplasmic reticulum;
- Fas2
Fasciclin 2;
- HC
hippocampal commissure;
- KO
knockout;
- MKI67/KI67
antigen identified by monoclonal antibody Ki 67;
- NCAM
neural cell adhesion molecule 1;
- P
postnatal day;
- PBS
phosphate-buffered saline;
- PFA
paraformaldehyde;
- PSPB
pallial-subpallial boundary;
- RB1CC1/FIP200/ATG17
RB1 inducible coiled-coil protein 1;
- SATB2
special AT-rich sequence binding protein 2;
- SEC16A
SEC16 homolog A, endoplasmic reticulum export factor;
- SLC17A6/VGLUT2
solute carrier family 17 (sodium-dependent inorganic phosphate cotransporter), member 6;
- SLC6A4/SERT
solute carrier family 6 (neurotransmitter transporter, serotonin), member 4;
- SQSTM1/p62
sequestosome 1;
- TBR1
T-box brain gene 1;
- TBST
Tris-buffered saline with Tween 20;
- TCA
thalamocortical axon;
- ULK
unc-51 like kinase;
- unc-51
serine/threonine-protein kinase unc-51
Conflict of interest
The authors declare that they have no competing financial interests.
Acknowledgements
We are grateful to Dr. Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science) for sharing the Atg7flox/flox mouse line. We are grateful to the staff in the following shared resource facilities for their expertise and technical assistance: St. Jude Veterinary Pathology Core (Dr. Peter Vogel), and St. Jude Cell & Tissue Imaging Center (Dr. Victoria Frohlich). We are grateful to Dr. Angela McArthur for editing the manuscript.
References
- [1].Wang B, Kundu M. Canonical and noncanonical functions of ULK/Atg1. Curr Opin Cell Biol. 2017;45:47–54. doi: 10.1016/j.ceb.2017.02.011. PMID:28292700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. PMID:18305538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–7. doi: 10.1038/nrm2245. PMID:17712358. [DOI] [PubMed] [Google Scholar]
- [4].Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–305. doi: 10.1074/jbc.M900573200. PMID:19258318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. PMID:19225151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al.. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. PMID:19211835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang CC, Sheffler DJ, et al.. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell. 2015;59:285–97. doi: 10.1016/j.molcel.2015.05.031. PMID:26118643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741–50. doi: 10.1038/ncb2757. PMID:23685627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Park JM, Jung CH, Seo M, Otto NM, Grunwald D, Kim KH, Moriarity B, Kim YM, Starker C, Nho RS, et al.. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy. 2016;12:547–64. doi: 10.1080/15548627.2016.1140293. PMID:27046250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhou C, Ma K, Gao R, Mu C, Chen L, Liu Q, Luo Q, Feng D, Zhu Y, Chen Q. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 2017;27:184–201. doi: 10.1038/cr.2016.146. PMID:27934868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A, Hansmann I, Pfaffenwimmer T, Kijanska M, Stoffel I, et al.. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell. 2014;53:471–83. doi: 10.1016/j.molcel.2014.01.024 . PMID:24440502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, Hailey DW, Lippincott-Schwartz J, Tooze SA. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006;119:3888–900. doi: 10.1242/jcs.03172. PMID:16940348. [DOI] [PubMed] [Google Scholar]
