

Abstract
Chronic kidney disease of unknown aetiology (CKDu) refers to the epidemic level of incidence of CKD in several low- and middle-income countries, usually near the equator, for which the aetiology has not been identified. CKDu represents a form of CKD hotspot, defined as a country, region, community or ethnicity with a higher than average incidence of CKD. In terms of the number of persons affected, the so-called hypertensive nephropathy of African Americans probably represents the largest CKD hotspot, which is largely driven by variants of the APOL1 gene, questioning the very existence of hypertensive nephropathy and illustrating how kidney disease driven by genetic predisposition may underlie some forms of hypertension. For CKDu, hard physical work leading to dehydration (the first global warming-related disease?) and local toxins are leading aetiological candidates. Meso-American nephropathy is probably the best-characterized CKDu. In this issue of CKJ, a systematic review and meta-analysis by Gonzalez et al. identified positive associations between Meso-American nephropathy and male gender, family history of CKD, high water intake and lowland altitude. We now discuss the potential relationship of family history to genetic predisposition and how a better understanding of CKDu may help advance the aetiological characterization of the nearly 50% of end-stage renal disease patients worldwide that have no known cause for CKD or have been assigned non-specific diagnoses.
Keywords: chronic kidney disease of unknown aetiology, CKD hotspot, genetics, Meso-American nephropathy
The term chronic kidney disease (CKD) hotspot was recently coined to define a country, region, community or ethnicity with a higher than average incidence of CKD [1]. Meso-American nephropathy is the best characterized CKD of unknown aetiology (CKDu) corresponding to a CKD hotspot in the Pacific Coast lowlands of El Salvador, Nicaragua, Guatemala and Costa Rica [1, 2]. In this issue of CKJ, the first systematic review and meta-analysis of this disease identified positive associations with male gender, family history of CKD, high water intake and lowland altitude [2]. The association with male gender and lowland altitude is well known and thought to be a surrogate for hard physical work under warm and humid conditions resulting in repeated dehydration episodes [3, 4]. As correctly pointed out by Gonzalez-Quiroz et al. [2], the association with high water intake is difficult to interpret. It may be interpreted as arguing against the hypothesis that dehydration is a driver of CKD in this population, but also as supporting this hypothesis by illustrating high water ingestion needs that may fail to completely replace fluid lost during strenuous physical work. Complicating matters further, polyuria resulting from partial antidiuretic hormone resistance (isosthenuria) reflecting the loss of the urine concentration function of the kidneys is a manifestation of many forms of CKD. Thus the association with a high water intake may simply reflect the existence of CKD. As interesting as the positive associations are the lack of association found for pesticide exposure, alcohol consumption, non-steroidal anti-inflammatory drugs and heat stress, although for the later, a non-significant 50% increase in risk was found [2]. A prior systematic review encompassing worldwide sites of CKDu only shared with the present systematic review the association of CKDu with family history and male sex [5]. We will focus on the association observed in both systematic reviews, the Meso-American and the worldwide CKDu reviews, of CKDu with a family history of CKD, given the potential conceptual impact on our understanding of CKDu beyond CKD hotspots [2, 5]. The association with a family history was reported from both Guatemala and El Salvador, as well as in Southeast Asia [2, 5]. In this regard, three causes of end-stage renal disease (ESRD) in major registries may be considered inaccurate aetiological coding since diagnostic criteria are unclear and the percentage of patients with ESRD attributed to them varies wildly from country to country but overall represents a relatively constant percentage of ESRD patients: unknown aetiology, vascular/hypertensive and chronic pyelonephritis/chronic tubulointerstitial non-cystic disease [6, 7]. Clearly advances in early aetiological diagnosis and therapy are needed if the current worldwide increase in CKD-related premature mortality is to be contained [8]. Genetic factors are obvious candidates, given recent data and advances in the field.
