

Abstract
Organic cation transporter (OCT) 2 mediates the entry step for organic cation secretion by renal proximal tubule cells and is a site of unwanted drug-drug interactions (DDIs). But reliance on decision tree–based predictions of DDIs at OCT2 that depend on IC50 values can be suspect because they can be influenced by choice of transported substrate; for example, IC50 values for the inhibition of metformin versus MPP transport can vary by 5- to 10-fold. However, it is not clear whether the substrate dependence of a ligand interaction is common among OCT2 substrates. To address this question, we screened the inhibitory effectiveness of 20 µM concentrations of several hundred compounds against OCT2-mediated uptake of six structurally distinct substrates: MPP, metformin, N,N,N-trimethyl-2-[methyl(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)amino]ethanaminium (NBD-MTMA), TEA, cimetidine, and 4–4-dimethylaminostyryl-N-methylpyridinium (ASP). Of these, MPP transport was least sensitive to inhibition. IC50 values for 20 structurally diverse compounds confirmed this profile, with IC50 values for MPP averaging 6-fold larger than those for the other substrates. Bayesian machine-learning models of ligand-induced inhibition displayed generally good statistics after cross-validation and external testing. Applying our ASP model to a previously published large-scale screening study for inhibition of OCT2-mediated ASP transport resulted in comparable statistics, with approximately 75% of “active” inhibitors predicted correctly. The differential sensitivity of MPP transport to inhibition suggests that multiple ligands can interact simultaneously with OCT2 and supports the recommendation that MPP not be used as a test substrate for OCT2 screening. Instead, metformin appears to be a comparatively representative OCT2 substrate for both in vitro and in vivo (clinical) use.
Introduction
It is estimated that approximately 40% of all prescribed drugs are positively charged at physiologic pH and that the kidney plays a significant role in the elimination of these “organic cations” (OCs) from the body (Neuhoff et al., 2003; Hagenbuch, 2010). The proximal tubule is the site for the secretion of OCs via a two-step process that involves the OC transporter (OCT) 2 and multidrug and toxin extrusion transporter (MATE) 1. It is widely accepted that in humans the initial step in OC secretion is mediated by the electrogenic uniporter OCT2, which relies on the inside negative potential of renal proximal tubule cells to drive the net movement of OCs from the blood into the renal proximal tubule (Holohan and Ross, 1980; Budiman et al., 2000); together, OCT2 and MATE1 help to define the pharmacokinetics of structurally diverse OCs that share these transporters as a common pathway for elimination. Competition for the limited number of transport sites in this shared pathway can result in altered pharmacokinetics of prescribed OCs, potentially resulting in adverse drug-drug interactions (DDIs) (Somogyi et al., 1987; Stage et al., 2015; Yin et al., 2016). Additionally, in drug development there is a significant cost in both time and money invested in assessing the DDI risk of new molecular entities (NMEs). Hence, the prediction and prevention of DDIs through an understanding of the selectivity of OCT2/MATE1 would reduce costs in drug development in addition to improving health care.
OCT2 has been a particular focus of efforts to predict the likelihood that an NME will inhibit the activity of the OC secretory process. The primary approach for assessing the selectivity of OCT2 uses cultured cells that express the transporter to determine the extent of inhibition of transport activity produced by each of a set of test agents (Nies et al., 2011b). But, whereas the set of inhibitory agents may be large and structurally diverse (Kido et al., 2011), transport activity is generally assessed by monitoring the uptake of a single, presumably representative, substrate. Interestingly, the pharmacophores generated by different groups to describe the molecular determinants of ligand interaction with OCT2, although qualitatively similar in their inclusion of several common structural characteristics (including hydrophobicity, hydrogen-bonding features, and positive charge), differ from one another with respect to the three-dimensional placement of these elements (Suhre et al., 2005; Zolk et al., 2009; Nies et al., 2011a; Xu et al., 2013; Liu et al., 2016). This led to the suggestion that ligand interaction with OCT2 may be influenced by the choice of substrate used to assess transport activity (Belzer et al., 2013), an idea supported by the observation that IC50 values for the inhibition of OCT2 activity produced by six commonly prescribed drugs were, on average, 9- to 10-fold greater when using MPP as a substrate than when using metformin as a substrate (Zolk et al., 2009; Belzer et al., 2013). Additionally, a follow-up study (Hacker et al., 2015) that used 125 commonly prescribed drugs reported that OCT2-mediated metformin transport is significantly more sensitive to inhibition than is MPP transport. These observations support the hypothesis that OCT2 has a complex binding surface where ligands may interact simultaneously at different sites (Volk et al., 2003; Minuesa et al., 2009; Harper and Wright, 2013). They also decrease confidence in the validity of recommendations to pursue (or not) clinical studies of DDIs produced by a suspected inhibitor that are based on the inhibition of a single substrate (Giacomini et al., 2010). But, whereas differences in the inhibitory profiles for MPP and metformin have been reported repeatedly (Zolk et al., 2009; Belzer et al., 2013; Hacker et al., 2015; Yin et al., 2016), the extent of such differential substrate interactions with OCT2 is not clear. An assessment of the susceptibility of other OCT2 substrates to transport inhibition may provide insight into the mechanism of ligand interaction at OCT2 and clarify whether there is an “ideal” substrate for characterizing drug affinity at OCT2, or whether testing multiple substrates is the more prudent course.