- [13].Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112:1493–502. doi: 10.1182/blood-2008-02-137398. PMID:18539900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cheong H, Wu JM, Gonzales LK, Guttentag SH, Thompson CB, Lindsten T. Analysis of a lung defect in autophagy-deficient mouse strains. Autophagy. 2014;10:45–56. doi: 10.4161/auto.26505. PMID:24275123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yan J, Kuroyanagi H, Tomemori T, Okazaki N, Asato K, Matsuda Y, Suzuki Y, Ohshima Y, Mitani S, Masuho Y, et al.. Mouse ULK2, a novel member of the UNC-51-like protein kinases: unique features of functional domains. Oncogene 1999;18:5850–9. doi: 10.1038/sj.onc.1202988. PMID:10557072. [DOI] [PubMed] [Google Scholar]
- [16].Yan J, Kuroyanagi H, Kuroiwa A, Matsuda Y, Tokumitsu H, Tomoda T, Shirasawa T, Muramatsu M. Identification of mouse ULK1, a novel protein kinase structurally related to C. elegans UNC-51. Biochem Biophys Res Commun 1998;246:222–7. doi: 10.1006/bbrc.1998.8546. [DOI] [PubMed] [Google Scholar]
- [17].Ogura K, Wicky C, Magnenat L, Tobler H, Mori I, Muller F, Ohshima Y. Caenorhabditis elegans unc-51 gene required for axonal elongation encodes a novel serine/threonine kinase. Genes Dev 1994;8:2389–400. doi: 10.1101/gad.8.20.2389. PMID:7958904. [DOI] [PubMed] [Google Scholar]
- [18].Toda H, Mochizuki H, Flores R, 3rd Josowitz R, Krasieva TB, Lamorte VJ, Suzuki E, Gindhart JG, Furukubo-Tokunaga K, Tomoda T. UNC-51/ATG1 kinase regulates axonal transport by mediating motor-cargo assembly. Genes Dev. 2008;22:3292–307. doi: 10.1101/gad.1734608. PMID:19056884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mochizuki H, Toda H, Ando M, Kurusu M, Tomoda T, Furukubo-Tokunaga K. Unc-51/ATG1 controls axonal and dendritic development via kinesin-mediated vesicle transport in the Drosophila brain. PLoS One. 2011;6:e19632. doi: 10.1371/journal.pone.0019632. PMID:21589871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].O'Donnell M, Chance RK, Bashaw GJ. Axon growth and guidance: receptor regulation and signal transduction. Annu Rev Neurosci. 2009;32:383–412. doi: 10.1146/annurev.neuro.051508.135614. PMID:19400716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Joo JH, Wang B, Frankel E, Ge L, Xu L, Iyengar R, Li-Harms X, Wright C, Shaw TI, Lindsten T, et al.. The Noncanonical Role of ULK/ATG1 in ER-to-Golgi Trafficking Is Essential for Cellular Homeostasis. Mol Cell 2016;62:491–506. doi: 10.1016/j.molcel.2016.05.030 . PMID:27203176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tomoda T, Bhatt RS, Kuroyanagi H, Shirasawa T, Hatten ME. A mouse serine/threonine kinase homologous to C. elegans UNC51 functions in parallel fiber formation of cerebellar granule neurons. Neuron. 1999;24:833–46. doi: 10.1016/S0896-6273(00)81031-4. PMID:10624947. [DOI] [PubMed] [Google Scholar]
- [23].Zhou X, Babu JR, da Silva S, Shu Q, Graef IA, Oliver T, Tomoda T, Tani T, Wooten MW, Wang F. Unc-51-like kinase 1/2-mediated endocytic processes regulate filopodia extension and branching of sensory axons. Proc Natl Acad Sci U S A. 2007;104:5842–7. doi: 10.1073/pnas.0701402104. PMID:17389358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, et al.. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–5. PMID:17589504. [DOI] [PubMed] [Google Scholar]
- [25].Lv X, Jiang H, Li B, Liang Q, Wang S, Zhao Q, Jiao J. The crucial role of Atg5 in cortical neurogenesis during early brain development. Sci Rep. 2014;4:6010. doi: 10.1038/srep06010. PMID:25109817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vazquez P, Arroba AI, Cecconi F, de la Rosa EJ, Boya P, de Pablo F. Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy. 2012;8:187–99. doi: 10.4161/auto.8.2.18535. PMID:22240590. [DOI] [PubMed] [Google Scholar]