GENETIC PREDISPOSITION TO CKD: NOT LIMITED TO THE YOUNG
Traditionally nephrologists have thought of familial kidney disease in children and younger patients or when family history was present. Indeed, genetic diseases cause 17% (the third most common cause) of ESRD in children and 69–85% (in younger children) and 11% (in older children) of steroid-resistant nephrotic syndrome children [9, 10]. Autosomal dominant polycystic kidney disease (ADPKD) was thought to be the only familial disease that led to ESRD in considerable numbers of more mature patients, with a mean age at the start of dialysis of 55–60 years (11, 12). However, ADPKD is characterized by large, cystic kidneys, a feature that allowed the diagnosis even before the underlying genetic defects were described. We may hypothesize that genetic diseases without such a striking phenotype may have gone unnoticed, especially if the condition accelerated renal aging, resulting in the potential development of ESRD in the aged. If this is the case, only the identification of the underlying genetic defects will allow the diagnosis of such conditions in the future. In this regard, in Europe a second peak of renal replacement therapy (RRT) incidence at 70–79 years of age, likely due to mutations in PKD2, is almost as high [15–20 per million age-related population (pmarp)] as the classical 55–59 years peak of incidence likely related to PKD1 mutations [12]. Furthermore, the incidence of RRT because of ADPKD at >85 years of age, likely due to PKD2 mutations, is around ∼6 pmarp, similar to the incidence in the age range 40–44 years. This high age range may lead physicians to dismiss a genetic cause for kidney disease (Figure 1). The fact that a bona fide monogenic kidney disease has a natural history of up to nine decades is an eye-opener when exploring the potential impact of the genetic background not only on the need for RRT in the aged, but also in the so-called age-associated physiological decrease of estimated glomerular filtration rate (eGFR) (Figure 2). In this regard, the age range at which genetic screening for steroid-resistant nephrotic syndrome may be informative has increased in recent years. A recent targeted next-generation sequencing panel identified a likely genetic cause in 20% of paediatric and 21% of adult nephrotic cases [13]. Additionally, genetic studies have been informative in adults with CKDu in Western countries. Whole-exome sequencing provided a diagnosis in 24% of 92 adults with CKDu or familial nephropathy or hypertension, encompassing 13 distinct genetic disorders [14]. Nephronophthisis is the most prevalent genetic cause of ESRD in children. However, among 5606 patients with adult-onset ESRD, 0.5% had homozygous deletions in the nephronophthisis-causing gene NPHP1. The median age at ESRD onset was 30 years and the age ranged to 61 years. Most (88%) patients were diagnosed of CKDu [15].
FIGURE 1.
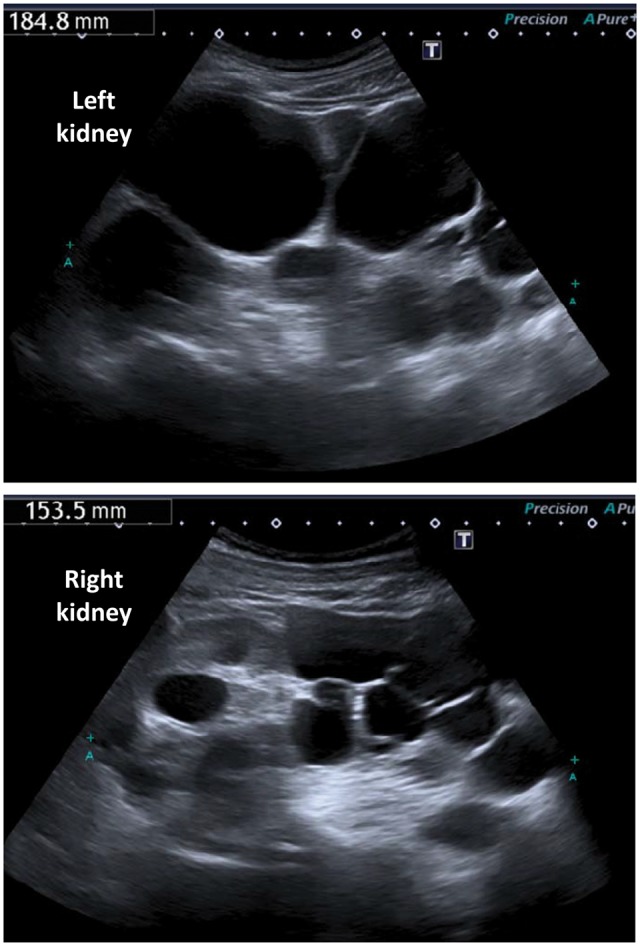
Genetic predisposition to CKD in the elderly. Kidney sonography of an 85-year-old male who had been diagnosed with primary hypertension and CKD due to hypertensive nephropathy at the ages of 74 and 77 years, respectively. Sonography at age 81 years showed multiple bilateral renal cysts and enlarged kidneys without parameters for polycystic kidney disease. At age 85 years, the sonography shown in the image was reviewed by a second radiologist, with evidence of polycystic kidney disease. The final diagnosis was a primary nephropathy (polycystic kidney disease) with secondary hypertension and was incorporated into the patient's records at age 88 years. This case illustrates the potential misdiagnosis of primary hypertension in patients with hypertension secondary to kidney disease, especially at early CKD stages when renal function is relatively well preserved, as well as the potential for genetic kidney disease to lead to advanced CKD in the ninth decade of life and the fact that physicians do not think of hereditary kidney disease at this age even when imaging provides the diagnosis.
