

ABSTRACT
Programmed necrosis, necroptosis, is considered to be a highly immunogenic activity, often mediated via the release of damage-associated molecular patterns (DAMPs). Interestingly, enhanced macroautophagic/autophagic activity is often found to be accompanied by necroptosis. However, the possible role of autophagy in the immunogenicity of necroptotic death remains largely obscure. In this study, we investigated the possible mechanistic correlation between phytochemical shikonin-induced autophagy and the shikonin-induced necroptosis for tumor immunogenicity. We show that shikonin can instigate RIPK1 (receptor [TNFRSF]-interacting serine-threonine kinase 1)- and RIPK3 (receptor-interacting serine-threonine kinase 3)-dependent necroptosis that is accompanied by enhanced autophagy. Shikonin-induced autophagy can directly contribute to DAMP upregulation. Counterintuitively, among the released and ectoDAMPs, only the latter were shown to be able to activate the cocultured dendritic cells (DCs). Interruption of autophagic flux via chloroquine further upregulated ectoDAMP activity and resultant DC activation. For potential clinical application, DC vaccine preparations treated with tumor cells that were already pretreated with chloroquine and shikonin further enhanced the antimetastatic activity of 4T1 tumors and reduced the effective dosage of doxorubicin. The enhanced immunogenicity and vaccine efficacy obtained via shikonin and chloroquine cotreatment of tumor cells may thus constitute a compelling strategy for developing cancer vaccines via the use of a combinational drug treatment.
KEYWORDS: autophagy, dendritic cell-based cancer vaccine, ectoDAMPs, necroptosis, stage IV mammary carcinoma
Introduction
Recently, cell demise in certain contexts has been shown to elicit immune responses against specific cellular determinants, a phenomenon described as immunogenic cell death (ICD).1,2 ICD can be characterized by the expression of damage-associated molecular patterns (DAMPs). DAMPs are a group of molecules that can confer specific cellular functions in normal live cells but are actively released from the stressed or injured cells or exposed on their cell surface, mediating the onset of a spectrum of cell-mediated immune responses.3 ICD can be found in 3 types of cellular activities, apoptosis, cell death with/by autophagy, or necrosis. Necrosis has been previously considered to be a result of accidental cell death. However, recent genetic and biochemical studies have revealed that there are multiple regulatory pathways for necrosis.4,5 Regulated necrosis, also termed necroptosis, was originally considered as a form of nonapoptotic cell death that takes place upon treatment of specific test cells with TNF in the presence of CASP/caspase inhibition.2,6,7 Other studies have also suggested that necroptosis could be mediated via different cell events and signaling pathways.8 Irrespective of the possible difference in the contributing pathways, necroptosis is commonly characterized by the assembly of receptor interacting protein (RIPK1)- and RIPK3-expressing necrosomes that could be inhibited by necrostatin-1 (NEC-1).9
Necroptosis has been suggested to generate immunogenic activity which is conventionally defined as a consequence of the release of DAMPs upon secondary necrosis.10 Conversely, the process of necroptosis has also been shown to be closely associated with enhanced autophagic activity.11-13 Although some evidence has demonstrated the indispensable roles of autophagy in the release of DAMPs and DAMP-mediated apoptotic ICD activity,14–17 the direct involvement of autophagy in DAMP-mediated immunogenicity in necroptotic death is lacking.
Hundreds of candidate or commercialized cancer drugs have been screened for their ability to induce ICD.18,19 Among these ICD drugs, shikonin (SK) is classified as a type I ICD inducer that promotes either apoptosis or necroptosis depending on the cell types and treatment regimens.20–27 In this study, we show that SK can induce RIPK1/3-mediated necroptosis in mouse mammary tumor cells, 4T1-luc2 cells. The resultant necroptotic cell death was accompanied by enhanced autophagy, which was in turn generated by an elevated level of reactive oxygen species (ROS). In addition, the ectolocalization of DAMPs was upregulated in an autophagy-dependent manner. The SK-treated mammary tumor cells were a very efficient treatment model when used in a DC-based cancer vaccine formulation. Most interestingly, this increase in immunogenicity can only be attributed to the surface DAMPs (ectoDAMPs), but not the soluble DAMPs. Moreover, the ectoDAMP activities in test cells were further enhanced by interrupting the autophagic flux, and the SK treatment with blockage of autophagic flux also effectively stimulated DC activation and the derived vaccine efficacy. Together, our findings demonstrate that the onset of immunogenicity by necroptosis can be attributed to autophagy-dependent DAMP expression. Our results also revealed a specific and alternative mechanism for mediating the immunogenicity of necroptosis in tumor cells, and provide a strategy for improving the DC-based cancer vaccine.
Results
Shikonin induced necroptosis in 4T1-luc2 tumor cells
Shikonin (SK) has recently been classified as an ICD inducer.18,19 that can instigate cell apoptosis and necroptosis, depending on the test concentrations and treated cell types.23,24,28,29 To determine the cell death mode of SK-treated, mouse stage IV mammary carcinoma 4T1-luc2 cells, increasing dosage of SK was applied and cell viability was determined at 24 h by MTT assay. Test 4T1-luc2 cells remained completely viable until the SK concentration reached 1 μM, at which point approximately 70% cells were still viable. Cell survival rate continued to decrease in a dose-dependent manner, and complete cell demise was detected at 16 μM SK. The IC50 value was determined to be 1.58 μM (Fig. 1A). Furthermore, our results showed that expression of both cleaved CASP3 (caspase 3) and CASP8 (caspase 8) were only detectable in the TNF plus cycloheximide (TC) treatment and these activities were not detectable with any of the other SK treatments (Fig. 1B). Results obtained from the time course experiment were also consistent with the notion that 4T1-luc2 cells treated with SK under our test conditions did not undergo apoptosis (Fig. S1A). In accordance, SK-mediated cell death was also not rescued by the pancaspase inhibitor, zVAD-fmk (Fig. 1C). In addition, SK-induced cell death was not affected by treatment with the autophagy inhibitor, 3-methyladenine (3-MA) (Fig. 1C) and, consistently, when expression of ATG5 and BECN1/beclin-1 was suppressed by treatment with specific siRNA (Fig. 1E). Together, these results demonstrate that 4T1-luc2 cell demise resulting from SK treatment could not have been attributed to apoptosis or excessive autophagic activities. In contrast, as the inhibitors that specifically diminish necroptotic activity via binding to RIPK1 and RIPK3, necrostain-1 (NEC-1)7 and GSK'87230 were indeed able to protect test 4T1-luc2 cells from death rendered by such SK treatment (Fig. 1C). In addition, specific siRNAs, such as siRipk1 and siRipk3, were used to inhibit necroptosis and were applied to further support the possible mechanism of necroptosis signaling in SK-induced cell death by using ANXA5/annexin V and propidium iodide (PI) double-staining assay. As Fig. 1E shown, SK mainly induced ANXA5- and PI-double-positive cells, and much fewer ANXA5-positive and PI-negative cells. This result suggests that 4T1 cells treated with SK underwent necrosis, rather than early apoptosis. In cells treated additionally with siRipk1 or siRipk3, SK-induced cell death was drastically inhibited, i.e., with SK treatment ANXA5- and PI-double-positive cells were drastically reduced from 75% to around 20% or even 15%. We also detected cell death in treatment with a known necroptotic stimulus, TNF + cycloheximide + zVAD-fmk (TCZ), in combination. The level of ANXA5- and PI-double-positive cells with TCZ treatment was similar to that observed for SK treatment. In cells treated additionally with siRipk1 or siRipk3, SK-induced cell death was drastically inhibited, i.e., with SK treatment ANXA5- and PI-double-positive cells were drastically reduced from 75% to around 20% or even 15%. This result suggests that SK-induced necrotic cell death can be regulated by the RIPK1 and RIPK3 signaling pathway system. According to RIPK3-dependent necroptosis, SK also increased the expression of phosphorylated MLKL in a dosage-dependent way, and this effect was also detected in necroptotic cells (via TCZ treatment) (Fig. S1B). Together, our results strongly suggest that a complex mode of RIPK1/3 involving necrosome activity is necessary for the onset of necroptosis in 4T1-luc2 cells when treated by 5 μM SK for 24 h. When subjected to morphological examination revealed by transmission electron microscopy (TEM), 4T1-luc2 cells treated as above displayed severe vacuolation and mitochondria swelling without chromatin condensation, and all of which were absent in untreated control counterparts (Fig. 1F). Based on these results, we thus conclude that 4T1-luc2 cells treated with SK for 24 h exhibited strong necroptotic activity under our experimental conditions.
Figure 1.
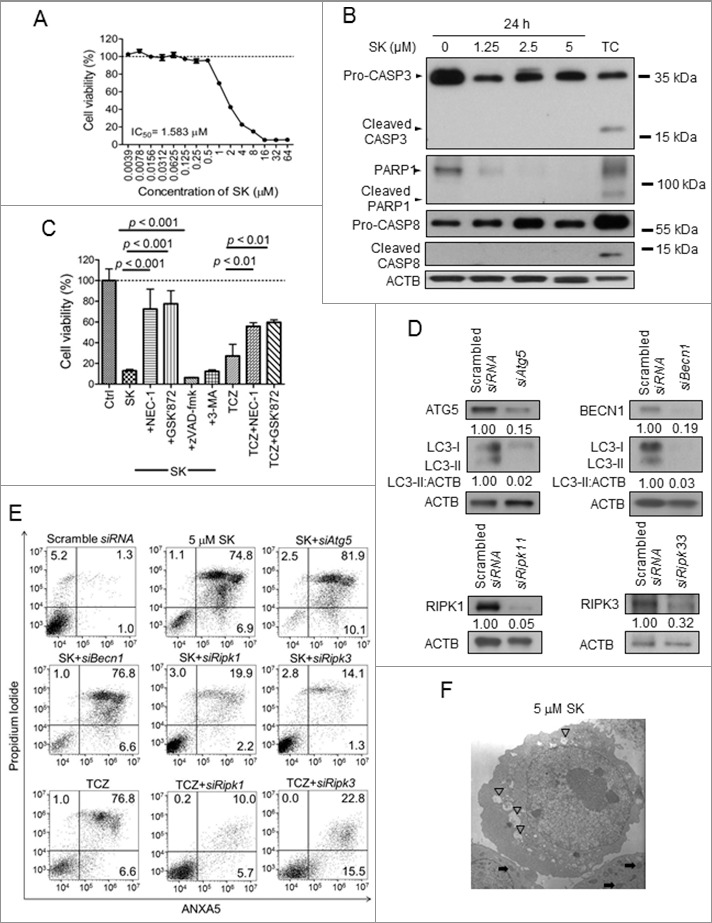
Shikonin (SK) induced necroptosis in 4T1-luc2 tumor cells. (A) The effect of SK on cell viability. 4T1-luc2 cells were treated with increasing concentrations of SK for 24 h and cell survival was determined by MTT assay. (B) Effect of SK on enzyme markers. Test cells were treated with 0, 1.25, 2.5, and 5 µM SK for 24 h and total cell lysates were harvested and subject to SDS-PAGE. Combinational treatment with 20 ng/mL TNF and 10 µg/mL cycloheximide (TC) for 24 h was applied as the positive control for apoptosis. Expression of CASP8, CASP3, and PARP1 was determined by western blot analysis. (C) Effect of NEC-1, GSK'872, zVAD-fmk, and 3-MA on SK-induced cell death and TCZ (TNF+cycloheximide+zVAD-fmk)-induced necroptosis. 4T1-luc2 cells were treated with 50 µM SK for 24 h and in presence or absence of 50 µM NEC-1, 40 µM zVAD-fmk, 5 mM 3-MA, or 3 µM GSK'872. Test cells also were treated with 20 ng/mL TNF, 10 µg/mL cycloheximide, and 40 µM zVAD-fmk in combination for 24 h as the necroptotic stimulus. Cell viability was determined using the MTT assay. (D) Efficacy of specific siRNAs for Atg5, Becn1, Ripk1, and Ripk3 in 4T1-luc2 tumor cells. 4T1-luc2 cells were transfected with siAtg5, siBecn1, siRipk1, and siRipk3 for 24 h and interference efficacy was determined by using western blot at 72 h post transfection. Numbers below each strip indicate the relative staining intensities of test proteins. (E) Effect of knocking down siAtg5, siBecn1, siRipk1, and siRipk3 expression on SK-mediated cytotoxicity and TCZ-induced necroptosis. 4T1-luc2 cells with or without treatment with siAtg5, siBecn1, siRipk1, or siRipk3 siRNA were then treated with 5 µM SK or TCZ for 24 h and cell viability was determined by ANXA5 and PI staining. (F) Subcellular morphology of SK-treated cells. Ultrastructure of typical 4T1-luc2 cells treated with 5 μM SK for 24 h was visualized by transmission electron microscopy. Numerous swollen mitochondria ( ) and vacuoles (▽) were observed as indicated. Data are expressed as mean SD of triplicate determinations. Data presented are from one of 3 representative experiments.