- [27].Wang C, Liang CC, Bian ZC, Zhu Y, Guan JL. FIP200 is required for maintenance and differentiation of postnatal neural stem cells. Nat Neurosci. 2013;16:532–42. doi: 10.1038/nn.3365. PMID:23542691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al.. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. PMID:16625204. [DOI] [PubMed] [Google Scholar]
- [29].Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al.. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. PMID:16625205. [DOI] [PubMed] [Google Scholar]
- [30].Liang CC, Wang C, Peng X, Gan B, Guan JL. Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem. 2010;285:3499–509. doi: 10.1074/jbc.M109.072389. PMID:19940130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–57. doi: 10.1038/nrn3961. PMID:25991442. [DOI] [PubMed] [Google Scholar]
- [32].Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci U S A. 2011;108:11121–6. doi: 10.1073/pnas.1107969108. PMID:21690395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Current Opinion in Cell Biology. 2010;22:132–9. doi: 10.1016/j.ceb.2009.12.004. PMID:20056399. [DOI] [PubMed] [Google Scholar]
- [34].Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32:2–11. doi: 10.1128/MCB.06159-11. PMID:22025673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lopez-Bendito G, Molnar Z. Thalamocortical development: how are we going to get there? Nat Rev Neurosci. 2003;4:276–89. doi: 10.1038/nrn1075. PMID:12671644. [DOI] [PubMed] [Google Scholar]
- [36].Ogura K, Goshima Y. The autophagy-related kinase UNC-51 and its binding partner UNC-14 regulate the subcellular localization of the Netrin receptor UNC-5 in Caenorhabditis elegans. Development. 2006;133:3441–50. doi: 10.1242/dev.02503. PMID:16887826. [DOI] [PubMed] [Google Scholar]
- [37].Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, et al.. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15:406–16. doi: 10.1038/ncb2708. PMID:23524951. [DOI] [PubMed] [Google Scholar]
- [38].Noda NN, Fujioka Y. Atg1 family kinases in autophagy initiation. Cell Mol Life Sci. 2015;72:3083–96. doi: 10.1007/s00018-015-1917-z. PMID:25948417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Alers S, Loffler AS, Paasch F, Dieterle AM, Keppeler H, Lauber K, Campbell DG, Fehrenbacher B, Schaller M, Wesselborg S, et al.. Atg13 and FIP200 act independently of Ulk1 and Ulk2 in autophagy induction. Autophagy. 2011;7:1423–33. doi: 10.4161/auto.7.12.18027. PMID:22024743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hieke N, Loffler AS, Kaizuka T, Berleth N, Bohler P, Driessen S, Stuhldreier F, Friesen O, Assani K, Schmitz K, et al.. Expression of a ULK1/2 binding-deficient ATG13 variant can partially restore autophagic activity in ATG13-deficient cells. Autophagy. 2015;11:1471–83. doi: 10.1080/15548627.2015.1068488. PMID:26213203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liang Q, Yang P, Tian E, Han J, Zhang H. The C. elegans ATG101 homolog EPG-9 directly interacts with EPG-1/Atg13 and is essential for autophagy. Autophagy. 2012;8:1426–33. doi: 10.4161/auto.21163. PMID:22885670. [DOI] [PubMed] [Google Scholar]
- [42].Tian E, Wang F, Han J, Zhang H. epg-1 functions in autophagy-regulated processes and may encode a highly divergent Atg13 homolog in C. elegans. Autophagy. 2009;5:608–15. doi: 10.4161/auto.5.5.8624. PMID:19377305. [DOI] [PubMed] [Google Scholar]