Here we test the inhibitory effectiveness of 400 or more compounds against the OCT2-mediated uptake of six structurally distinct substrates [metformin, cimetidine, TEA, MPP, and the fluorescent probes 4–4-dimethylaminostyryl-N-methylpyridinium (ASP) and N,N,N-trimethyl-2-[methyl(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)amino]ethanaminium (NBD-MTMA)]. We also provide a quantitative comparison of our own OCT2 inhibition profiles to that reported by Kido et al. (2011) in their single-substrate (ASP) screen of the inhibition of OCT2 activity produced by 900 prescription drugs. These data sets were used for Bayesian machine-learning analysis and the development of predictive algorithms, which we share herein, thereby allowing others to generate predictions of ligand interaction with OCT2 prior to in vitro testing.
Materials and Methods
Chemicals.
[3H]MPP [specific activity (S.A.), 80 Ci/mmol] was purchased from PerkinElmer (Waltham, MA); [3H]cimetidine (S.A., 80 Ci/mmol) and [3H]TEA (S.A., 54 Ci/mmol) were purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO); and [14C]metformin (S.A., 90 mCi/mmol) was purchased from Moravek, Inc. (Brea, CA). Unlabeled cimetidine and metformin were purchased from Sigma-Aldrich (St. Louis, MO) and AK Scientific, Inc. (Union City, CA), respectively. The fluorescent compound ASP was purchased from Invitrogen (Carlsbad, CA); the fluorescent compound NBD-MTMA (Aavula et al., 2006) (purity, >97%) and the non–radioactively labeled MPP (purity, >99.5%) were synthesized by the Department of Chemistry and Biochemistry, University of Arizona (Tucson, AZ). Ham’s F-12 medium Kaighn’s modification, and Dulbecco’s modified Eagle’s medium were obtained from Sigma-Aldrich. The National Institutes of Health Clinical Collection (NCC), a plated array of approximately 900 small molecules that have a history of use in human clinical trials, was acquired from Evotec (San Francisco, CA). Other reagents were of analytical grade and were commercially obtained.
Drug Screening.
Compounds from the NCC, distributed in 80 wells of 96-well plates [100 nmol/well in 10 µl of dimethylsulfoxide (DMSO)], were screened for their inhibitory effectiveness against the transport activity of six model substrates of OCT2. A total of 480 compounds were used for MPP, TEA, NBD-MTMA, and metformin; 400 compounds were used for cimetidine and ASP. Each compound was diluted to a concentration of 20 µM, pH 7.4, to a final concentration of 2% DMSO using a VIAFLO Multichannel Electronic Pipette (Integra Biosciences Corp., Hudson, NH) (Martínez-Guerrero et al., 2016).
Cell Culture.
Chinese hamster ovary (CHO) cells with a single integrated Flp-In recombination site were obtained from Invitrogen. Methods for the generation of cell lines that stably express human OCT2 (hOCT2) were described previously (Pelis et al., 2007). Cells were passed every 3–4 days and maintained at 37°C in a humidified environment with 5% CO2. The expression of hOCT2 in cells was maintained through hygromycin (200 µg/ml; Invitrogen) selective pressure. When seeded into 96-well plates (Greiner Bio-One; VWR, Arlington Heights, IL) for transport assays, these cells were grown to confluence in antibiotic-free media.
Transport Experiments.
Cells were seeded in 96-well plates with 200 µl of cell media containing 550,000 or 275,000 cells/ml, and experiments were typically performed 24 or 48 hours later, respectively. To begin an experiment, media was aspirated and the wells were washed for three cycles with 300 µl of room temperature Waymouth Buffer (WB; 135 mM NaCl, 13 mM HEPES, 2.5 mM CaCl2 · 2H2O, 1.2 mM MgCl2, 0.8 mM MgSO4 · 7H2O, 5 mM KCl, and 28 mM d-glucose, pH 7.4) using an automatic fluid aspirator/dispenser (model 406; BioTek, Winooski, VT). Transport was then initiated by the addition of 60 µl of WB containing a radiolabeled or fluorescent substrate and other compounds as needed. For time-course experiments using ASP as a substrate, transport buffer was added using the automatic fluid aspirator/dispenser, and for all other experiments transport buffer was added using the VIAFLO 96-well multichannel pipette. After selected time intervals, transport was terminated by rinsing with three cycles of cold WB (300 µl) or for the measurement of OCT2-mediated ASP uptake, with a continuous rinse of 900 µl of cold WB. For radiolabeled substrates, uptake was quantified by adding 200 µl of scintillation cocktail per well and sealing the plates (Topseal-A; PerkinElmer). After allowing the plates to sit for at least 2 hours, radioactivity was determined in a 12-channel multiwell scintillation counter (Wallac Trilux 1450 MicroBeta; PerkinElmer). The uptake of fluorescent substrates was assessed by adding 60 µl of DMSO (for NBD-MTMA) or 100 µl of 1 mM SDS (for ASP), and, after sitting for at least 2 hours, fluorescence was measured using a CLARIOStar microplate reader (BMG LABTECH, Ortenberg, Germany) with emission and excitation filters set to 490 and 540 nm for NBD-MTMA, or to 460 and 575 nm for ASP.
Transport Data Analysis.