) and vacuoles (▽) were observed as indicated. Data are expressed as mean SD of triplicate determinations. Data presented are from one of 3 representative experiments.
SK-treated 4T1-luc2 cells effectively immunized mice against primary tumors
One key criterion for effecting ICD activity is the capability of the in vitro treated tumor cells to elicit an immune protection response in mice against a subsequent challenge with the untreated tumor cell counterparts in the absence of any adjuvant treatment.31,32 To examine whether the SK-treated 4T1-luc2 cells can die from the ICD pathway, we then carried out the following experiments. Two groups of 10 wild-type mice each were immunized via subcutaneous injection with either 105 or 5 × 105 4T1-luc2 cells treated by 5 μM SK for 24 h. Sham operation and mice immunized with the same number of 4T1-luc2 cells that underwent freeze and thaw (F/T) cycles were included as control mice. At 7 d postvaccination, mice were orthotopically implanted into mammary fat pad with 5 × 105 live 4T1 tumor cells. Tumor growth was measured every 3 d and mice survival was monitored starting at 7 d post-tumor implantation. As shown in Fig. 2A, in comparison with the control mice groups and F/T treatment groups, mice treated with half a million, dying SK-treated 4T1 cells showed significantly less activity in tumor growth (Fig. 2A). In accordance, this group of vaccinated mice also showed a lower rate of tumor formation (Fig. 2B) and a prolonged survival time (Fig. 2C). The bioluminescence imaging (BLI) data further demonstrated the substantial effect on primary tumor growth (Fig. 2D). Human breast cancers with triple-negative (TN) characteristics, the estrogen receptor-negative, progesterone receptor-negative, and human epidermal growth factor receptor 2-negative phenotypes, are resistant to target therapies and possess the highest relapse and metastasis rates among breast cancers.33,34 Therefore, we next investigated whether SK-treated 4T1 cells could mediate a therapeutic benefit on distant visceral metastasis. In this treatment model, mammary tumors orthotopically implanted were removed at 18 d postimplantation. One day post tumor resection, mice were subjected to vaccination via intraperitoneal (i.p.) injection of 5 × 105 SK-treated 4T1-luc2 cells once a wk for 2 consecutive wk. Mice with sham operation and 4T1-luc2 cells with F/T were used as controls. Post-tumor-resection metastasis was determined by bioluminescence imaging (BLI) results (Fig. S2A) and survival rates were recorded. As shown in Fig. S2B, after tumor resection, tumor metastasis in test mice developed with virtually identical kinetics regardless of the treatments. In addition, survival benefits were also not observed (Fig. S2B and S2C). Therefore, whereas SK-instigated ICD activity may be effective against the primary mammary tumor formation, it failed to confer protection against tumor metastasis under the specific experimental conditions shown in Fig. S2B and S2C. As direct vaccination with necroptotic 4T1-luc2 cells failed to protect test mice from tumor metastasis, we next attempted to evaluate the capacity of a DC-based vaccine regimen.
Figure 2.
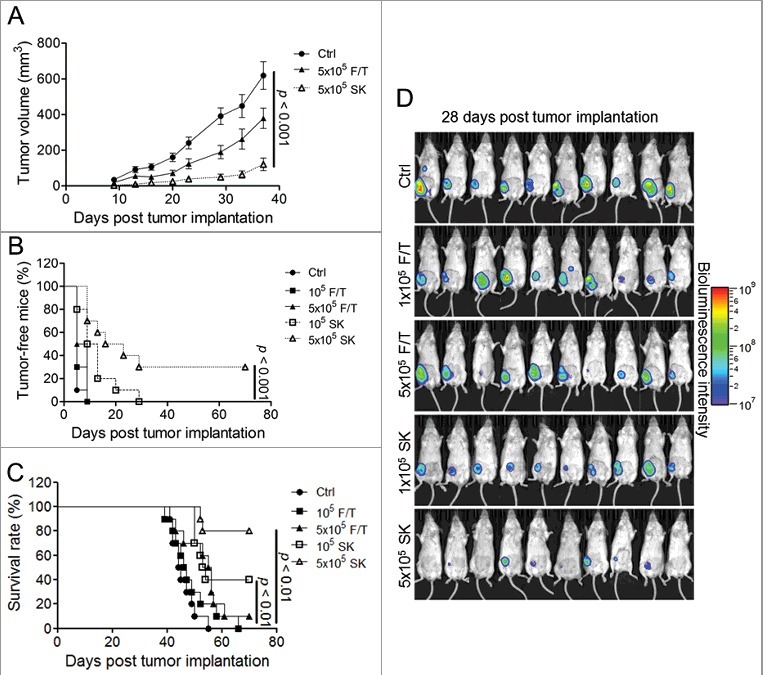
SK-treated 4T1-luc2 cells effectively immunized mice against primary tumors. Test mice (n = 10) were vaccinated with varying dosages (cell numbers) of F/T treated 4T1-luc2 cells or test cells treated with 5 μM SK for 24 h. At 7 d post-vaccination, live 4T1-luc2 tumor cells were implanted. (A) Tumor growth rate. Tumor volume was monitored until 37 d post tumor implantation. (B) Tumor-free incidence and (C) mouse survival rates were recorded until 70 d post tumor implantation. (D) Tumor progression in test mice visualized by bioluminescence imaging (BLI). BLI was determined at 28 d post tumor implantation. Fluorescence images are shown to present data on tumor growth.
SK-DC vaccine is effective in preventing metastasis
To demonstrate the in vivo efficacy of a DC-based vaccine we generated as described above, 10-d BM-DCs samples were cocultured with 4T1-luc2 cells which were treated with 5 μM SK for 24 h or underwent 4 repeats cycles of F/T. Naïve DCs, F/T-DCs, and pulsed DCs (SK-DCs) were then washed and isolated and 1 × 106 cells were i.p. injected into test mice, starting at one d post tumor resection. Mice were vaccinated weekly for 2 wk. Mice of the counterpart group were treated with 5 mg/kg doxorubicin (Dox), as scheduled with DC vaccine. In addition, a therapy regimen combining the DC vaccination with Dox (Dox + SK-DCs) was also performed with identical scheduling. The percentages of tumor-free mice and survival rates were monitored and recorded up to 80 days post tumor implantation. Results of this 4T1 tumor-resection experiment showed that as compared with phosphate-buffered saline (PBS)-treated, naïve DCs (DCs), and F/T-DCs mouse group, all treatment groups showed a significantly lower level of metastasis and prolonged survival time (Fig. 3A and 3B). Dox is known to be quite effective in suppression of tumor metastasis, which was conferred a 66% metastasis-free rate at the end of the experiment (P < 0.001), and the DC vaccine treatment was comparatively effective in this regard (P < 0.01). In comparison with the mock-treated mice that die with a median survival of 33 d, experimental treatment groups all showed significant improvement in median survival (Fig. 3B). The survival time of Dox-treated mice (P < 0.01), SK-DCs (P < 0.01), and combinational therapy (P < 0.001) were all significantly different from F/T-DCs treated mice. Furthermore, the bioluminescence imaging (BLI) data at 40 d post-tumor implantation further showed the antimetastatic effect of all treatment groups as compared with the PBS-treated group (Fig. 3C). In our other similar experiment, tumor-free mice and survival rates were monitored and recorded up to 160 d post-tumor implantation. Interestingly, this result showed that DC vaccine and combinational therapy could maintain the higher survival rates and low metastasis incidence of test mice (50%) at the experimental endpoint as compared with Dox-treated group (20%) (Fig. S3A and S3B). Based on our findings, we consider that this vaccine could be more benefit in prolonging survival rate of test mice than the chemotherapy alone under our experiment conditions.
Figure 3.
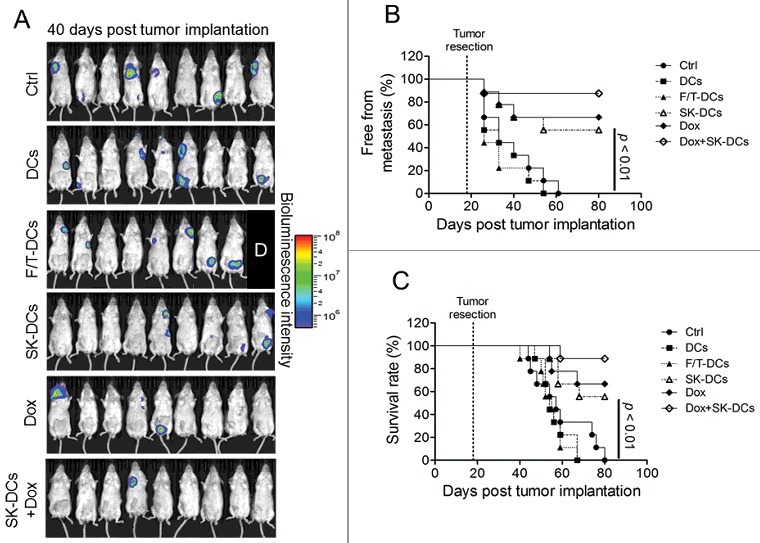
SK-DC vaccine is effective in preventing metastasis. Mice (n = 9) were implanted with 4T1-luc2 cells and tumors were surgically removed at 18 d postimplantation. Test mice were then vaccinated 1 d later via i.p. injection with 1 × 106/100 μL naïve DCs, F/T-treated 4T1 cell-pulsed DCs, or SK-treated 4T1 cell-pulsed DCs (SK-DCs). Two groups of mice were also treated intravenously with 5 mg/kg Dox, but with or without i.p. injection of test DC vaccine, named the Dox and Dox + SK-DCs groups. (A) Bioluminescence imaging of metastatic tumor progression in mice followed. Levels of tumor metastasis in test mice at 40 d post tumor implantation were determined by BLI. The label “D” indicates the mouse died before imaging. The metastasis-free incidence (B) and survival rate (C) of test mice were recorded until 80 d post- tumor implantation. Log-rank test was applied for statistical analyses of survival rate and metastasis-free incidence analysis.
SK-induced DAMP release and DAMP ectolocalization by 4T1-luc2 cells
The success of a DC vaccine regimen has been reported to be most likely attributed to the upregulated expression of DAMPs by treated tumor cells that in turn activate the cocultured DCs.35 To examine the release of DAMPs by SK-treated 4T1-luc2 cells, conditioned culture media were collected from test cells in culture and subjected to western blot analysis. As shown in Fig. 4A, the release of DAMPs including HSP90AA1, HSPA1A and CALR (calreticulin) was found to be gradually increased when cells were treated with increasing concentrations of SK. The levels of both LDHA (lactate dehydrogenase A) and the examined DAMPs, except CALR, in test media from SK-treated cells were similar to those collected from F/T treated cell counterparts. The extracellular expression of test DAMPs was verified by using correspondent ELISA, which was consistent to our results in western blot analysis (Fig. S4). These results suggested that at 24 h post SK treatment, 4T1-luc2 cells were basically intact in comparison to F/T-treated cells. To our surprise, expression of ectoDAMPs that have been previously reported only for apoptotic cells36 was also readily detected in our test cells. SK-treated tumor cells were visualized for expression of ectoDAMPs by confocal microscopy through indirect immunofluorescent staining. The expression of DAMPs including HSPA1A, HSP90AA1, and CALR in SK-treated tumor cells was mainly located on the cell surface as compared with untreated tumor cells (Fig. 4B). At 1 h post treatment with 2 μM or 5 μM SK, approximately 20% and > 60% 4T1-luc2 cells were detected to induce the ectolocalization of HSP90AA1, respectively (Fig. 4C, upper panel). At 2 μM SK, the population of test cells having ecto-HSP90AA1 kept increasing to approximately 50% over a time course of 9 h of treatment, and then remained at a plateau level. At the test concentration of 5 μM, the ectolocalization of HSP90AA1 was reached and maintained at a maximal level of approximately 80% of test cells (Fig. 4C, upper panel), beginning at 3 h post-treatment and was then sustained there after (Fig. 4C, upper panel). The ectolocalization kinetic of HSPA1A also followed a similar dose- and time-dependent pattern, but considerably lower percentages (approximately 38% to 56%) of test cells were involved (Fig. 4C, middle panel). CALR was known to provide the “eat me” signal for phagocytes to pick up the apoptotic cells. In this study, we were able to detect the upregulation of ecto-CALR on SK-treated cells, though at a much lower level (14 to 31%) with fewer test cells responding (Fig. 4C, lower panel). As for the expression intensity at the cellular level, 5 μM SK elicited the highest ectolocalization level of 3 DAMPs among all test dosages (Fig. 4D).