FIGURE 2.
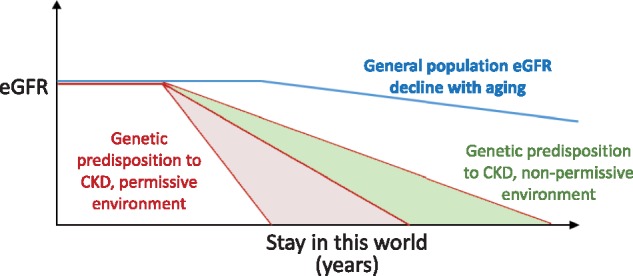
Genetic predisposition and ESRD—impact of the environment and acquired factors. We hypothesize that the genetic background may predispose to CKD development and that this is not limited to ADPKD or other well-characterized genetic kidney diseases. It is likely that the clinical manifestation consists of acceleration of so-called physiological renal aging. Environmental factors or acquired conditions may be protective (shaded in green) or accelerate (shaded in red; permitting CKD progression) renal function loss. The fact that CKD in the elderly is not usually considered when studying the familiar incidence of CKD may have kept this potential genetic influence under the radar. In fact, families are usually only aware of dialysis or transplantation in the family, but are not aware of serum creatinine levels.
HOW A FAMILY HISTORY OF CKD HELPED IDENTIFY GENETIC PREDISPOSITION TO CKD IN WHAT WAS THOUGHT TO BE KIDNEY DISEASE SECONDARY TO HYPERTENSION
Over the years, medicine has tried to explain the pathophysiology of disease well before the real cause was identified. A major example is the concept of hypertensive nephropathy. Despite its high frequency as a diagnosis for incident RRT patients, the diagnostic criteria are unclear and anchored in tradition and have not been adapted to the evolving concept of CKD. Mann and Hilgers [16] state that the diagnosis of hypertensive nephrosclerosis is generally inferred from the characteristic clinical features: usually a long history of hypertension, a relatively normal urine sediment, small kidneys, and, if previous information is available, slowly progressive renal insufficiency with gradually increasing proteinuria. They further indicate that most important from a clinical viewpoint is that hypertension precedes the development of either proteinuria or renal insufficiency. Finally, there should be no other obvious cause of renal disease [16]. Thus it is a diagnosis of exclusion, made only when no other cause is found: diagnosing hypertensive nephropathy or other nephropathy depends on how hard physicians search for a cause for CKD. Furthermore, we now know that both pathological proteinuria and renal insufficiency are late events in the course of CKD. CKD can be diagnosed when renal function, as assessed by the GFR, and proteinuria are both normal, as long as there is other evidence of kidney injury, such as pathological albuminuria. Thus even when hypertension predates both proteinuria and renal insufficiency, CKD may have predated hypertension. The most dramatic example showing that nephrologists may be mistakenly attributing diverse forms of CKD, including genetic CKD, to hypertension is the demonstration that APOL1 gene variants underlie the high frequency of CKD of diverse aetiology in African Americans, including human immunodeficiency virus–associated nephropathy, diabetic nephropathy, focal segmental glomerulosclerosis and, crucially, hypertensive nephropathy [17, 18].