- [43].Miceli S, Negwer M, van Eijs F, Kalkhoven C, van Lierop I, Homberg J, Schubert D. High serotonin levels during brain development alter the structural input-output connectivity of neural networks in the rat somatosensory layer IV. Front Cell Neurosci. 2013;7:88. doi: 10.3389/fncel.2013.00088. PMID:23761736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen X, Ye R, Gargus JJ, Blakely RD, Dobrenis K, Sze JY. Disruption of Transient Serotonin Accumulation by Non-Serotonin-Producing Neurons Impairs Cortical Map Development. Cell Rep. 2015;10:346–58. doi: 10.1016/j.celrep.2014.12.033. PMID:25600870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].van Kleef ES Gaspar P, Bonnin A. Insights into the complex influence of 5-HT signaling on thalamocortical axonal system development. Eur J Neurosci. 2012;35:1563–72. doi: 10.1111/j.1460-9568.2012.8096.x. PMID:22607002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lai T, Garriga G. The conserved kinase UNC-51 acts with VAB-8 and UNC-14 to regulate axon outgrowth in C. elegans. Development. 2004;131:5991–6000. doi: 10.1242/dev.01457. PMID:15539493. [DOI] [PubMed] [Google Scholar]
- [47].Watari-Goshima N, Ogura K, Wolf FW, Goshima Y, Garriga G. C. elegans VAB-8 and UNC-73 regulate the SAX-3 receptor to direct cell and growth-cone migrations. Nat Neurosci. 2007;10:169–76. doi: 10.1038/nn1834. PMID:17237778. [DOI] [PubMed] [Google Scholar]
- [48].Ogura K, Shirakawa M, Barnes TM, Hekimi S, Ohshima Y. The UNC-14 protein required for axonal elongation and guidance in Caenorhabditis elegans interacts with the serine/threonine kinase UNC-51. Genes Dev. 1997;11:1801–11. doi: 10.1101/gad.11.14.1801. PMID:9242488. [DOI] [PubMed] [Google Scholar]
- [49].Tomoda T, Kim JH, Zhan CX, Hatten ME. Role of Unc51.1 and its binding partners in CNS axon outgrowth. Genes Dev. 2004;18:541–58. doi: 10.1101/gad.1151204. PMID:15014045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Delcroix JD, Valletta JS, Wu C, Hunt SJ, Kowal AS, Mobley WC. NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron. 2003;39:69–84. doi: 10.1016/S0896-6273(03)00397-0. PMID:12848933. [DOI] [PubMed] [Google Scholar]
- [51].Karagogeos D. Neural GPI-anchored cell adhesion molecules. Front Biosci. 2003;8:s1304–20. doi: 10.2741/1214. PMID:12957835. [DOI] [PubMed] [Google Scholar]
- [52].Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10:19–26. doi: 10.1038/nn0207-263b . PMID:17189949. [DOI] [PubMed] [Google Scholar]
- [53].Fukamauchi F, Aihara O, Wang YJ, Akasaka K, Takeda Y, Horie M, Kawano H, Sudo K, Asano M, Watanabe K, et al.. TAG-1-deficient mice have marked elevation of adenosine A1 receptors in the hippocampus. Biochem Biophys Res Commun. 2001;281:220–6. doi: 10.1006/bbrc.2001.4334. PMID:11178983. [DOI] [PubMed] [Google Scholar]
- [54].Malhotra JD, Tsiotra P, Karagogeos D, Hortsch M. Cis-activation of L1-mediated ankyrin recruitment by TAG-1 homophilic cell adhesion. J Biol Chem. 1998;273:33354–9. doi: 10.1074/jbc.273.50.33354. PMID:9837910. [DOI] [PubMed] [Google Scholar]
- [55].Pavlou O, Theodorakis K, Falk J, Kutsche M, Schachner M, Faivre-Sarrailh C, Karagogeos D. Analysis of interactions of the adhesion molecule TAG-1 and its domains with other immunoglobulin superfamily members. Mol Cell Neurosci. 2002;20:367–81. doi: 10.1006/mcne.2002.1105. PMID:12139915. [DOI] [PubMed] [Google Scholar]
- [56].Komatsu M, Wang QJ, Holstein GR, Friedrich VL Jr., Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104:14489–94. doi: 10.1073/pnas.0701311104. PMID:17726112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.