Figure 4.
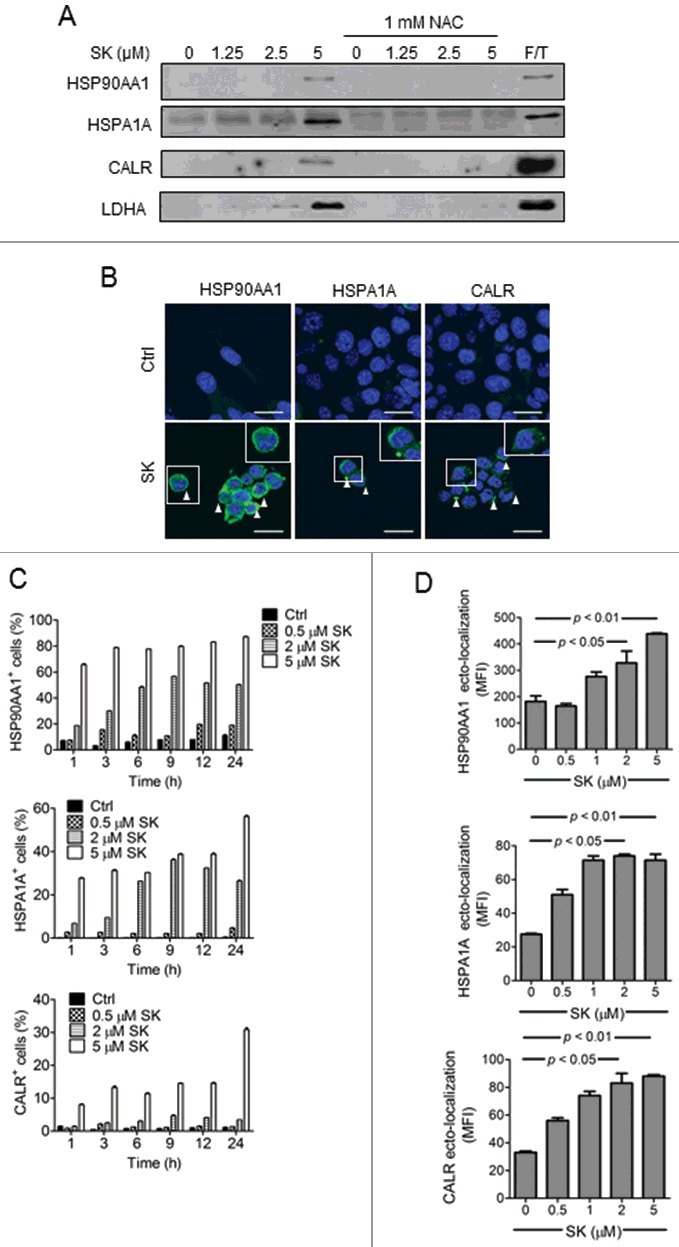
SK induced DAMP release and DAMP ectolocalization by 4T1-luc2 cells. (A) Effect of SK on the release of test DAMPs and LDHA from tumor cells. 4T1-luc2 cells were treated with increasing concentrations of SK for 24 h and test culture media were harvested. Proteins in conditioned media were fractionated by SDS-PAGE and subjected to western blotting using anti-HSP90AA1, HSPA1A, and CALR Ab. LDHA was used as a control. (B) Imaging of DAMP ectolocalization in SK-treated cells. The expression levels of a single surface DAMP (white triangle) were visualized by confocal microscopy and compared among treatment groups. Ectolocalizations of HSP90AA1, HSPA1A, and CALR were compared. Bar: 20 µm. (C) Time course and dosage effect of SK on 4T1 cells. Test cells treated with different concentrations of SK for 1 to 24 h were subjected to the corresponding antibody staining and subjected to flow cytometry analysis. Percentages of cells expressing respective ectoDAMP were determined. (D) The dosage-dependent effect of SK on the ectolocalization of HSPA1A/90 and CALR. Test cells treated for 24 h with the indicated SK dosage were analyzed for the ectolocalization of DAMPs. Data are expressed as mean ± SEM of triplicate determinations. P value was determined by a one-way ANOVA with Tukey's test. Data presented are from one of 3 representative experiments. MFI, mean fluorescence intensity.
Immunogenicity of SK-induced 4T1-luc2 ICD occurs in a cell-to-cell-interaction-dependent manner
The immunogenicity of cell necroptosis has been previously attributed to passively secreted or released DAMPs. Given that both the secreted and the surface forms of DAMPs were upregulated by SK-treated 4T1-luc2 cells, we decided to examine their respective contribution to the action of DC activation. Test 4T1-luc2 cells were treated with 5 µM SK for 24 h, cells were pelleted and the supernatants were collected. Pelleted tumor cells were resuspended and cocultured with 10-d bone marrow-derived DCs (BM-DCs) for 24 h. Another set of BM-DCs were cultured with the supernatant. Test 4T1-luc2 cells were also cultured with BM-DCs by using transwell apparatus. BM-DCs were harvested at 24 h postculturing and the expression of CD40, CD80, CD86, and H2-I molecules was quantified by flow cytometry analysis. Fig. 5A shows that BM-DCs constitutively expressed basal levels of the 4 specific markers examined. Culturing DCs in the DAMP-containing, conditioned medium collected from necroptotic 4T1-luc2 cells did not result in any change in the expression of test markers (Fig. 5B, upper panel). These results were also supported by the results obtained from the transwell experiments, which showed that the release of DAMPs into the culture medium was unable to efficiently activate test DCs (Fig. 5B, middle panel). Interestingly, expression of 4 test markers on test DCs were reduced in coculture with untreated 4T1-luc2 cells. In contrast, the upregulation of 4 test markers was able to detect on DCs in coincubation with SK-treated 4T1-luc2 cells as comparison with untreated 4T1-luc2 cells (Fig. 5B, lower panel). To determine the role of ectoDAMPs in tumor cells on DC activation, treated tumor cells were incubated with anti-HSPA1A, anti-HSP90AA1, and anti-CALR antibodies for 2 h prior to coculture with BM-DCs. As shown in Fig. 5C, the expression of 4 test molecules were reduced to a similar level of untreated tumor cells as the ectolocalization of DAMPs in SK-treated tumor cells was retarded by the antibody treatment. Importantly, these apparent cellular interaction-mediated activities were substantially increased upon direct interaction of test DCs with SK-treated 4T1-luc2 cells. In accordance, significantly higher levels of IL6 (interleukin 6), IL1B (interleukin 1 beta), and IL12B (interleukin 12B) were also produced by the cocultured BM-DCs (Fig. 5D). We also conducted an in vivo experiment to block the function of ectoDAMPs with neutralizing antibodies in a DC-based therapy model, and investigated the contribution of ectoDAMPs to immunogenicity in necroptotic tumor cells. Our results show that the SK-pulsed DC vaccine (SK-DCs) was able to significantly reduce 75% of test tumor metastasis and prolonged survival time (75% prolonged survival) of test mice at the end of experimental period (at 83 d post-tumor implantation). Furthermore, when DCs were pulsed with SK-treated tumor cells in which the function of SK-induced ectoDAMPs was disrupted by the specific antibodies treatment, the antimetastasis efficacy of the test vaccine (SK+Abs-DCs) was effectively reduced to the level of the control group (Ctrl). From bioluminescence imaging on 47 d post tumor implantation, 6 and 4 test mice (n = 8) had visible tumor metastasis in Ctrl and SK+Abs-DCs groups. In contrast, the SK group had only 2 test mice with tumor metastasis (Fig. S5A). In addition, the median of survival time for the SK+Abs-DCs group (71.5 days) was similar to that of the Ctrl group (69 days) (Fig. S5C). These results together strongly suggest that it is the presence of cell surface ectoDAMPs, instead of the passively released DAMPs secreted from treated 4T1-luc2 cells, that instigated the in vitro activation of testing DCs.
Figure 5.
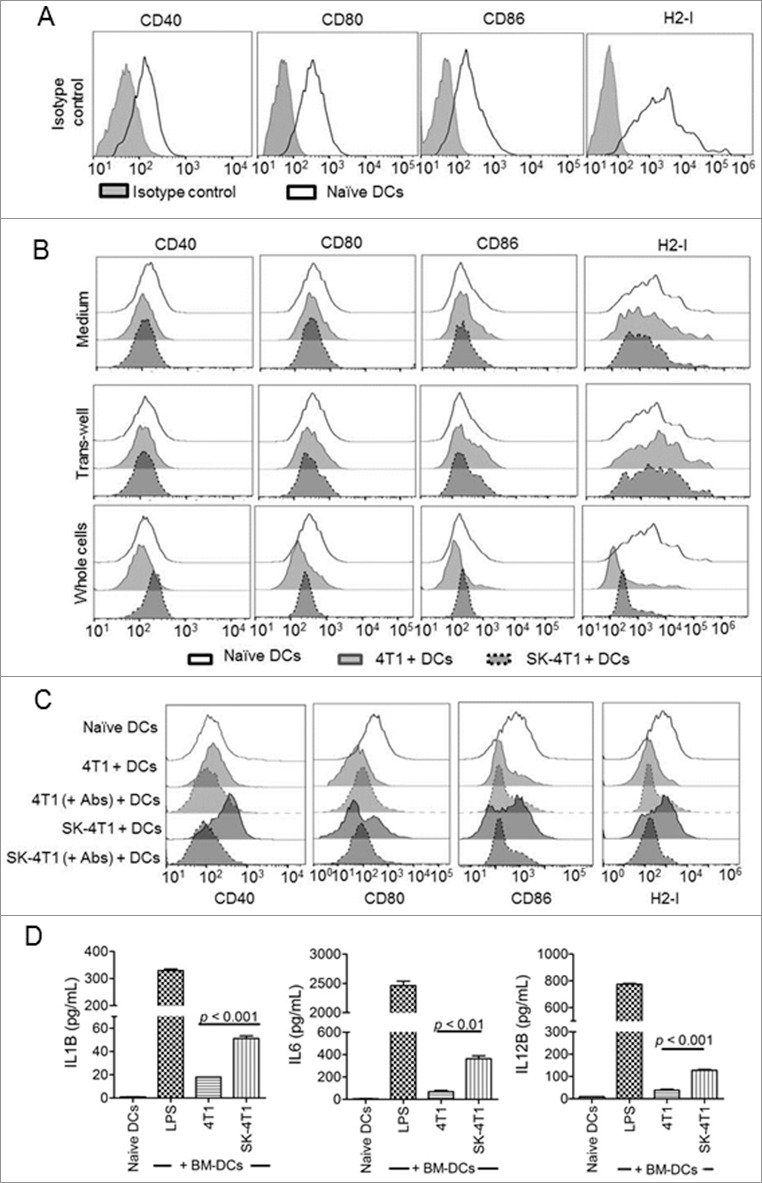
Immunogenicity of SK-induced 4T1-luc2 ICD occurs in a cell-to-cell-interaction-dependent manner. (A) Expression of test activation markers on naïve DCs. The expression of CD40, CD80, CD86, and H2-I molecules was determined on 10-d-old freshly purified BM-DCs (thin line/white area). The corresponding isotype controls were also used (gray area). (B) The effect of ectoDAMPs versus the release of DAMPs on DC activation. 4T1-luc2 cells treated in the presence or absence of 5 μM SK for 24 h were centrifuged. The resulting supernatants were collected and used as conditioned media for BM-DC culturing. The expression of 4 activation markers in DCs was analyzed at 24 h postcoculturing with tumor cells (upper panel). 4T1-luc2 cells that were collected were washed, resuspended, and used to coculture BM-DCs using a transwell apparatus. DCs were harvested 24 h after incubation and the cell activation status determined (middle panel). Aliquots of collected 4T1-luc2 cells were also mixed with BM-DCs at a 1:1 ratio and expression of CD40, CD80, CD86, and H2-I molecules were determined by flow cytometry analysis (lower panel). (C) Antibody blocking of ectoDAMP expressions in SK-treated tumor cells suppressed DC activation. SK-treated 4T1 cells were incubated with or without anti-HSPA1A, anti-HSP90AA1, and anti-CALR antibodies (+ Abs) for 2 h before they were cocultured with BM-DCs. After incubation for 24 h, test cells were harvested and the expression of CD40, CD80, CD86 and H2-I molecules on BM-DCs was determined via flow cytometry analysis. (D) The effect of SK-treated cells on expression of specific proinflammatory cytokines in DCs. Conditioned coculture medium of DC + 4T1 cells was harvested and the levels of secreted IL1B, IL6, and IL12B were determined by ELISA. Data are expressed as mean SEM of triplicate determinations. P value was determined by a one-way ANOVA with Tukey's test. Data presented are from one of 3 representative experiments.