Kinetic parameters were based on estimates of the initial rate of uptake derived from either 30-second (radiolabeled substrates) or 2-minute (fluorescent substrates) determinations of the net accumulation in CHO cells that stably expressed OCT2. For all six substrates, the kinetics of uptake reflected the sum of two processes: 1) OCT2-mediated uptake that was described by Michaelis-Menten kinetics; and 2) a nonsaturable, first-order component that was dominated by extracellular substrate left after the rapid rinsing procedure. Although the first-order component was evident in parallel determinations of transport in wild-type (WT) (non–OCT2-expressing) CHO cells (see Supplemental Fig. 1), we found that subtracting WT accumulation from total accumulation measured in OCT2-expressing cells frequently introduced errors into kinetic analyses, particularly at the high end of the substrate concentration employed for some substrates (e.g., metformin). These errors reflected modest differences in the efficiency of rinsing extracellular substrate from the OCT2 and WT CHO cell lines, which are evident in differences in the retention of an extracellular space marker after rinsing (data not shown). Consequently, kinetic parameters were determined from total uptake (mediated plus nonsaturable) using the following relationship:
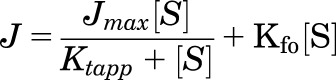 |
(1) |
where J is the rate of mediated uptake from a substrate concentration of [S], Jmax is the maximal rate of mediated substrate uptake, Ktapp is the apparent Kt (Michaelis-Menten constant) value (i.e., the substrate concentration in the bulk medium that resulted in half-maximal mediated uptake), and Kfo is a first-order rate constant describing the nonsaturable (nonmediated) component of total net substrate accumulation. Rates of transport were expressed in moles per minute, normalized to the surface area of the confluent monolayer. For the purpose of comparison with rates reported in studies that normalize transport to cell protein, we find the factor of 0.035 mg cell protein/cm2 to be reasonably accurate (Schömig et al., 2006).
For screening the influence of test inhibitors on OCT2 activity, it proved adequate to determine mediated uptake by subtracting the accumulation in WT CHO cells from total uptake (30 seconds or 2 minutes, respectively, for radiolabeled or fluorescent substrates) in OCT2-expressing cells, measured in the presence and absence of test inhibitor. Resulting OCT2-mediated transport was expressed relative to that determined in the absence of inhibitor (i.e., the percentage of control uptake). In addition, for some compounds, the IC50 value was determined by measuring the rate of OCT2-mediated substrate transport as a function of increasing inhibitor concentration, as described by:
 |
(2) |
where J* is the rate of OCT2-mediated transport of labeled substrate from a concentration of substrate equal to [S*] (which was selected to be at least three times less than the Ktapp value for transport of that substrate), and Japp is a constant that includes the maximal rate of substrate transport times the ratio of the inhibitor IC50 and the Ktapp value for the transport of the labeled substrate (Groves et al., 1994).
IC50 values were also predicted (IC50-pred) from the screening inhibition measurements using the approach described by Kido et al. (2011):
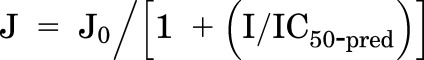 |
(3) |
where J and J0 represent OCT2-dependent transport activity determined in the presence and absence of the inhibitor, respectively, and I is the fixed inhibitor concentration (in this case, 20 μM). This approach gives reasonable inhibitor affinity estimates when the screening concentration is within the linear part of the IC50 curve (i.e., approximately 10%–90% inhibition) (Gao et al., 2002; Kido et al., 2011; Hu et al., 2016); in the present study, this corresponded to IC50 values between 2 and 180 μM.
Results are presented as the mean ± S.E. Unless otherwise noted, statistical analyses were performed using a two-tailed paired Student’s t test. Curve fitting used algorithms in Prism version 6.07 (GraphPad Software, San Diego, CA).
Computational Modeling.
We generated and validated Laplacian-corrected naive Bayesian classifier models using Discovery Studio version 4.1 (Biovia, San Diego, CA). The values of the AlogP; molecular weight; number of rotatable bonds, rings, aromatic rings, hydrogen bond acceptors, and hydrogen bond donors; molecular fractional polar surface area; and molecular function class fingerprints of maximum diameter 6 [extended connectivity fingerprint 6 (ECFP_6)] were used as the molecular descriptors. Compounds that reduced transport to less than 50% of control were classed as actives, and everything else was classed as inactive. Computational models were validated using leave-one-out cross-validation, in which each sample was left out one at a time. A model was built using the remaining samples, and that model was used to predict the left-out sample. Each model was internally validated, receiver operating characteristic (ROC) curve plots were generated, and the cross-validated ROC “area under the curve” was calculated. Then, 5-fold cross-validation (i.e., leave out 20% of the data set, and repeat five times) was also performed.
Sixteen Bayesian models were built with the ECFP_6 descriptor only, using Assay Central (Collaborations Pharmaceuticals, Inc., Raleigh, NC) (Clark and Ekins, 2015; Clark et al., 2015), consisting of either training data only or combined with testing data for each probe mentioned previously. Chemical structures were examined for valence errors, anionic charges were neutralized, salts were removed, and certain molecules, such as mixtures (e.g., dimenhydrinate) or non–drug-like compounds (e.g., zinc-chloride), were omitted prior to building a respective model. Structures were also checked for accuracy against four common, reliable resources: CompTox (https://comptox.epa.gov/dashboard), ChemSpider (http://www.chemspider.com/), Merck Index (https://www.rsc.org/merck-index), Pubchem (https://pubchem.ncbi.nlm.nih.gov/). When there was not agreement across these resources, consistency was ensured across similar structures by removing any conflicting stereochemistry. The same threshold was used (50% inhibition or greater) as well as the same method of 5-fold cross-validation and ROC calculation. Testing data sets consisting of 80 compounds were collated to measure the predictive capability of training data and generate statistics.
Results
Kinetic Characterization of OCT2 Test Substrates.