MALE GENDER AND GENETIC KIDNEY DISEASE
The fact that male gender is a risk factor for Meso-American CKD may be explained by environmental factors such as the triggering effect of hard physical work in a warm humid environment. However, beyond X-linked diseases, such as Alport and Fabry disease [19–21], other autosomal genetic kidney diseases, such as ADPKD, progress faster in males than in females, despite more severe liver disease in females [22]. Whether this is part of a more generalized predisposition of males to faster CKD progression is hotly debated [23, 24]. In this regard, when assessed by equations to estimate GFR, CKD diagnosed as a GFR <60 mL/min/1.73 m2 is more frequent in females, although in some studies an eGFR <30 mL/min/1.73 m2 and RRT incidence are more frequent in males [23]. The higher frequency of Meso-American nephropathy in males was found using the GFR <60 mL/min/1.73 m2 definition and thus the predominance of males was even more striking [2].
A FAMILY HISTORY OF CKD AS A RISK FACTOR FOR MESO-AMERICAN NEPHROPATHY: SUGGESTIVE OF A GENETIC PREDISPOSITION TO KIDNEY DISEASE?
Aetiological factors may differ from CKD hotspot to CKD hotspot. Thus different environmental factors may be impacting on a genetic background that may in turn have differences or similarities between different geographical locations.
Despite the higher risk of Meso-American nephropathy when there is a family history of CKD, the authors rapidly dismiss a potential genetic influence and support the hypothesis of a shared environmental factor, such as the hard work of sugarcane collection. In our view, the dismissal of genetic factors is premature. The authors further argue that genetic association studies have failed so far to explain the high familial clustering of CKD [2]. In this regard, genetic eGFR signals identified so far have been estimated to account for <4% of eGFR phenotypic variance in the general population [25]. However, it is unclear whether studies have been performed in the specific populations affected by Meso-American nephropathy. Some genetic determinants of CKD risk can be ethnicity-specific, like APOL1 and individuals of African descent [18]. Furthermore, some of the known gene variants associated with CKD may be present in Meso-American populations. In the USA, 2% of self-identified Hispanics or Latin Americans carry the APOL1 risk alleles as compared with 14% of African Americans and 0.05% of European Americans [26]. There is also the possibility that additional genetic risk factors, not yet described in other populations, underlie the family aggregation of CKD is Meso-America. Only specifically targeted studies are likely to uncover them. In this regard, APOL1–environment interactions may be of greater clinical importance in triggering nephropathy in African Americans than APOL1 interactions with other single-nucleotide polymorphisms [27]. Similarly, a perfect storm of a specific environment and specific genetic background may contribute to the high prevalence of CKD in Meso-America.
There is some evidence potentially supporting a pre-existing predisposition to Meso-American nephropathy. Urinary neutrophil gelatinase–associated lipocalin (NGAL) is a marker of both acute kidney injury and CKD [28]. In Meso-American nephropathy, sugarcane workers with the largest increases in urinary NGAL:creatinine ratios during the 6-month harvest season had decreasing eGFR levels [29]. Interestingly, in healthy Nicaraguan adolescents 12–18 years of age with no prior work history, urinary NGAL, but not proteinuria, was increased in those regions with the highest Meso-American nephropathy incidence [30]. If the absence of work history is correct, this may imply a genetic predisposition or environmental factors beyond hard working conditions.
THE WAY FORWARD
Although Gonzalez-Quiroz et al. [2] identified limitations in the studies included in the meta-analysis, the first meta-analysis in search of risk factors for Meso-American nephropathy provides interesting insights that should be used to drive research. The fact that a family history of CKD is a risk factor for Meso-American nephropathy and that it is the only other risk factor, together with male sex, that is shared by this specific form of CKDu and by other worldwide forms of CKDu is highly suggestive of a potential genetic predisposition. Indeed, it should be considered sufficiently suggestive to perform carefully designed genome-wide association studies and/or whole exome sequencing studies in search of predisposing gene variants that may provide insights into the pathogenesis of the disease and may also be informative about the drivers of CKD in general. The age of genomics is changing many concepts regarding hereditary kidney disease and we hypothesize that it will also become informative for forms of CKD outside the bona fide hereditary kidney diseases [31].