SK induced autophagy in 4T1-luc2 cells
A number of recent studies have illustrated the indispensable role of autophagy in DAMP release and the subsequent cell death-mediated immune responses.37 However, the involvement of autophagy in the ectolocalization of DAMPs during cell necroptosis has not been previously revealed. To examine if autophagy can be triggered in tumor cells by SK, cell lysates were prepared from 4T1-luc2 cells that were treated for 24 h with increasing concentrations of SK, then harvested and analyzed for autophagic activities. As shown in Fig. 6A, de novo formation of the ATG12-ATG5 complex and synthesis of ATG7 were found to be gradually increased at relatively high concentrations of SK.31 Lipidated MAP1LC3B (MAP1LC3B-II/LC3B-II) level was also increased upon SK treatment with a drastically similar dose-dependent pattern. In addition, time course studies of 5 μM SK-treated cells showed that autophagic activity was enhanced in a temporal manner, starting at 6 h and continuing to increase throughout the 24-h experimental period (Fig. 6A, right panel). At the last time point tested, LC3-II level was further enhanced when 4T1-luc2 cells were treated with 5 μM SK by combination with chloroquine (CQ), and this is a unique characteristic of an autophagic flux (Fig. 6B).37 These results were further analyzed using confocal microscopy, which showed LC3B-positive green punctate staining in the SK-treated and CQ-treated cells and in cells cotreated with both SK and CQ (Fig. 6C, left panel), but not in control sample. The level of puncta in test cells was then quantitatively analyzed, and the results showed that the numbers of LC3B-positive puncta were significantly higher than those presented in control counterparts (Fig. 6C, right panel). Treatment of cells with SK in combination with CQ was observed to further enhance the formation of LC3B activity in this assay. In addition, 4T1-luc2 cells were then transfected with plasmids containing mCherry-EGFP-LC3B; mCherry+ GFP+ and stable cell clones were selected. Valid mCherry+ GFP+ stable clones were then treated with 5 μM SK, and the effect examined by confocal microscopy to characterize the sequential cellular activities of autophagy. As shown in Fig. 6D, all green fluorescent puncta in small sizes were found to coemit red fluorescence, indicating the bona fide autophagosome nature of treated cells.38 In contrast, the majority of puncta emitted only the red fluorescence. These red-only puncta were known to be characteristic of the more acidic autolysosomes. In addition, such SK-treated 4T1-luc2 cells were examined by TEM to observe the double-membrane structure, which is the critical ultrastructure of autophagosomes.37,39 The result shown in Fig. 6E indicated that SK induced autophagosomes that enclosed some membranous organelles. Together, our results thus indicate that compared to the untreated cells, readily detectable autophagic activities were induced in SK-treated cells at all tested concentrations.
Figure 6.
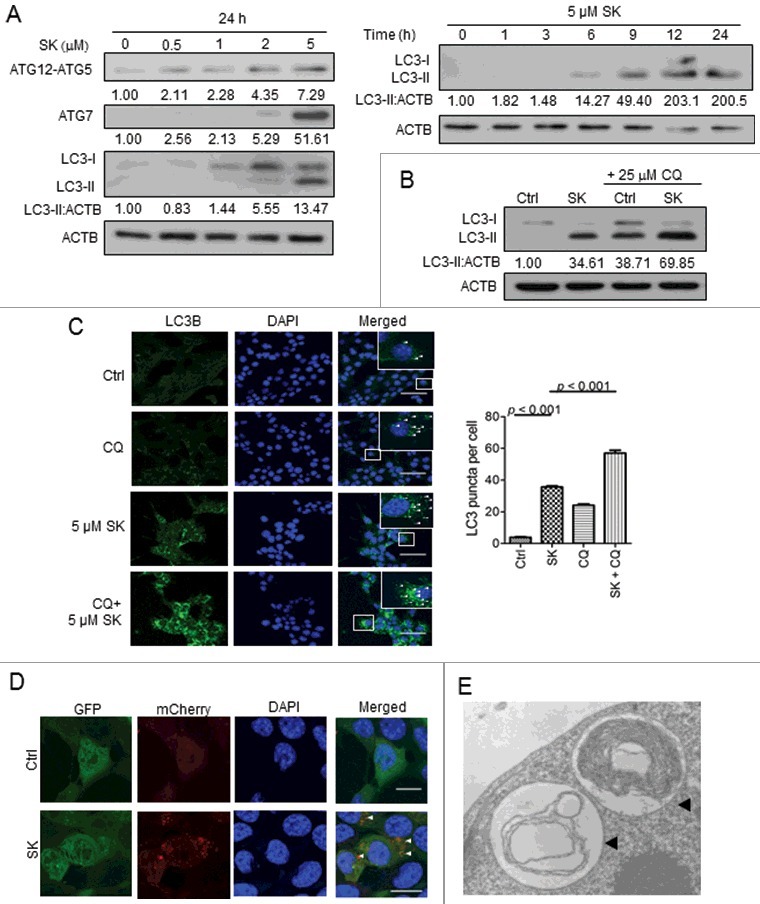
SK induced autophagy in 4T1-luc2 cells. (A) Effect of SK on the expression of autophagy-related proteins in test cells. Expression of the ATG12–ATG5 complex, ATG7 and LC3B in 4T1-luc2 cells treated with increasing concentrations of SK for 24 h were sequentially analyzed by SDS-PAGE and western blot analysis. The numbers under each strip represent the relative protein densities of each treatment compared to the 0 h control (left panel). Relative expression ratios of LC3-II in cells treated with 5 µM SK were examined in a time course experiment (right panel). (B) Effect of SK on autopagic flux. LC3-I and LC3-II expression in 4T1-luc2 cells treated with 5 µM SK for 24 h in the presence or absence of CQ, determined by western blot analysis. (C) Effect of SK on LC3B expression at the subcellular level. LC3B proteins stained as fluorescent puncta were detected by indirect immunofluorescence using anti-LC3B monoclonal antibody and resolved by confocal microscopy in 4T1-luc2 cells treated with SK as stated above, in the presence or absence of CQ (left column, left panel). Nuclei were stained with DAPI (in blue color, middle column). Subcellular localization of puncta was indicated (right column) and scored. Bar: 60 µm. The puncta (right panel) value indicates the mean SEM scored from at least 30 cells. Data are expressed as mean SEM of triplicate determinations. P values were determined based on a one-way ANOVA analysis with Tukey's test. (D) Effect of SK on autophagic flux as visualized through the expression of mCherry-LC3B-GFP. 4T1-luc2 cells stably expressing transgenic mCherry-LC3B-GFP were subjected to SK treatment as above and resolved by confocal microscopy. Cells expressing mCherry-LC3B or GFP-LC3B are indicated. Subcellular location was assessed by DAPI (in blue). Colocalization of mCherry- with GFP-LC3B puncta is also shown. Bar: 15 µm. Data presented are representative one of 3 different experiments. (E) Subcellular morphology of SK-induced autophagy in test cells. Ultrastructure of 5 µM SK-treated 4T1-luc2 cells was revealed by transmission electron microscopy. The double-membrane structures of autophagosomes (black arrowheads) are indicated.
SK-induced DAMP ectolocalization is closely associated with the enhanced autophagic activity
As shown above in this study, we clearly demonstrated that SK elicited autophagic activity in 4T1-luc2 cells. We then investigated the association of this autophagy with DAMP ectolocalization. The expression of ATG5 and BECN1 in 4T1-luc2 cells was first diminished through siRNA knockdown. Test cells were then subjected to treatment with 5 μM SK for 24 h, a time point at which the ectolocalization of DAMPs was found to have plateaued. As shown in Fig. 7A, ectolocalization of examined DAMPs, including CALR and HSP90AA1, was substantially (> 70%) suppressed and ectolocalization of HSPA1A was partially inhibited (> 40%) upon the inhibition of autophagy by Atg5 and Becn1 siRNAs, suggesting that their expression is an autophagy-dependent activity. Similarly, these effects on inhibition of SK-induced DAMP ectolocalization could also be found in necroptosis-deficient cells (siRipk1 and siRipk3). To further characterize the association of autophagy formation with the expression of DAMP, mCherry-GFP-LC3B transfected stable cell clones were treated as above and cell lysates were prepared. Proteins conjugated with mCherry that remained attached to the autophagy-related vesicles of different stages were then pulled down by immunoprecipitation. Interestingly, HSPA1A, HSP90AA1, and CALR were all readily co-immunoprecipitated with mCherry-labeled LC3-II in SK + CQ treated cells (Fig. 7B), suggesting a close association between the levels of DAMPs and autophagic activity. This close association was further supported by the significantly enhanced expression of ectoDAMPs upon blockage of the fusion between autophagosome and lysosome (Fig. 7C). To reflect the role of autophagy and necroptosis on subsequent DC activation, SK-treated tumor cells with autophagy and necroptosis deficiency were applied to co-incubate with DCs and examined DC activation. As long as necroptosis signaling or autophagy activities were hindered, the DC activation of SK-mediated immunogenicity was significantly decreased to the level of the naïve DCs group (Fig. S6A). This finding indicated the critical role of autophagy activity and necroptosis signaling on SK-mediated immunogenic cell death. Conversely, 4T1-luc2 cells treated with SK in combination with CQ activated DCs with a much higher potency (Fig. S6B).
Figure 7.
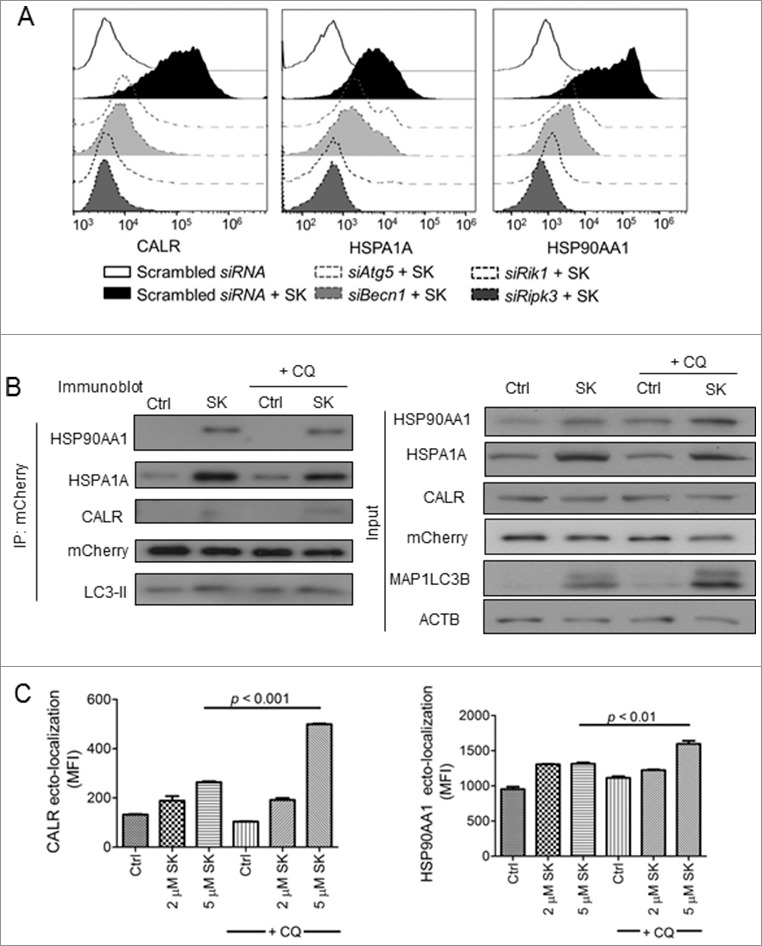
SK-induced DAMP ectolocalization is closely associated with enhanced autophagic activity. (A) Effect of knocking down ATG5, BECN1, RIPK1 and RIPK3 expression on SK-mediated DAMPs ectolocalization. 4T1-luc2 cells were transfected with a knockdown siRNA for Atg5, Becn1, Ripk1, and Ripk3. At 72 h post-transfection, cells were treated with 5 μM SK for 24 h and ectolocalization of several DAMPs were determined by flow cytometry analysis. (B) Cell lysates were prepared from SK-treated mCherry-GFP-LC3B-transfected stable cell clones. Protein complexes were pulled down via immunoprecipitation by using anti-mCherry antibody and subjected to SDS-PAGE and western blotting analyses. (C) The effect of CQ on SK-mediated DAMP ectolocalizations. 4T1-luc2 cells were treated by 2 or 5 μM SK in the presence or absence of CQ. Ectolocalizations of DAMP were determined by flow cytometry analysis. Data are expressed as mean SEM of triplicate determinations. P values were determined based on a one-way ANOVA analysis with Tukey's test. All data are from one experiment, representative of at least 3 independent experiments. MFI, mean fluorescence intensity.