OCT2-mediated transport activity was determined using six substrates: metformin, cimetidine, MPP, TEA, ASP, and NBD-MTMA. These compounds were chosen because they are: 1) known substrates of OCT2; 2) structurally diverse (Fig. 1; Supplemental Table 1); and 3), in the case of metformin and cimetidine, clinically relevant (Nies et al., 2011b). Two-minute time courses showing OCT2-mediated net uptake of all six substrates are shown in Fig. 1. The time courses for MPP, TEA, metformin, and cimetidine were curvilinear and adequately described by one-phase association (first-order exponential rise to steady state; Prism 5; GraphPad); NBD-MTMA and ASP uptakes were described by simple linear regression (Fig. 1). Subsequent kinetic analyses used 30-second uptakes for the radiolabeled substrates metformin, cimetidine, MPP, and TEA, resulting in 5%–25% underestimates of the initial rates of transport (as predicted from the slopes at time zero of the one-phase association curves) (Supplemental Fig. 1). The initial rates of transport of the fluorescent substrates NBD-MTMA and ASP were based on 2-minute uptakes, which were within the apparent linear phase of transport.
Fig. 1.

Time course of OCT2-mediated uptake of 0.31 µM [3H]MPP (A), 13.9 µM [14C]metformin (B), 0.0218 µM [3H]TEA (C), 0.0134 µM [3H]cimetidine (D), 3 µM NBD-MTMA (E), and 10 µM ASP (F). Uptakes are reported as clearance (microliters per square centimeter). These data represent OCT2-mediated transport (i.e., uptake in wild-type CHO cells was subtracted from total substrate uptake measured in OCT2-expressing cells). Each data point is the mean ± S.E. determined in two experiments (MPP, metformin, cimetidine, TEA, and ASP) or three experiments (NBD-MTMA), each using three to five replicate wells. The lines fit to the data for MPP, metformin, TEA, and cimetidine were calculated using an exponential one-phase association function (Prism; GraphPad); the uptakes of NBD-MTMA and ASP were described by simple linear regression.
Figure 2 shows the kinetic profiles for OCT2-mediated transport of the test substrates and highlights another basis for their inclusion in this study, namely, the wide range of their kinetic parameters. The kinetic values determined in two to eight separate experiments for each substrate are summarized in Table 1. Jmax values ranged from 17 (for MPP) to 656 pmol/cm2 per minute (for metformin), and Ktapp values ranged from 5 µM (for MPP) to 285 µM (for metformin); the kinetic parameters for all substrates were within the range reported previously by us and others (Suhre et al., 2005; Nies et al., 2011b; Belzer et al., 2013; Harper and Wright, 2013; Severance et al., 2017).
Fig. 2.

The kinetics of OCT2-mediated transport of MPP (A), metformin (B), TEA (C), cimetidine (D), NBD-MTMA (E), and ASP (F). Rates of uptake presented here were corrected for the first-order component of transport (see eq. 1 and Supplemental Fig. 1). Each data point represents the mean ± S.E. rate of mediated (saturable) transport based on 30-second (MPP, metformin, TEA, and cimetidine) or 2-minute (NBD-MTMA and ASP) net accumulation in five to eight replicate wells with 8–12 different substrate concentrations, determined in three to eight individual experiments. Lines and kinetic parameters were derived from fitting the Michaelis-Menten equation to these data.
TABLE 1.
Kinetics of OCT2-mediated transport for the six test substrates
| Substrate |
Ktapp |
±S.E. |
Jmax |
±S.E. |
n |
|---|---|---|---|---|---|
| μM | pmol/cm2 per minute | ||||
| Metformin | 285.2 | 67.5 | 656.2 | 42.0 | 3 |
| MPP | 4.6 | 1.5 | 17.2 | 1.5 | 3 |
| NBD | 8.8 | 2.1 | 33.2 | 1.6 | 3 |
| TEA | 57.7 | 18.3 | 99.9 | 10.1 | 2 |
| Cimetidine | 15.1 | 3.5 | 18.9 | 0.7 | 2 |
| ASP | 37.6 | 21.0 | 51.1 | 8.5 | 8 |
Screening of Inhibition of OCT2-Mediated Transport.
We initially determined the effect of between 320 and 400 (depending on the substrate) compounds from the NCC on the transport of each of the six test substrates. These compounds were used as the “training set” for the development of Bayesian models discussed later in this report. To validate these models, a test set of an additional 80 compounds was used against the OCT2-mediated transport of these substrates, resulting in a total of 400–480 tested compounds. These 480 compounds are structurally diverse and included molecules that are predicted to carry a net positive (∼31.2%), negative (∼19.4%), or neutral (∼49.4%) charge at physiologic pH (Supplemental Table 2). Figure 3A shows the effect of a 20 µM concentration of each of 480 compounds (Supplemental Table 3) on OCT2-mediated transport of 12 µM [14C]metformin. Presented in rank order of increasing inhibitory potency, 87 of these compounds (18%) blocked metformin transport by at least 50%. These same compounds were also tested against the transport of [3H]MPP (15 nM), but only 43 (9%) blocked transport by at least 50% (Fig. 3B). The inset of Fig. 3B shows the profile of inhibition of MPP transport with the test agents rank ordered according to their inhibition of metformin; although the rank order of effectiveness differed somewhat, the overall profile of inhibition was similar to that observed for metformin. The difference in apparent sensitivity to the inhibition of OCT2 activity between the transport of metformin and MPP was particularly evident when the results for individual inhibitors of each substrate were compared in a pairwise fashion (Fig. 3C). Focusing on the 225 compounds that inhibited metformin transport by at least 10% (indicative of IC50 values <200 µM), 89% of these compounds were more effective inhibitors of metformin transport than of MPP transport. Overall (by paired t test), these compounds were more effective inhibitors of metformin transport than of MPP transport (P < 0.05), and on average reduced metformin transport by about 34% more than they did MPP transport.