ACKNOWLEDGEMENTS
Grant support was provided by ISCIII and FEDER funds, PI16/02057, Sociedad Española de Nefrologia, ISCIII-RETIC REDinREN RD016/009, Rio Hortega (to M.V.P.G.), Comunidad de Madrid CIFRA2 B2017/BMD-3686.
CONFLICT OF INTEREST STATEMENT
None declared.
Footnotes
This article should be listed in pubmed as comment on: Gonzalez-Quiroz M, Pearce N, Caplin B, Nitsch D. What do epidemiological studies tell us about chronic kidney disease of undetermined cause in Meso-America? A systematic review and meta-analysis. Clin Kidney J 2018
REFERENCES
- 1. Martín-Cleary C, Ortiz A.. CKD hotspots around the world: where, why and what the lessons are. A CKJ review series. Clin Kidney J 2014; 7: 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. González-Quiroz M, Pearce N, Caplin B.. et al. What do epidemiological studies tell us about chronic kidney disease of undetermined cause in Meso-America? A systematic review and meta-analysis. Clin Kidney J 2018; 11: 496–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Correa-Rotter R, Wesseling C, Johnson RJ.. CKD of unknown origin in Central America: the case for a Mesoamerican nephropathy. Am J Kidney Dis 2014; 63: 506–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roncal-Jimenez CA, García-Trabanino R, Wesseling C.. et al. Mesoamerican nephropathy or global warming nephropathy? Blood Purif 2016; 41: 135–138 [DOI] [PubMed] [Google Scholar]
- 5. Lunyera J, Mohottige D, Von Isenburg M.. et al. CKD of uncertain etiology: a systematic review. Clin J Am Soc Nephrol 2016; 11: 379–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pippias M, Kramer A, Noordzij M.. et al. The European Renal Association—European Dialysis and Transplant Association Registry annual report 2014: a summary. Clin Kidney J 2017; 10: 154–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.ERA-EDTA Registry. ERA-EDTA Registry Annual Report 2015 Amsterdam: Academic Medical Center, Department of Medical Informatics, 2017. https://www.era-edta-reg.org/files/annualreports/pdf/AnnRep2015.pdf (21 June 2018, date last accessed)
- 8. Sanchez-Niño MD, Sanz AB, Ramos AM.. et al. Translational science in chronic kidney disease. Clin Sci 2017; 131: 1617–1629 [DOI] [PubMed] [Google Scholar]
- 9. Noone DG, Iijima K, Parekh R.. Idiopathic nephrotic syndrome in children. Lancet 2018; 392: 61–74 [DOI] [PubMed] [Google Scholar]
- 10. ESPN/ERA-EDTA Registry. An Update on the Registry-October 2017 Amsterdam: Academic Medical Center, Department of Medical Informatics 2017. https://www.espn-reg.org/files/AR_2015_dec.pdf (21 June 2018, date last accessed)
- 11. Rodriguez-Osorio L, Vanessa Perez-Gomez M, Ortiz A.. Decreasing incidence of renal replacement therapy over time at the critical 50-59-year age range suggests a role for nephroprotective therapy in ADPKD. Kidney Int 2015; 88: 194. [DOI] [PubMed] [Google Scholar]
- 12. Spithoven EM, Kramer A, Meijer E.. et al. Analysis of data from the ERA-EDTA Registry indicates that conventional treatments for chronic kidney disease do not reduce the need for renal replacement therapy in autosomal dominant polycystic kidney disease. Kidney Int 2014; 86: 1244–1252 [DOI] [PubMed] [Google Scholar]
- 13. Sen ES, Dean P, Yarram-Smith L.. et al. Clinical genetic testing using a custom-designed steroid-resistant nephrotic syndrome gene panel: analysis and recommendations. J Med Genet 2017; 54: 795–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lata S, Marasa M, Li Y.. et al. Whole-exome sequencing in adults with chronic kidney disease: a pilot study. Ann Intern Med 2018; 168: 100–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Snoek R, van Setten J, Keating BJ.. et al. NPHP1 (Nephrocystin-1) gene deletions cause adult-onset ESRD. J Am Soc Nephrol 2018; 29: 1772–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mann J, Hilgers K.. Clinical Features, Diagnosis, and Treatment of Hypertensive Nephrosclerosis In: UpToDate, Waltham, MA. Topic last updated: Aug 28, 2017. http://www.uptodate.com (21 June 2018, date last accessed)