Given that SK triggers ROS production22,40,41 and subsequent proteasome damage42,43 in a variety of cell types tested, we examined the possibility for these sequential events in SK-treated 4T1-luc2 cells. One h after SK treatment, ROS levels in test cells were significantly elevated in a concentration-dependent manner (Fig. S7A). This effect was completely reversed when SK-induced ROS production was inhibited by the ROS scavenger N-acetylcysteine (NAC). Compared to the untreated cells, proteasome activity was reduced to approximately 70% and 20% when cells were treated with 1 and 5 μM SK, respectively (Fig. S7B, left panel). Consistent with this result, the increased accumulation of ubiquitinated proteins in SK-treated cell lysate was also reverted by treatment with NAC (Fig. S7B, right panel). Moreover, we show that this ROS generation in SK treatment was able to directly contribute to the detected cell death, because NAC prevented the 4T1-luc2 cells from SK-instigated cell death (Fig. S7C, SK 91.3% vs. NAC + SK 2.1%). In an attempt to verify the compensatory role of selective autophagy during proteasome dysfunction as previously reported,44 we then evaluated the activity of ubiquitinated proteins and SQSTM1 (sequestosome 1) in SK-treated cells. As revealed in Fig. S7D, 4T1-luc2 cells treated with SK displayed drastically more colocalization of ubiquitinated proteins and SQSTM1 proteins as compared with these of the untreated controls. In addition, whereas CQ alone induced only puncta formation of SQSTM1 proteins, cells treated concomitantly with SK and CQ exhibited significantly more accumulation of ubiquitinated proteins and SQSTM1 proteins (Fig. S7D, SK + CQ). These results hence suggest that SK can induce selective autophagy that can be further enhanced by blocking the fusion activity between lysosomes and autophagosomes. Together, our findings demonstrate that SK can elicit ROS production and result in proteasome damage in treated tumor cells. SK-induced ROS generation was apparently also involved directly in the elicitation of autophagy and the ectolocalization of DAMPs, as revealed by the fact that NAC not only diminished the test autophagic activity, but also the released forms (Fig. 4A) and ectolocalization of DAMPs (Fig. S7E). These findings, we consider, revealed important new insight into the mechanism(s) with which cells undergoing necroptosis can orderly exhibit ROS-mediated, autophagy-dependent ectolocalization of DAMP and then activate DCs.
SK + CQ benefitted DC vaccine
To further verify the effect of autophagy and necroptosis on SK-mediated immunogenicity and subsequently antitumor activity on the DC-based cancer vaccine, we executed an in vivo experiment in which autophagy or necroptosis activities in 4T1 tumor cells were interrupted by specific siRNA or knockout of Ripk1 (Fig. S8A). Consistently, the pulsed DC vaccine (SK-DCs) was able to strongly inhibit tumor metastasis and prolong the survival of test mice as compared with control group (P < 0.05). However, when autophagy activity or necroptotic signaling in tumor cells was hindered, the antimetastasis effect and the prolonged survival time of the SK-meditated immunogenicity on DCs would be significantly reduced as compared with those of the SK-DCs group (P < 0.05) (Fig. S8B, S8C, and S8D). SK-induced autophagy activity does play an important role on the necroptosis activity and the subsequent immunogenicity on the derived DC-based cancer vaccine. Additionally, we demonstrated above that tumor cells upregulated ectolocalization of DAMPs and significantly enhanced the potential to activate DCs when the fusion activity of autophagosomes and lysosomes was blocked by SK treatment.
We hence further investigated whether such effects could result in subsequent immunogenicity. Mice implanted with 4T1 tumor cells and later receiving tumor resection were then treated with Dox as described above. Additional counterpart mouse groups were treated with 2 mg/kg and 5 mg/kg Dox, 2 mg/kg Dox in combination with DC vaccine pulsed with SK-treated 4T1-luc2 tumor cells (2 mg/kg Dox + SK-DCs), and 2 mg/kg Dox in combination with DC vaccine cells pulsed with SK and CQ-treated 4T1-luc2 tumor cells, where autophagosome-lysosome fusion was blocked (2 mg/kg Dox + SK + CQ-DCs). Interestingly, 2 mg/kg Dox could not significantly reduce tumor metastasis and prolong survival as compared with the untreated counterpart mice in the 80-d observation period (Fig. 8A). In contrast, mice receiving 5 mg/kg Dox exhibited significant benefit as compared with the untreated mice and mice treated with 2 mg/kg Dox in metastasis-free rates (2 mg/kg Dox 30% vs. 5 mg/kg Dox 60%) and survival rates (2 mg/kg Dox 30% vs. 5 mg/kg Dox 60%) at the endpoint of this experiment. Although the 2 mg/kg Dox + SK-DCs-treated group did not provide an advantage over the 2 mg/kg Dox-treated group alone, the antimetastasis efficacy and survival rates of the 2 mg/kg Dox + SK + CQ-DC-treated group was significantly increased to the level equivalent to that of the 5 mg/kg Dox treatment and much improved as compared with 2 mg/kg Dox + SK-DC (2 mg/kg Dox + SK-DCs 40% vs. 2 mg/kg Dox + SK + CQ-DCs 70%). The bioluminescence imaging (BLI) data further suggested the substantial effect on tumor metastasis at 33 d post-tumor implantation (Fig. 8B).
Figure 8.
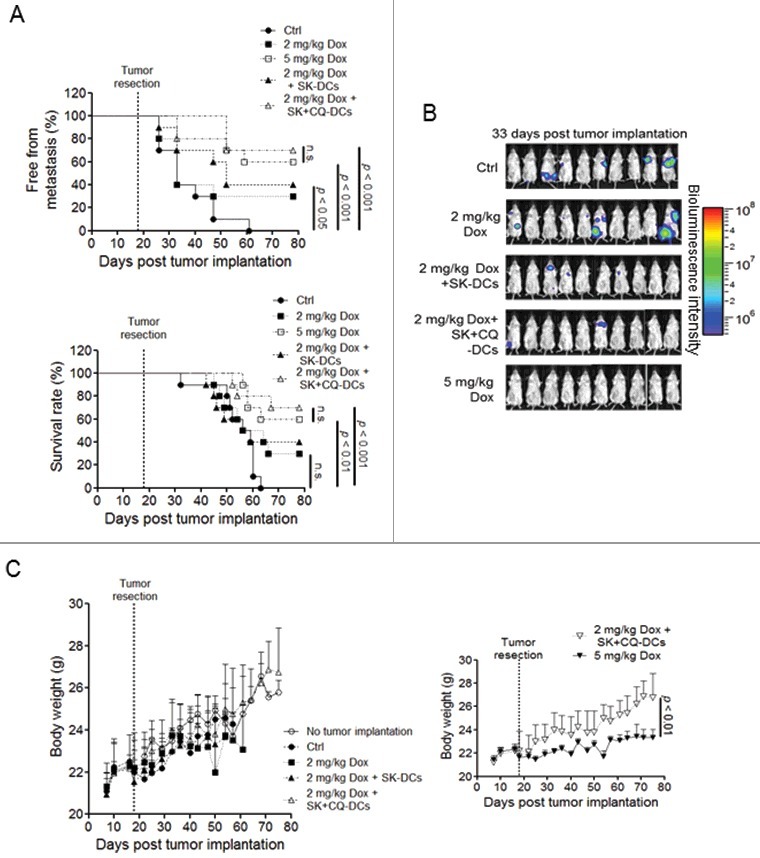
SK + CQ treatment benefited DC vaccine. Test mice (n = 10) were implanted with 4T1-luc2 tumor cells and the primary tumors were resected as described in Fig. 3. Mice were then vaccinated with 5 mg/kg Dox, 2 mg/mL Dox, 2 mg/kg Dox plus SK-treated, 4T1-pulsed DCs (2 mg/mL Dox + SK-DCs), or 2 mg/kg Dox plus SK-treated 4T1 cells where the autophagosome-lysosome fusion activity was blocked (i.e., 2 mg/kg Dox + SK + CQ-DCs). (A) The metastasis-free incidence and survival rate of test mice were recorded until 78 d post-tumor implantation. (B) Representative bioluminescence images were shown at 33 d post tumor implantation. (C) Body weight was measured for the treated mice. Data are expressed as mean ± SEM. P values for the metastasis and survival rates were determined by the log-rank test. A one-way ANOVA analysis with Tukey's test was applied to body weight.
We then evaluated whether there is a beneficial effect in body weight loss of combinational therapy as seen in Fig. 8C. The body weight of test mice in all treatment groups except 5 mg/kg Dox showed no significant difference as compared with mice without tumor implantation. The 2 mg/kg Dox + SK + CQ-DCs-treated group gave the least side effects as revealed by the degree of weight loss ((26.7-23.35)/26.7≌13% less) caused by 5 mg/kg Dox treatment (Fig. 8C, right panel). Together, these results suggest that SK can stimulate anticancer immune activities and effectively reduce the dosage needed for Dox to reach the similar antitumor effect conferred by employing Dox alone for treatment.
Discussion
Necrotic cell death has long been recognized as a trigger for various inflammatory activities. Specific passively released DAMP molecules from necroptotic cells have been proposed to play a critical role in the induction of immunogenicity10,45,46; however, exactly how cell death mediated by necroptosis becomes immunogenic cell death remains unclear. Based on our in vitro and vivo results (Fig. 1, 4, and 5 and Fig. S5), the immunogenicity of SK-induced necroptotic 4T1 tumor cells for DC activation in vitro can be attributed to the upregulation of ectolocalization of specific DAMP molecules. And this upregulation of ectoDAMP activities effectively contributed to the high potency of the resultant DC-based vaccine activity (Fig. S5). In the future, it may be interesting to evaluate the possible importance of released DAMPs on the immunogenicity of SK-induced necroptosis attributed to other immune cells types. Importantly, blockage of the autophagic activity at the stage of autophagosome processing further enhanced the ectolocalization of DAMPs and that is very conductive for developing immunogenicity against tumor cells.
The interplay between autophagic activity and expression of DAMPs has been previously contemplated to play important roles in progression and metastasis of cancerous cells.47,48 These crosstalk activities are thought to assist the shaping of the immune responses against dying cells upon onset of ICD.49 In an attempt to address this complex model of molecular mechanisms, our current studies showed that the phytochemical SK, isolated from medicinal plant Lithospermum erythrorhizon, was highly efficacious in instigating 4T1-luc2 tumor cell demise, and this cellular mechanism was mediated by necrosis via ROS production and sequential proteasome inhibition. As a result, high levels of ubiquitinated misfolded proteins became accumulated,50,51 and this was followed by SQSTM1-mediated selective autophagy.52 As such autophagic activity is enhanced in treated cells, levels of both secreted DAMPs and its ectocounterparts, including HSPA1A, HSP90AA1, and CALR, were increased, demonstrating their intimate association with the tested autophagy.
The close association between autophagy and DAMP ectolocalization was further demonstrated by the colocalization of HSPA1A, HSP90AA1, and mCherry-GFP-LC3B proteins in transfected 4T1-luc2 cells that were treated by SK. The dramatic drop in the ectolocalization levels of DAMPs on BECN1 and ATG5 knockdown, SK-treated 4T1-luc2 cells firmly illustrates the dependency of ectoDAMP upregulation on autophagic activity. It has previously been shown that when both proteasome and lysosome are inhibited, peptide intermediates bound on HSP90AA1 proteins would become encapsulated into autophagosomes and such a complex could serve as an immunogenic substrate for DC-based cross-presentation.53,54 In complete accordance with these previous findings, and in comparison with SK-treated cells, the autophagic activity of cells treated with SK + CQ in combination was strongly enhanced, accompanied by further upregulated ectoDAMP, resulting in additional DC activation and further increase in DC vaccine efficacy. Therefore, our findings together may suggest that SK-induced aggresomes and DAMPs were encompassed by autophagosomes and whose presentation on the antigen donor cell surface could be drastically enhanced by lysosome inhibition in cells undergoing necroptosis.