Fig. 3.

The inhibitory effect of 480 test compounds from the National Clinical Collection on the OCT2-mediated transport of ∼12 µM [14C]metformin (A) and ∼15 nM [3H]MPP (B). The 30-second accumulation of the two substrates was measured in the presence of a 20 µM concentration of each test agent. The height of the shaded gray region indicates the average ± S.E. (black lines) accumulation (expressed relative to uptake measured in the absence of inhibitor; i.e., control) determined in two separate experiments, each measured in triplicate and corrected for uptake measured in wild-type CHO cells. The histograms are arranged from no inhibition (left side) to complete inhibition (right side), whereas the inset in (B) represents the MPP histogram using the rank order derived from the metformin screen in (A). The horizontal red dashed lines in (A and B) indicate 50% inhibition, whereas the vertical red dashed lines divide the active inhibitors (<50% of control uptake; to the right) from the “inactive” inhibitors (>50% control, to the left). (C) is the pairwise comparison of the inhibitory profiles of MPP and metformin produced by the test compounds from the NCC. The dashed red lines represent 90% of remaining transport; compounds that fall in the top right quadrant produced little to no inhibition for either substrate. The solid red line is the line of unity, and data points that fall on this line represent compounds that inhibited MPP and metformin uptake equally.
Figure 4 shows the rank-ordered inhibition profiles for OCT2-mediated transport of cimetidine, TEA, NBD-MTMA, and ASP (with insets showing profiles that used the metformin rank order) (Supplemental Table 3). These profiles were qualitatively similar to those observed for metformin and MPP, with the 50% inhibition standard produced by 74 (15%), 90 (19%), 81 (20%), or 92 (23%) compounds, against the transport of TEA, NBD-MTMA, cimetidine, or ASP, respectively. Figure 5 shows the pairwise comparisons of inhibition produced by these compounds versus the inhibition of metformin (Fig. 5, A, C, E, and G) and MPP (Fig. 5, B, D, F, and H); MPP was consistently the least sensitive to inhibition when compared with the inhibitory profiles of these other substrates. Pairwise comparisons of these other substrates against metformin revealed smaller, albeit statistically significant, differences among the inhibitory profiles of TEA, NBD-MTMA, and ASP. Metformin transport was approximately 3% more sensitive to inhibition than TEA and NBD-MTMA, and 7% less sensitive to inhibition than ASP (P < 0.0001 for TEA, P < 0.001 for NBD-MTMA, P < 0.0126 for ASP). With a 0.6% difference between the average observed inhibition, the inhibitory profile for cimetidine was not significantly different from the inhibitory profile of metformin (P = 0.45).
Fig. 4.

The effect of 400–480 compounds from the NCC on the OCT2-mediated transport of NBD-MTMA (A), TEA (B), cimetidine (C), and ASP (D). The 30-second accumulation of TEA and cimetidine, and the 2-minute accumulation of NBD-MTMA and ASP, were measured in the presence of a 20 µM concentration of each test agent. In each histogram, the height of the shaded gray region indicates the average ± S.E. (black lines) accumulation (expressed relative to uptake measured in the absence of inhibitor (i.e., control) determined in two separate experiments, each measured in triplicate and corrected for uptake measured in wild-type CHO cells, displayed in order of increasing (left-to-right) inhibition of OCT2-mediated transport (inset histograms are arranged in the same order as for metformin inhibitory effectiveness shown in Fig. 3A). Blank spaces that interrupt the inset histograms for cimetidine and ASP represent compounds that were tested against metformin uptake but were not tested against ASP and cimetidine uptake (80 compounds). The horizontal red dashed lines represent 50% inhibition, whereas the vertical red dashed lines divide the active inhibitors (<50% of control uptake) from the inactive inhibitors (>50% control).
Fig. 5.

Pairwise comparisons of the percentage of remaining OCT2-mediated transport activity (from Fig. 4) for NBD-MTMA, TEA, cimetidine, and ASP (y-axes) vs. the remaining transport activity for metformin (A, C, E, and G) and MPP (B, D, F, and H) (x-axes). The lines of unity are represented by solid red lines, and dashed lines represent 90% of remaining transport activity (top right quadrant is composed of inhibitors that inhibited the transport of both substrates by less than 10%).
The systematic influence of substrate identity on the inhibition of OCT2 activity was examined in more detail by determining IC50 values for a subset of the NCC test compounds. Figure 6A shows the effect of increasing concentrations of a representative test inhibitor (irinotecan) on the OCT2-mediated transport of MPP and metformin. Consistent with the general observation evident from the single-concentration screening data presented in Fig. 3, the IC50 value for irinotecan inhibition of MPP transport was 4-fold larger than that for the inhibition of metformin transport (13.8 vs. 3.4 µM) (Table 2). Figure 6B shows the composite IC50 data for irinotecan inhibition of the transport of three additional substrates, with IC50 values of TEA (4.7 µM), NBD-MTMA (4.0 µM), and ASP (3.0 µM). The IC50 value for the inhibition of MPP transport was consistently 3- to 4-fold greater than those for the other test substrates (Supplemental Fig. 2; Table 2). Figure 7A shows the pairwise comparison of IC50 values for the inhibition of MPP and metformin transport for 20 compounds selected from the screening set. IC50 values for the inhibition of MPP transport were, on average, 5.7-fold greater (570%) than those for the inhibition of metformin transport. It is relevant to note that the IC50 values for these compounds calculated from the screening data, using control uptake (100%) and the single point reflecting transport measured in the presence of a 20 µM inhibitor, correlated closely with the measured IC50 values. Figure 7, B and C show pairwise comparisons of “measured versus predicted” IC50 values for MPP and metformin, respectively; the predicted IC50 values differed from the measured values by less than 40%. This degree of concordance suggested that a comparison of the predicted IC50 values from the screening data would permit an expanded view of the relative inhibitory profiles for OCT2-mediated transport of MPP and metformin that reflected a broader range of substrate structure than that afforded by the subset of compounds included in Fig. 7A, while also providing a pharmacological context afforded by IC50 values (rather than “percentage of control”). Figure 7D shows the pairwise comparisons of predicted IC50 values for inhibition of metformin transport against those for MPP transport (estimated from percentage control values between 10% and 90%, a total of 162 compounds). Of these predicted IC50 values, 29% differed by more than 3-fold (designated by the dashed lines in Fig. 7D; Supplemental Table 3).