- 17. Genovese G, Friedman DJ, Ross MD.. et al. Association of trypanolytic APOL1 variants with kidney disease in African Americans. Science 2010; 329: 841–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Freedman BI, Sedor JR.. Hypertension-associated kidney disease: perhaps no more. J Am Soc Nephrol 2008; 19: 2047–2051 [DOI] [PubMed] [Google Scholar]
- 19. Kruegel J, Rubel D, Gross O.. Alport syndrome—insights from basic and clinical research. Nat Rev Nephrol 2013; 9: 170–178 [DOI] [PubMed] [Google Scholar]
- 20. Ortiz A, Oliveira JP, Waldek S.. et al. Nephropathy in males and females with Fabry disease: cross-sectional description of patients before treatment with enzyme replacement therapy. Nephrol Dial Transplant 2008; 23: 1600–1607 [DOI] [PubMed] [Google Scholar]
- 21. Ortiz A, Cianciaruso B, Cizmarik M.. et al. End-stage renal disease in patients with Fabry disease: natural history data from the Fabry Registry. Nephrol Dial Transplant 2010; 25: 769–775 [DOI] [PubMed] [Google Scholar]
- 22. Gansevoort RT, Arici M, Benzing T.. et al. Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: a position statement on behalf of the ERA-EDTA Working Groups on Inherited Kidney Disorders and European Renal Best Practice. Nephrol Dial Transplant 2016; 31: 337–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fernandez-Prado R, Fernandez-Fernandez B, Ortiz A.. Women and renal replacement therapy in Europe: lower incidence, equal access to transplantation, longer survival than men. Clin Kidney J 2018; 11: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Piccoli GB, Alrukhaimi M, Liu Z-H.. et al. Women and kidney disease: reflections on World Kidney Day 2018. Clin Kidney J 2018; 11: 7–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Limou S, Vince N, Parsa A.. Lessons from CKD-related genetic association studies-moving forward. Clin J Am Soc Nephrol 2018; 13: 140–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Udler MS, Nadkarni GN, Belbin G.. et al. Effect of genetic african ancestry on eGFR and kidney disease. J Am Soc Nephrol 2015; 26: 1682–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Langefeld CD, Comeau ME, Ng MCY.. et al. Genome-wide association studies suggest that APOL1-environment interactions more likely trigger kidney disease in African-Americans with nondiabetic nephropathy than strong APOL1—second gene interactions. Kidney Int 2018; doi: 10.1016/j.kint.2018.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Castillo-Rodriguez E, Fernandez-Prado R, Martin-Cleary C.. et al. Kidney injury marker 1 and neutrophil gelatinase-associated lipocalin in chronic kidney disease. Nephron 2017; 136: 263–267 [DOI] [PubMed] [Google Scholar]
- 29. Laws RL, Brooks DR, Amador JJ.. et al. Biomarkers of kidney injury among Nicaraguan sugarcane workers. Am J Kidney Dis 2016; 67: 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ramírez-Rubio O, Amador JJ, Kaufman JS.. et al. Urine biomarkers of kidney injury among adolescents in Nicaragua, a region affected by an epidemic of chronic kidney disease of unknown aetiology. Nephrol Dial Transplant 2016; 31: 424–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ars E, Torra R.. Rare diseases, rare presentations: recognizing atypical inherited kidney disease phenotypes in the age of genomics. Clin Kidney J 2017; 10: 586–593 [DOI] [PMC free article] [PubMed] [Google Scholar]