In our present study, we showed that dying 4T1-luc2 tumor cells which lose their cell integrity could be employed for pulse-triggering the DC activation and to establish the pulsed DC-based tumor vaccine (Fig. 5); however, ectoDAMPs were still found to remain attached to the plasma membrane. The transwell experiment (Fig. 4B) together with antibody blocking experiments (Fig. 4C) clearly demonstrated that the immunogenicity of necroptotic 4T1-luc 2 cells was exclusively due to ectoDAMPs, but not their released or secreted counterpart versions, or any equivalents that constituted the tumor cell lysates. The role for ectoDAMPs in eliciting the cell immunogenicity proposed here may differ from that of the primitive antigen peptide chaperoning activity55; however, the involvement of ecto-HSPs in tumor cells in activation of DCs and subsequent tumor-specific CD8+ T cell cytotoxicity has indeed recently been demonstrated in myeloma cells and ovarian cancer cells that are subjected to death via apoptosis.56,57 Our results thus suggest that the ectolocalization of DAMPs could act as the bona fide, key immunogenic contributor to the mode of action of necroptosis.
A number of studies have reported that in order to effectively induce necroptosis, the system needs to trigger TNF activity in the absence of CASP/caspase activation and this is the current major opinion for this role of TNF.2,58,59 To investigate possible potential pathways that can lead to 4T1-luc2 cell death, we first examined the expression of a variety of cytokines secreted by SK-treated 4T1-luc2 cells and the possible role of NFKB activation in SK treatment. Although little or no expression of several proinflammatory cytokines including TNF, IL1B, IL6, and IL12B (Fig. S9A) was detected, NFKB activation was readily detected transiently during a short time period (30 min) post SK treatment (Fig. S9B). This finding provides evidence of possible involvement of NFKB signaling pathways in the SK-elicited 4T1-luc2 cell death.60-62 In addition, as seen in Fig. S7A, our results echoed previous results suggesting that ROS is the major contributor to cell death by necroptosis.63,64 In addition, our findings also suggest the important role of autophagy in necroptosis, which correlates with the results of a recent study reported by Goodall et al.65
In this study, we investigated the effects of SK on mouse 4T1 mammary carcinoma cells. We conclude that SK can trigger necroptosis through a ROS-damaged proteasome pathway, and this activity can render cells with enhanced autophagic activities. The instigated autophagy can mediate a specific mode of DAMP expression, where the ectoDAMPs, instead of the secreted or released counterpart DAMPs, were found to act in a cell-to-cell interaction mode, that is responsible for the subsequent activation of DCs, resulting in a high cancer vaccine efficacy. This vaccine formulation, the SK-treated, 4T1 cell-pulsed DCs, not only helped protect test mice from primary tumor growth but were also quite effective against the metastasis after the tumor resection protocol. Together, our findings demonstrated that the expression of autophagy-related ectoDAMPs appeared to be critical for immunogenicity in cells undergoing necroptosis against metastasis (Fig. 9). Recently, it has been shown that some tumor cells detached from primary tumor(s) can circulate in the bloodstream, namely the circulating tumor cells.66 These circulating tumor cells have been considered as an important factor on tumor metastasis and drug resistance.67,68 Prospectively, one may consider that the increase of such ectolocalization of DAMPs on circulating tumor cells, through a SK-mediated necroptosis, may provide a more concentrated effect against tumor metastasis as compared to the release of DAMPs from tumor cells into the bloodstream. If this hypothesis could be proven in the future, our findings may have application for targeting specifically on circulating tumor cells for an antimetastatic effect.
Figure 9.
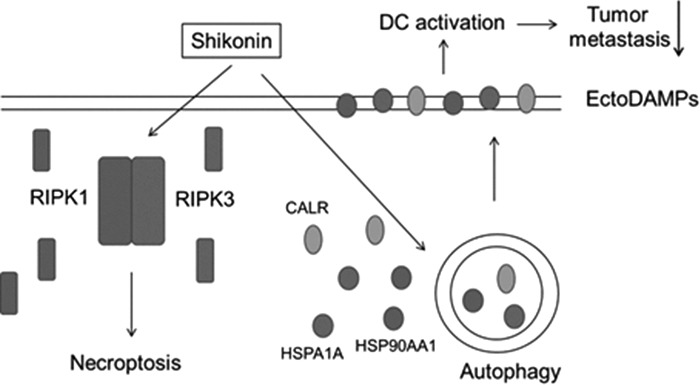
Hypothetical model depicting key molecular mechanisms of autophagy-related ectoDAMP expression in SK-mediated necroptosis in our test tumor cell system.
Materials and methods
Cell lines
4T1-luc2 cells (a kind gift from Dr. Pei-Wen Hsiao, Academia Sinica, Taipei, ROC) were derived from mouse mammary tumor 4T1 cells (ATCC, CRL-2539) that were stably transfected with a luciferase transgene. 4T1-luc2 cells were cultured and maintained in RPMI-1640 (Invitrogen, 31800) supplemented with 10 µg/mL blasticidin S (Invivogen, ant-bl), 10% fetal bovine serum (Gibco™, 10082147), 1 mM penicillin-streptomycin (Gibco™, 15140122) and 1 mM sodium pyruvate (Gibco™, 11360070) at 37°C in 5% CO2 and 95% humidity.
Mice
Female 6- to 8-wk-old BALB/c mice were purchased from the National Laboratory Animal Breeding and Research Center (Taipei, Taiwan). Test mice were maintained in a standardized laminar airflow cabinet under specific pathogen-free conditions at the Animal Room facility of Agricultural Biotechnology Research Center, Academia Sinica. Experimental protocols involving animals were approved by the IACUC office of Academia Sinica (Taipei, Taiwan).
Cell viability assay
Optimally growing 4T1-luc2 tumor cells at the density of 1 × 104 cells/100 µL were treated with SK (Nacalai Tesque, 04056) for the indicated time in the presence or absence of 20 μM zVAD-fmk (Selleckchem, S8102), 5 mM 3-MA (Enzo Life Science, BML-AP502), 20 µM NEC-1 (Selleckchem, S8037), or GSK'872 (BioVision, 2673). Necroptotic cells are induced by the combination of 20 µM zVAD-fmk, 20 ng/mL TNF (Peprotech, 315-01A), and 10 µg/mL cycloheximide (Sigma-Aldrich, C7698). Cells were then washed with 1 × PBS (137 mM sodium chloride, 2.7 mM potassium chloride, 10 mM sodium phosphate dibasic, and 2 mM potassium dihydrogen orthophosphate) and incubated with 0.5 mg/mL MTT (Sigma-Aldrich, M5655) in complete medium at 37°C for 3 h. Test cells were then pelleted at 800 × g and the supernatant was removed. The resulting pellets were dissolved in 100 µL 100% DMSO (Sigma-Aldrich, D4540) and quantified at a wavelength of 570 nm using the Biotek PowerWave HT spectrophotometer (Biotek, Winooski, VT, USA). In some experiments, dying cells were determined by staining cells with ANXA5-FITC and PI (BD Biosciences, 556547) according to the manufacturer's protocol. In brief, 1 × 105 cells were resuspended in 100 µL binding buffer and then 2 µL ANXA5-FITC and 5 µL PI were added to test cells, incubated for 15 min at room temperature in the dark. An additional 400 µL binding buffer was then added to the reaction prior to analysis.
Cell lysate preparation
For SDS-PAGE and proteasome activity assays, treated 4T1-luc2 tumor cells were harvested and washed with 1 × PBS. Washed cells were lysed with M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific, 78501) as described by the manufacturer. Protein contents were determined with the Bradford colorimetric method using the Quick Start™ Bradford Protein Assay Kit (Bio-Rad, 5000201).
Western blotting
Proteins in test cell lysate samples were fractionated by 5% to 20% SDS-PAGE (20 µg protein per lane). After electrophoresis, proteins were transferred onto Hybond-ECL membranes (GE-Healthcare, RPN2020D) and immunoblotted with primary anti-CASP8/caspase-8 (Cell Signaling Technology, 4790), anti-HSPA1A (Proteintech, 10995-1-AP), anti-HMGB1 (Abcam, ab18256), and anti-CALR (Cell Signaling Technology, 2891) antibodies, and monoclonal antibody against PARP1 (Cell Signaling Technology, 9532), LC3B (Cell Signaling Technology, 3868), HSP90AA1 (Cell Signaling Technology, 4877), ubiquitin (Santa Cruz Biotechnology, sc-271289), and ACTB/actin, beta (Santa Cruz Biotechnology, sc-47778). After removal of nonspecific binding, target proteins were detected by using the corresponding secondary antibodies. Protein bands were detected by enhanced chemiluminescence (Bio-Rad, 1705061) and developed by autoradiography. An Image Studio Lite Version 5.2 Software (Licor, Lincoln, NE) was applied in the densitometric ananlysis of target protein bands.
Knockdown experiments
To abolish specific protein expression in 4T1-luc2 tumor cells, 2 × 104 cells (4 mL) were transfected with 100 nM siRNA using Lipofectamine 2000 (Thermo Fisher Scientific, 1166819) according to the manufacturer's instructions. Transfected cells were incubated for 48 or 72 h as specifically indicated in the text. The sequences of siRNA oligonucleotides were designed as follows: siRipk1: sense: 5′-GUCUACUUACAUGACAATT-3′, siRipk3: sense: 5′-GCUCU GGUGACAAGAUUCATT-3′, siAtg5: sense: 5′-GCUACCCAGAUAACUUUCUTT- 3′, and siBecn1: sense: 5′- GGAGCCAUUUAUUGAAACUTT-3′. The efficacy of siRNA knockdown for protein expression was further confirmed by western blotting analysis.
Immunoprecipitation
Test tumor cells were lysed in RIPA buffer (150 mM NaCl, 20 mM Tris-Cl, pH 7.5, 1% NP-40 [Calbiochem, 9016-45-9], 1 mM EGTA [Sigma-Aldrich, E3889], 5 mM sodium pyrophosphate [Sigma-Aldrich, 221368], and 0.1% sodium deoxycholate [Sigma-Aldrich, D6750]) and the protein content was determined. The constitutively present antibodies were removed by subjecting 0.5 mg/500 μL cell lysate to protein G microbeads (Miltenyi Biotec, 130-071-101) and removed by magnetic bead sorting. The resulting run-through was then incubated with monoclonal antibody against mCherry (Thermo Fisher Scientific, M11217) overnight at 4°C. Immune complexes that formed were then enriched as above and eluted with 1 x Laemmli sample buffer and analyzed by western blotting.
Detection of DAMP ectolocalization
Aliquots of 2 × 105 test cells were harvested and washed with 1 × PBS. Cells were then stained with APC-conjugated anti-HSPA1A mAb (Miltenyi Biotec, 130-105-550), Alexa Fluor 488-conjugated anti-CALR Ab (Abcam, ab196158) and PE-conjugated anti-HSP90AA1 Ab (Abcam, ab65171), and subjected to flow cytometry analysis.
BM-DC preparation
BM-DCs were cultured and prepared as previously described.69-70 Briefly, total BM cells were collected from the femurs and tibias of BALB/c mice and depleted of red blood cells with ACK lysis buffer (0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM EDTA, pH 7.2). Cells were then cultured in complete RPMI-1640 medium supplemented with 20 ng/mL CSF2 (Peprotech, 315-03), 10% fetal bovine serum, 50 µM 2-ME (Gibco™, 21985023), 100 unit/mL penicillin, and 100 µg/mL streptomycin (Gibco™, 15140122) in a humidified 5% CO2 incubator at 37°C. On d 3, 30 mL fresh media with 20 ng/mL CSF2 was added. On d 6 and 8, 30 mL culture media were replaced with the equal volume of fresh media containing 20 ng/mL CSF2. On d 10, the nonadherent and loosely adherent DCs were harvested. Alternatively, 10-d BM-DCs were further labeled with magnetic bead-conjugated antibodies against ITGAX (Miltenyi Biotec, 130-097-059) and subjected to magnetic field for cell enrichment. Purity of ITGAXhi DCs was examined and the purity was routinely > 90%.