Fig. 6.

Influence of substrate on the inhibition of OCT2-mediated transport produced by irinotecan. (A) Effect of increasing the irinotecan concentration on OCT2-mediated transport of [14C] metformin (16 µM; solid squares) or [3H]MPP (15 nM; solid circles). Each point is the mean rate of transport measured at each inhibitor concentration (normalized to transport measured in the absence of inhibitor; “% control”) measured in two separate experiments (±S.E.; n = 2), each determined in four replicate wells. (B) Data from Fig. 4A, plus the IC50 profiles for irinotecan inhibition of [3H]TEA (20 nM; open diamonds), NBD-MTMA (3 µM; open circles), and ASP (10 µM; open triangles); error bars removed for clarity.
TABLE 2.
IC50 values for inhibition of OCT2 mediated transport of five substrates
All uptakes were corrected for the first-order (nonsaturable) component of total substrate uptake. Results are presented as the mean ± S.E.
| Inhibitor | Metformin |
MPP |
TEA |
NBD-MTMA |
ASP |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 | ±S.E. | IC50 | ±S.E. | IC50 | ±S.E. | IC50 | ±S.E. | IC50 | ±S.E. | |
| µM | ||||||||||
| Amantadine | 27.1 | 0.7 | 51.0 | 2.2 | 32.1 | 1.5 | 24.5 | 1.6 | 10.1 | 3.8 |
| Amitriptyline | 2.4 | 0.7 | 11.3 | 0.4 | 5.0 | 0.6 | 4.0 | 0.6 | 1.8 | 0.1 |
| Anastrozole | 51.1 | 0.5 | 327.6 | 2.6 | 113.4 | 9.4 | 86.0 | 6.8 | 39.2 | 3.0 |
| Atomoxetine | 6.6 | 0.1 | 20.4 | 2.3 | 12.8 | 2.0 | 7.6 | 0.9 | 3.5 | 0.0 |
| Atropine | 1.3 | 0.2 | 12.4 | 0.8 | 3.4 | 0.8 | 2.3 | 0.6 | 1.4 | 0.1 |
| Benztropine | 0.4 | 0.0 | 13.4 | 7.2 | 1.0 | 0.1 | 0.8 | 0.2 | 0.3 | 0.0 |
| Bupropion | 8.6 | 1.8 | 32.2 | 5.4 | 13.4 | 2.8 | 10.8 | 1.0 | 11.1 | 1.6 |
| Buspirone | 10.2 | 0.1 | 68.5 | 4.4 | 19.8 | 1.0 | 19.6 | 3.2 | 8.6 | 2.6 |
| Fluoxetine | 18.6 | 1.5 | 56.7 | 4.5 | 36.0 | 5.4 | 20.1 | 2.6 | 16.7 | 0.8 |
| Imipramine | 0.4 | 0.2 | 4.9 | 0.3 | 1.3 | 0.3 | 1.4 | 0.1 | 0.5 | 0.3 |
| Irinotecan | 3.3 | 0.1 | 15.6 | 1.4 | 4.5 | 0.3 | 3.2 | 0.7 | 1.7 | 0.0 |
| Mexiletine | 21.5 | 0.6 | 50.4 | 5.6 | 28.5 | 2.7 | 30.5 | 0.7 | 19.4 | 4.4 |
| Naloxone | 29.5 | 0.0 | 235.7 | 9.2 | 67.3 | 2.8 | 55.1 | 6.6 | 20.3 | 3.7 |
| Pilocarpine | 34.5 | 0.4 | 109.3 | 12.2 | 61.9 | 0.1 | 48.2 | 1.2 | 55.7 | 24.1 |
| Quinidine | 18.6 | 1.6 | 91.1 | 3.4 | 23.6 | 1.0 | 18.7 | 1.2 | 6.4 | 0.3 |
| Tacrine | 0.6 | 0.0 | 2.3 | 0.2 | 0.9 | 0.1 | 1.0 | 0.3 | 0.4 | 0.1 |
| Trimethoprim | 19.8 | 1.8 | 199.2 | 54.9 | 50.2 | 11.2 | 43.0 | 2.7 | 14.1 | 1.0 |
| Tripelennamine | 2.1 | 0.2 | 12.1 | 2.0 | 5.0 | 0.7 | 5.7 | 1.0 | 3.7 | 0.9 |
| Tropicamide | 94.3 | 2.4 | 691.5 | 31.3 | 205.4 | 4.6 | 189.0 | 1.6 | 141.4 | 42.5 |
| Verapamil | 10.3 | 1.0 | 49.7 | 3.3 | 17.8 | 2.3 | 9.7 | 1.9 | 4.6 | 1.5 |
Fig. 7.