Assay for DC activation
For determination of DC activation, optimally growing 4T1-luc2 tumor cells were exposed to 5 µM SK for 24 h and the media were harvested and centrifuged at 800 × g for 3 min. The supernatants were collected as conditioned media and used to incubate BM-DCs at a density of 1 × 105 cells/2 mL at 37°C. Alternatively, aliquots of 1 × 105 test cells were washed with 1 × PBS, resuspended and coincubated with 10-day BM-DCs at a ratio of 1:1 in a total volume of 1 mL. For some tests, aliquots of resuspended cells (1 × 105 test tumor cells/600 µL) were seeded at the bottom of a 0.4−µm pore size transwell apparatus (Sigma-Aldrich, CLS3460). Samples of 1 × 105 BM-DCs suspended in 100 µL of fresh media were added to the top well and incubated at 37°C for 24 h. After 24 h incubation, cells were detached, washed, and stained with APC-conjugated monoclonal Ab against ITGAX Ab (BioLegend, 117310) in combination with FITC-conjugated monoclonal Ab against CD40 (BioLegend, 124608), FITC-conjugated CD86 (BioLegend, 105006), PE-conjugated anti-TLR4 (BioLegend, 145404), PE-conjugated anti-H2-I molecule (BioLegend, 107608), and PE-conjugated anti-CD80 antibodies (BioLegend, 104708), according to the manufacturer's instructions. The cocultured media were also collected and checked for the secretion of IL1B (R&D Systems, DY401), IL12B (R&D Systems, DY499) and IL6 (R&D Systems, DY406) by ELISA, according to the manufacturer's instructions. Cytokine production was quantified using a Biotek PowerWave HT spectrophotometer (Biotek, Winooski, VT, USA).
Flow cytometry
All cells were stained by 3-color analysis as previously described.71 Briefly, cells were stained with respective antibodies in 1% BSA (Sigma-Aldrich, A9418) and 0.1% NaN3 (Merck, 26628-22-8) containing Mg2+- and Ca2+-free Dulbecco PBS (Thermo Fisher Scientific, 14190144). Data were acquired via LCR-II flow cytometer (BD Biosciences, San Jose, CA, USA) and analyzed by Flow Jo (Treestar, Costa Mesa, CA, USA).
Confocal microscopy
For fluorescent immunostaining, cells were fixed with formaldehyde (3.7%) in 1 × PBS, permeabilized with 100% ice-cold methanol, and then blocked with 1% BSA in 1 × PBS for 1 h. Cells were then incubated with primary Ab against SQSTM1 (GeneTex, GTX100685), LC3B (Cell Signaling Technology, 3868), or ubiquitin (Santa Cruz Biotechnology, sc-271289). The stained cells were washed 3 times with PBST (1% Tween-20 in 1 × PBS), incubated with fluorescently-labeled secondary antibody, washed 3 times with 1 × PBS. Finally, cell were stained with 300 nM DAPI (Sigma-Aldrich, D9542) for 10 min at room temperature and washed 3 times with 1 × PBS. Cells were then examined under a confocal microscope (Zeiss LSM510Meta NLO DuoScan; Carl Zeiss, Jena, Germany).
Construction of plasmid mCherry-EGFP-LC3B
Plasmid mCherry-EGFP-LC3B was constructed from plasmids encoding EGFP-LC3B (21073, deposited by Tamotsu Yoshimori) and plasmids encoding mCherry-LC3B (21075, deposited by Tamotsu Yoshimori) purchased from Addgene. The CMV_fwd_primer (5′-CGCAATGGGCGGTAGGCGTG-3′) and dsRed1_C_primer (5′-AGCTGGACATCACC TCCCACAACG-3′) with NheI and AgeI site exogenously added respectively were used to amplify mCherry cDNA in pmCherry-LC3B by PCR. The PCR product was digested by both restriction endonucleases and subsequently cloned into the NheI/AgeI site immediate upstream of the EGFP coding region in plasmid encoding EGFP-LC3B.
Transfection and stable pool generation
Plasmid mCherry-EGFP-LC3B was transfected into 4T1-luc2 tumor cells for stable pool generation. Transfection was performed using Lipofectamine 2000 according to the manufacturer's protocol. Stable transformants were selected in complete medium containing 500 μg/mL G418 (BioVision, 1557).
Animal model
For prophylactic vaccination, mice were first immunized subcutaneously with 4T1-luc2 tumor cells treated with 5 M SK for 24 h (105 and 5 × 105 cells/100 µL 1 × PBS). Mice injected with 1 × PBS were used as an experimental control. Alternatively, 4T1-luc2 cells were subjected to the following freeze and thaw (F/T) cycles: freezing cell suspension in liquid nitrogen for 90 sec and followed by then thawing cells at 37°C in a water bath, and the F/T cycles were repeated 4 times. At 7 d postvaccination, 5 × 105 live 4T1-luc2 tumor cells in 100 µ L PBS were orthotopically implanted into mammary fat pad. Tumor size was monitored by caliper measurements and tumor volume calculated using the following formula: 0.5 × length (mm) × width.2 (mm2) For the antitumor metastasis effect, 4T1-luc2 tumor cells (5 × 105 cells in 100 µL PBS) were orthotopically implanted into mammary fat pad of test mice. At 18 d post tumor implantation, the tumor mass was gently removed surgically.72 One d after tumor resection, mice were intraperitoneally injected with 1 × 105 and 5 × 105 F/T- and 5 μM SK-treated 4T1-luc2 cells once a wk for 2 wk. For DC vaccination, 5 × 106 BM-DCs were pulsed with 4T1-luc2 cells that were treated with 5 μM SK for 24 h at a 1:1 ratio for 24 h. BM-DCs were then purified via magnetic cell sorting (Miltenyi Biotec, 130-097-059). Mice were then vaccinated via i.p. injection with 1 × 106 cells/100 μL BM-DCs at the first day post tumor resection and consecutively vaccinated once a week for 2 wk. For doxorubicin (Enzo, BML-GR319) treatment, test mice were injected intravenously with 2 or 5 mg/kg Dox at 2 d post tumor resection and continuously treated twice a wk for 3 wk. Bioluminescence imaging of tumors was monitored by using a noninvasive in vivo imaging system once a wk. Test mice were injected intraperitoneally with 15 mg/kg D-luciferin potassium (PerkinElmer, 122799) in 1 × PBS and waited for 15 min before imaging. Mice were then anesthetized with 2.5% isoflurane through the XGI-8 Gas Anesthesia System (PerkinElmer, Boston, MA, USA) and XENOGEN IVIS 50 (PerkinElmer, Boston, MA, USA) was applied.
Statistical analysis
Data are presented in fold changes or in percentages with mean ± SEM as indicated in the figure legends. All statistical analyses were determined using GraphPad Software. As a comparison between multiple data sets, a one-way ANOVA analysis with the Tukey-Kramer method was performed. The log-rank test was applied for survival analysis and metastatic incidence analysis.
Funding Statement
National Science Council (NSC101-2320-B001-025, MOST103-2320-B001-001 and NSC102-2320-B001-002), Ministry of Science and Technology, MOST105-2320-B001-008.
Abbreviations
- 3-MA
3- methyladenine
- ACTB
actin, beta
- ANXA5
annexin A5
- ATG5
autophagy-related 5
- BECN1
Beclin 1, autophagy related
- BM-DC
bone marrow-derived dendritic cells
- BLI
bioluminescence imaging
- CALR
calreticulin
- CASP3
caspase 3
- CASP8
caspase 8
- CD
cluster of differentiation molecules
- CQ
chloroquine
- CSF2
colony stimulating factor (granulocyte-macrophage)
- Ctrl
control
- DAMP
damage-associated molecular pattern
- Dox
doxorubicin
- F/T
freeze and thaw
- HSPA1A
heat shock protein 1A
- H2-I
H2 I region, includes H2-Aa, Ab, Bl, Ea, Eb, Eb2, Ob, Pb
- HSP90AA1
heat shock protein 90, alpha (cytosolic), class A member 1
- i.p.
intraperitoneal
- ICD
immunogenic cell death
- IL1B
interleukin 1 beta
- IL6
interleukin 6
- IL12B
interleukin 12B
- LDHA
lactate dehydrogenase A
- MAP1LC3B/LC3B
microtubule-associated protein 1 light chain 3 beta
- MLKL
mixed lineage kinase domain-like
- NAC
N-acetylcysteine
- NEC-1
necrostatin-1
- NFKB
nuclear factor of kappa light polypeptide gene enhancer in B cells
- PARP1
poly (ADP-ribose) polymerase family, member 1
- PI
propidium iodide
- RIPK1
receptor (TNFRSF)-interacting serine-threonine kinase 1
- RIPK3
receptor-interacting serine-threonine kinase 3
- ROS
reactive oxygen species
- SK
shikonin
- SQSTM1
sequestosome 1
- TCZ
TNF + cycloheximide + zVAD-fmk
- TEM
transmission electron microscopy
- TNF
tumor necrosis factor
Conflict-of-interest and financial disclosure statements
The authors declare that they have no conflict of interests or need for financial disclosure statements.
References
- [1].Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, et al.. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–701. doi: 10.1084/jem.20050915. PMID:16365148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al.. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–20. doi: 10.1038/cdd.2011.96. PMID:21760595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. PMID:8011301. [DOI] [PubMed] [Google Scholar]
- [4].Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–47. doi: 10.1038/nrm3737. PMID:24452471. [DOI] [PubMed] [Google Scholar]
- [5].Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187:1477–85. doi: 10.1084/jem.187.9.1477. PMID:9565639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J, et al.. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9. doi: 10.1038/nchembio711 . PMID:16408008. [DOI] [PubMed] [Google Scholar]
- [7].Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al.. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–21. doi: 10.1038/nchembio.83. PMID:18408713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P. RIP1, a kinase on the crossroads of a cell's decision to live or die. Cell Death Differ. 2007;14:400–10. doi: 10.1038/sj.cdd.4402085. PMID:17301840. [DOI] [PubMed] [Google Scholar]
- [9].Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–65. doi: 10.1056/NEJMra1310050. PMID:24476434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–23. doi: 10.1016/j.immuni.2013.02.003. PMID:23438821. [DOI] [PubMed] [Google Scholar]
- [11].Bray K, Mathew R, Lau A, Kamphorst JJ, Fan J, Chen J, Chen HY, Ghavami A, Stein M, DiPaola RS, et al.. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS One. 2012;7:e41831. doi: 10.1371/journal.pone.0041831. PMID:22848625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Basit F, Cristofanon S, Fulda S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013;20:1161–73. doi: 10.1038/cdd.2013.45. PMID:23744296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lalaoui N, Lindqvist LM, Sandow JJ, Ekert PG. The molecular relationships between apoptosis, autophagy and necroptosis. Semin Cell Dev Biol. 2015;39:63–9. doi: 10.1016/j.semcdb.2015.02.003. PMID:25736836. [DOI] [PubMed] [Google Scholar]
- [14].Thorburn J, Horita H, Redzic J, Hansen K, Frankel AE, Thorburn A. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 2009;16:175–83. doi: 10.1038/cdd.2008.143. PMID:18846108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, et al.. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–7. doi: 10.1126/science.1208347. PMID:22174255. [DOI] [PubMed] [Google Scholar]
- [16].Li Y, Wang LX, Yang G, Hao F, Urba WJ, Hu HM. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 2008;68:6889–95. doi: 10.1158/0008-5472.CAN-08-0161. PMID:18757401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Twitty CG, Jensen SM, Hu HM, Fox BA. Tumor-derived autophagosome vaccine: Induction of cross-protective immune responses against short-lived proteins through a p62-dependent mechanism. Clin Cancer Res. 2011;17:6467–81. doi: 10.1158/1078-0432.CCR-11-0812. PMID:21810919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S, Yamazaki T, Sukkurwala AQ, Michaud M, Mignot G, et al.. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med. 2012;4:143ra99. doi: 10.1126/scitranslmed.3003807. PMID:22814852. [DOI] [PubMed] [Google Scholar]
- [19].Dudek AM, Garg AD, Krysko DV, De Ruysscher D, Agostinis P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 2013;24:319–33. doi: 10.1016/j.cytogfr.2013.01.005. PMID:23391812. [DOI] [PubMed] [Google Scholar]
- [20].Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo J, Hu X. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther. 2007;6:1641–9. doi: 10.1158/1535-7163.MCT-06-0511. PMID:17513612. [DOI] [PubMed] [Google Scholar]
- [21].Yang H, Zhou P, Huang H, Chen D, Ma N, Cui QC, Shen S, Dong W, Zhang X, Lian W, et al.. Shikonin exerts antitumor activity via proteasome inhibition and cell death induction in vitro and in vivo. Int J Cancer. 2009;124:2450–9. doi: 10.1002/ijc.24195. PMID:19165859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wiench B, Eichhorn T, Paulsen M, Efferth T. Shikonin directly targets mitochondria and causes mitochondrial dysfunction in cancer cells. Evid Based Complement Alternat Med. 2012;2012:726025. doi: 10.1155/2012/726025. PMID:23118796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen HM, Wang PH, Chen SS, Wen CC, Chen YH, Yang WC, Yang NS. Shikonin induces immunogenic cell death in tumor cells and enhances dendritic cell-based cancer vaccine. Cancer Immunol Immunother. 2012;61:1989–2002. doi: 10.1007/s00262-012-1258-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Piao JL, Cui ZG, Furusawa Y, Ahmed K, Rehman MU, Tabuchi Y, Kadowaki M, Kondo T. The molecular mechanisms and gene expression profiling for shikonin-induced apoptotic and necroptotic cell death in U937 cells. Chem Biol Interact. 2013;205:119–27. doi: 10.1016/j.cbi.2013.06.011. PMID:23811387. [DOI] [PubMed] [Google Scholar]
- [25].Huang C, Luo Y, Zhao J, Yang F, Zhao H, Fan W, Ge P. Shikonin kills glioma cells through necroptosis mediated by RIP-1. PLoS One. 2013;8:e66326. doi: 10.1371/journal.pone.0066326. PMID:23840441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Duan D, Zhang B, Yao J, Liu Y, Fang J. Shikonin targets cytosolic thioredoxin reductase to induce ROS-mediated apoptosis in human promyelocytic leukemia HL-60 cells. Free Radic Biol Med. 2014;70:182–93. doi: 10.1016/j.freeradbiomed.2014.02.016. PMID:24583460. [DOI] [PubMed] [Google Scholar]
- [27].Wada N, Kawano Y, Fujiwara S, Kikukawa Y, Okuno Y, Tasaki M, Ueda M, Ando Y, Yoshinaga K, Ri M, et al.. Shikonin, dually functions as a proteasome inhibitor and a necroptosis inducer in multiple myeloma cells. Int J Oncol. 2015;46:963–72. doi: 10.3892/ijo.2014.2804. PMID:25530098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Han W, Xie J, Li L, Liu Z, Hu X. Necrostatin-1 reverts shikonin-induced necroptosis to apoptosis. Apoptosis. 2009;14:674–86. doi: 10.1007/s10495-009-0334-x. PMID:19288276. [DOI] [PubMed] [Google Scholar]
- [29].Lu L, Qin A, Huang H, Zhou P, Zhang C, Liu N, Li S, Wen G, Zhang C, Dong W, et al.. Shikonin extracted from medicinal Chinese herbs exerts anti-inflammatory effect via proteasome inhibition. Eur J Pharmacol. 2011;658:242–7. doi: 10.1016/j.ejphar.2011.02.043. PMID:21392503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, Mocarski ES, Pouliot K, Chan FK, Kelliher MA, et al.. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci U S A. 2014;111:7391–6. doi: 10.1073/pnas.1403477111. PMID:24799678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9:353–63. doi: 10.1038/nri2545. PMID:19365408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. PMID:23157435. [DOI] [PubMed] [Google Scholar]
- [33].Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13:674–90. doi: 10.1038/nrclinonc.2016.66. PMID:27184417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tang Y, Wang Y, Kiani MF, Wang B. Classification, Treatment Strategy, and Associated Drug Resistance in Breast Cancer. Clin Breast Cancer. 2016;16:335–43. doi: 10.1016/j.clbc.2016.05.012. PMID:27268750. [DOI] [PubMed] [Google Scholar]
- [35].Udono H, Srivastava PK. Comparison of tumor-specific immunogenicities of stress-induced proteins gp96, hsp90, and hsp70. J Immunol. 1994;152:5398–403. PMID:8189059. [PubMed] [Google Scholar]
- [36].Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–75. doi: 10.1038/nrc3380. PMID:23151605. [DOI] [PubMed] [Google Scholar]
- [37].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–26. doi: 10.1016/j.cell.2010.01.028. PMID:20144757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–60. doi: 10.4161/auto.4451. PMID:17534139. [DOI] [PubMed] [Google Scholar]
- [39].Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. Monitoring autophagy by electron microscopy in Mammalian cells. Methods Enzymol. 2009;452:143–64. doi: 10.1016/S0076-6879(08)03610-0. PMID:19200881. [DOI] [PubMed] [Google Scholar]
- [40].Chang IC, Huang YJ, Chiang TI, Yeh CW, Hsu LS. Shikonin induces apoptosis through reactive oxygen species/extracellular signal-regulated kinase pathway in osteosarcoma cells. Biol Pharm Bull. 2010;33:816–24. doi: 10.1248/bpb.33.816. PMID:20460760. [DOI] [PubMed] [Google Scholar]
- [41].Ahn J, Won M, Choi JH, Kim YS, Jung CR, Im DS, Kyun ML, Lee K, Song KB, Chung KS, et al.. Reactive oxygen species-mediated activation of the Akt/ASK1/p38 signaling cascade and p21(Cip1) downregulation are required for shikonin-induced apoptosis. Apoptosis. 2013;18:870–81. doi: 10.1007/s10495-013-0835-5. PMID:23546866. [DOI] [PubMed] [Google Scholar]
- [42].Trougakos IP, Sesti F, Tsakiri E, Gorgoulis VG. Non-enzymatic post-translational protein modifications and proteostasis network deregulation in carcinogenesis. J Proteomics. 2013;92:274–98. doi: 10.1016/j.jprot.2013.02.024. PMID:23500136. [DOI] [PubMed] [Google Scholar]
- [43].Hohn A, Jung T, Grune T. Pathophysiological importance of aggregated damaged proteins. Free Radic Biol Med. 2014;71:70–89. doi: 10.1016/j.freeradbiomed.2014.02.028. PMID:24632383. [DOI] [PubMed] [Google Scholar]
- [44].Myeku N, Figueiredo-Pereira ME. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J Biol Chem. 2011;286:22426–40. doi: 10.1074/jbc.M110.149252. PMID:21536669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, White MR, Dinarello CA, Apte RN. IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol. 2011;187:4835–43. doi: 10.4049/jimmunol.1102048. PMID:21930960. [DOI] [PubMed] [Google Scholar]
- [46].Ayna G, Krysko DV, Kaczmarek A, Petrovski G, Vandenabeele P, Fesus L. ATP release from dying autophagic cells and their phagocytosis are crucial for inflammasome activation in macrophages. PLoS One. 2012;7:e40069. doi: 10.1371/journal.pone.0040069. PMID:22768222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhang Q, Kang R, Zeh HJ, 3rd Lotze MT, Tang D. DAMPs and autophagy: Cellular adaptation to injury and unscheduled cell death. Autophagy 2013;9:451–8. doi: 10.4161/auto.23691. PMID:23388380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Garg AD, Martin S, Golab J, Agostinis P. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ. 2014;21:26–38. doi: 10.1038/cdd.2013.48. PMID:23686135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hou W, Zhang Q, Yan Z, Chen R, Zeh Iii HJ, Kang R, Lotze MT, Tang D. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. 2013;4:e966. doi: 10.1038/cddis.2013.493. PMID:24336086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Rodriguez-Gonzalez A, Lin T, Ikeda AK, Simms-Waldrip T, Fu C, Sakamoto KM. Role of the aggresome pathway in cancer: Targeting histone deacetylase 6-dependent protein degradation. Cancer Res. 2008;68:2557–60. doi: 10.1158/0008-5472.CAN-07-5989. PMID:18413721. [DOI] [PubMed] [Google Scholar]
- [51].Bruning A, Juckstock J. Misfolded proteins: From little villains to little helpers in the fight against cancer. Front Oncol. 2015;5:47. PMID:25759792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. PMID:17580304. [DOI] [PubMed] [Google Scholar]
- [53].Li Y, Wang LX, Pang P, Twitty C, Fox BA, Aung S, Urba WJ, Hu HM. Cross-presentation of tumor associated antigens through tumor-derived autophagosomes. Autophagy. 2009;5:576–7. doi: 10.4161/auto.5.4.8366. PMID:19333005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Li Y, Wang LX, Pang P, Cui Z, Aung S, Haley D, Fox BA, Urba WJ, Hu HM. Tumor-derived autophagosome vaccine: Mechanism of cross-presentation and therapeutic efficacy. Clin Cancer Res. 2011;17:7047–57. doi: 10.1158/1078-0432.CCR-11-0951. PMID:22068657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Srivastava PK, Menoret A, Basu S, Binder RJ, McQuade KL. Heat shock proteins come of age: primitive functions acquire new roles in an adaptive world. Immunity. 1998;8:657–65. doi: 10.1016/S1074-7613(00)80570-1. PMID:9655479. [DOI] [PubMed] [Google Scholar]
- [56].Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood. 2007;109:4839–45. doi: 10.1182/blood-2006-10-054221. PMID:17299090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chang CL, Hsu YT, Wu CC, Yang YC, Wang C, Wu TC, Hung CF. Immune mechanism of the antitumor effects generated by bortezomib. J Immunol. 2012;189:3209–20. doi: 10.4049/jimmunol.1103826. PMID:22896634. [DOI] [PubMed] [Google Scholar]
- [58].He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–11. doi: 10.1016/j.cell.2009.05.021. PMID:19524512. [DOI] [PubMed] [Google Scholar]
- [59].Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–6. doi: 10.1126/science.1172308. PMID:19498109. [DOI] [PubMed] [Google Scholar]
- [60].Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O'Reilly L, Mason K, Gross O, Ma S, Guarda G, et al.. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36:215–27. doi: 10.1016/j.immuni.2012.01.012. PMID:22365665. [DOI] [PubMed] [Google Scholar]
- [61].Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D'Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL, et al.. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282. doi: 10.1038/ncomms7282. PMID:25693118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, Green DR, Oberst A, Albert ML.. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science. 2015;350:328–34. doi: 10.1126/science.aad0395. PMID:26405229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Festjens N, Vanden Berghe T, Vandenabeele P. Necrosis, a well-orchestrated form of cell demise: Signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta. 2006;1757:1371–87. doi: 10.1016/j.bbabio.2006.06.014. PMID:16950166. [DOI] [PubMed] [Google Scholar]
- [64].Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–14. doi: 10.1038/nrm2970. PMID:20823910. [DOI] [PubMed] [Google Scholar]
- [65].Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM, Green DR, Morgan M, Cramer SD, Thorburn A. The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev Cell. 2016;37:337–49. doi: 10.1016/j.devcel.2016.04.018. PMID:27219062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Williams SC. Circulating tumor cells. Proc Natl Acad Sci U S A. 2013;110:4861. doi: 10.1073/pnas.1304186110. PMID:23533270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Pantel K, Speicher MR. The biology of circulating tumor cells. Oncogene. 2016;35:1216–24. doi: 10.1038/onc.2015.192. PMID:26050619. [DOI] [PubMed] [Google Scholar]
- [68].Gradilone A, Raimondi C, Naso G, Silvestri I, Repetto L, Palazzo A, Gianni W, Frati L, Cortesi E, Gazzaniga P. How circulating tumor cells escape from multidrug resistance: Translating molecular mechanisms in metastatic breast cancer treatment. Am J Clin Oncol. 2011;34:625–7. doi: 10.1097/COC.0b013e3181f94596. PMID:22101388. [DOI] [PubMed] [Google Scholar]
- [69].Chow KP, Lee JM, Qiu JT, Liao SK, Lin SC, Hsu SL, Wu NN, Lin YF, Wu TS.. Enhanced IL-10 production by CD4+ T cells primed in IL-15Ralpha-deficient mice. Eur J Immunol 2011;41:3146–56. doi: 10.1002/eji.201141746. PMID:21874651. [DOI] [PubMed] [Google Scholar]
- [70].Yin SY, Wang CY, Yang NS. Interleukin-4 enhances trafficking and functional activities of GM-CSF-stimulated mouse myeloid-derived dendritic cells at late differentiation stage. Exp Cell Res. 2011;317:2210–21. doi: 10.1016/j.yexcr.2011.06.013. PMID:21741972. [DOI] [PubMed] [Google Scholar]
- [71].Chow KP, Qiu JT, Lee JM, Hsu SL, Yang SC, Wu NN, Huang W, Wu TS.. Selective reduction of post-selection CD8 thymocyte proliferation in IL-15Ralpha deficient mice. PLoS One. 2012;7:e33152. doi: 10.1371/journal.pone.0033152. PMID:22448237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lin TJ, Lin HT, Chang WT, Mitapalli SP, Hsiao PW, Yin SY, Yang NS. Shikonin-enhanced cell immunogenicity of tumor vaccine is mediated by the differential effects of DAMP components. Mol Cancer. 2015;14:174. doi: 10.1186/s12943-015-0435-9. PMID:26403780. [DOI] [PMC free article] [PubMed] [Google Scholar]