Pairwise comparisons of IC50 values (micromolar) for the inhibition of OCT2-mediated transport of structurally distinct substrates. (A) Pairwise comparison of IC50 values for the inhibition of OCT2-mediated transport of metformin (x-axis) and MPP (y-axis) determined for a subset of 20 compounds selected from the NCC (Table 2) (note the pairwise comparison of IC50 values for irinotecan, shown as an open circle). Each IC50 value is the mean ± S.E. (n = 2) of values determined in two separate experiments using the experimental design used for the experiments presented in Fig. 6 (also Supplemental Fig. 2). In this comparison (and the others), the line of unity (equal IC50 values for the inhibition of both substrates) is represented by the solid red line, and dashed lines represent ratios of IC50 values that differ from one another by 3-fold. Pairwise comparison of measured (x-axes) and predicted IC50 values for the inhibition of MPP transport (B) or metformin transport (C). Measured values are those shown in (A) (Table 2); predicted values were calculated using the single-point method (see Materials and Methods). (D) Pairwise comparisons of predicted IC50 values for the inhibition of metformin transport against those for MPP transport for 162 compounds from the NCC that inhibited transport by between 10% and 90%.
Modeling OCT2 Selectivity.
Eliminating the inhibitory profiles for MPP and ASP, as substrates that are the least and most sensitive to the inhibition of transport activity, respectively, we generated a “consensus” profile based on the average of inhibition produced by each inhibitor against the transport of metformin, TEA, cimetidine, and NBD-MTMA. These data were used to calculate predicted IC50 values that reflected average inhibitory interactions with OCT2, ignoring those compounds that inhibited less than 10% of transport activity (i.e., predicted IC50 values of >180 µM). Employing methods that have been described previously (Martínez-Guerrero et al., 2016), these data were then used in an attempt to generate a pharmacophore highlighting the common structural features correlated with ligand interaction with OCT2. These efforts, however, failed to converge on a unique pharmacophore using Discovery Studio.
We then applied machine-learning methods to develop Bayesian models using the results of the 400 (metformin, MPP, TEA, NBD-MTMA) or 320 (cimetidine, ASP) drugs in the initial “training set” screening (Supplemental Table 3). Using 50% reduction of OCT2-mediated uptake as the cutoff for “active” inhibitors (the same criterion used by Kido et al., 2011, in their screening of the inhibition of OCT2-mediated ASP transport), the resulting Discovery Studio models had areas under the ROC (using 5-fold cross-validation) of between 0.768 (ASP) and 0.806 (NBD-MTMA) (Table 3). These models adequately described the training set with 68% (MPP) to 86% (metformin) concordance (the sum of correctly predicted inhibitors and noninhibitors divided by the total number of test compounds) for all of the test substrates (Table 3). The consensus profile of OCT2 inhibition (reflecting the average inhibition of metformin, TEA, cimetidine, and NBD-MTMA transport for each inhibitor in the training set) resulted in an ROC of 0.798 and a concordance of 87% (Supplemental Fig 3; Table 3). Use of the ECFP_6 descriptors with Discovery Studio Bayesian models allowed the identification of molecular features that favored inhibition (including basic nitrogens, tertiary amines, and multiple aromatic rings) (Supplemental Fig. 4A) as well as features that did not promote inhibition (including acid features, and hydroxyl and carbonyl groups) (Supplemental Fig. 4B). These models were validated by determining the inhibition of OCT2-mediated transport of each substrate produced by a test set of 80 novel compounds from the NCC. The resulting ROC values ranged from 0.806 (cimetidine) to 0.848 (metformin) with concordances from 68% (NBD-MTMA) to 81% (ASP) (except for the MPP model, which had an ROC for the test set of 0.619 and a concordance of 52%) (Table 3).
TABLE 3.
Discovery Studio Bayesian model training and test set statistics for each probe substrate and consensus of metformin, NBD-MTMA, TEA, and cimetidine
| Training/Test | ROC LOO | ROC (5-fold) | Sensitivity | Specificity | Concordance | |
|---|---|---|---|---|---|---|
| Metformin | Training | 0.840 | 0.802 | 0.875 | 0.854 | 0.858 |
| Test | 0.848 | 0.643 | 0.833 | 0.800 | ||
| MPP | Training | 0.815 | 0.779 | 0.919 | 0.658 | 0.683 |
| Test | 0.619 | 0.667 | 0.514 | 0.525 | ||
| NBD-MTMA | Training | 0.844 | 0.806 | 0.896 | 0.833 | 0.845 |
| Test | 0.837 | 0.857 | 0.636 | 0.675 | ||
| TEA | Training | 0.815 | 0.789 | 0.895 | 0.857 | 0.863 |
| Test | 0.800 | 0.867 | 0.754 | 0.775 | ||
| Cimetidine | Training | 0.827 | 0.784 | 0.969 | 0.758 | 0.800 |
| Test | 0.806 | 0.813 | 0.766 | 0.775 | ||
| ASP | Training | 0.806 | 0.768 | 0.959 | 0.741 | 0.791 |
| Test | 0.838 | 0.842 | 0.803 | 0.813 | ||
| Consensus | Training | 0.836 | 0.798 | 0.884 | 0.861 | 0.865 |
| Test | 0.823 | 0.857 | 0.667 | 0.700 |
LOO, leave-one-out.
In addition to using Discovery Studio, we also used Assay Central to generate 16 models which used Bayesian algorithm and ECFP_6 values alone (Supplemental Table 4). These models were built, cross-validated, and externally validated as described for the other models. Training model ROCs were between 0.775 (ASP training model) and 0.819 (metformin training model) with concordances of 75% and 80%, respectively. The consensus training model had an ROC of 0.810 and a concordance of 68%; the consensus testing set had an ROC of 0.797 and a concordance of 39%. These values were within 3% of those generated by Discovery Studio.
We took the opportunity to compare the profile for the inhibition of ASP transport that we observed with that reported by Kido et al. (2011) in their assessment of the inhibition of OCT2-mediated ASP transport produced by a 20 µM concentration of each member of a 900-compound drug library. The library of Kido et al. (2011) contained 656 compounds that were not included in the set of compounds we screened against ASP transport and 595 compounds that were not contained in the set of compounds in our consensus library of OCT2 inhibitors. Using the unique compounds of Kido et al. (2011) as a test set against our ASP and OCT2 consensus models with Assay Central analysis resulted in ROCs of 0.768 and 0.756, and concordances of 79% and 73%, respectively (Supplemental Fig. 5). In other words, the Bayesian models generated from the present results in Assay Central correctly identified 75% of the inhibitors and noninhibitors determined in a previously published, independent study of OCT2 selectivity.
The Assay Central Bayesian machine-learning models are included in the Supplemental Material. A major part of the value proposition of Assay Central is to have a growing number of target models, as well as models for ADME off-targets, in the same place and of similarly high quality, using the same data formats and technologies. We suggest that access to models like these will assist academia and industry to maximize the utility of data concerning OCT2 and other transporters that are accumulating in the literature.
Discussion
OCT2 is a target for clinically significant DDIs and, as such, has been the focus of studies investigating ligand specificity and decision tree–based assays to determine the likelihood that novel therapeutic compounds will act as perpetrators of unwanted interactions (Giacomini et al., 2010; Hillgren et al., 2013). The International Transport Consortium initially suggested that either MPP or metformin be used as OCT2 probe substrates to assess the DDI potential of NMEs (Giacomini et al., 2010), a recommendation subsequently supported by the Food and Drug Administration in their 2012 draft guidance to industry (Center for Drug Evaluation and Research, U. S. Food and Drug Administration, 2012). Ensuing evidence that substrate identity can exert a marked influence on the inhibition of transport activity (Belzer et al., 2013; Yin et al., 2016) challenged that suggestion by showing that the decision-tree criterion of Cmax/IC50 ratio (i.e., the ratio of unbound Cmax to the IC50 value of the drug as an inhibitor of OCT2 activity) can differ markedly depending on whether MPP or metformin is the probe substrate used for the determination of IC50.
In light of the growing body of evidence that substrate identity can influence the inhibition of drug transport, the Food and Drug Administration recently altered their recommendation, which now suggests that the inhibition constant of a test drug should be determined with a probe substrate that may also be used in later clinical studies (Center for Drug Evaluation and Research, U. S. Food and Drug Administration, 2017), and our results support this view. In the present study, we showed that IC50 values for the inhibition of OCT2-mediated MPP transport are, on average, ∼5-fold greater than those for the inhibition of metformin transport, thus confirming and extending several previous observations of differential inhibitory sensitivity of these substrates (Belzer et al., 2013; Thévenod et al., 2013; Hacker et al., 2015; Yin et al., 2016). In addition, we showed this to be a common characteristic of OCT2-mediated MPP transport; it proved less sensitive to inhibition than did transport of four additional, structurally diverse substrates (i.e., cimetidine, TEA, ASP, and NBD-MTMA). Moreover, inhibition of the transport of these additional substrates proved to be comparatively similar to inhibition of the transport of metformin. Taken together, these results support the view that metformin can serve as a representative OCT2 substrate capable of use in both in vitro and in vivo (i.e., clinical) settings.
The inhibitory profiles were used to generate Bayesian machine-learning models that identified structural features most commonly associated with active (i.e., comparatively high-affinity) binding to OCT2 (Supplemental Fig 3). Although these models are generally not appropriate for the accurate prediction of clinically relevant IC50 values, they proved effective in identifying active inhibitors in a test set of compounds selected from our library of inhibitors. Moreover, they proved equally effective at identifying active inhibitors in a library screened for the inhibition of OCT2 transport by a separate research group that used similar, but inevitably different, methods and protocols for one substrate probe (i.e., ASP) (Kido et al., 2011). This was a particularly valuable observation in light of growing concern expressed over the substantial variability noted for IC50 and kinetic parameters in reviews of studies with multidrug transporters (Bentz et al., 2013), including the OCTs (Wright and Dantzler, 2004; Klaassen and Aleksunes, 2010; Nies et al., 2011b), and we have commented on the problems this poses for combining data sets in the literature for the purpose of model development (Ekins et al., 2012). The present observations suggest that, in the future, results be quantitatively compared with values already in the literature as a means of establishing a validated database to further modeling efforts. Finally, it is worth emphasizing that the anticipated application of our machine learning–derived models is in early preclinical drug discovery where the virtual screening of large libraries of novel structures for their probable interaction with OCT2 can be performed cost-effectively to filter out potentially problematic compounds.
The present results also provide insight into the mechanism of ligand interaction with OCT2. If all substrates and inhibitors “compete” for a common binding site on or within OCT2, then the IC50 for inhibition of the transport activity produced by an individual compound should be independent of the identity of the substrate used to assess transport activity (providing that the concentration of substrate is well below the Michaelis constant value for its transport, which was the case for all experiments in the current study) (Segel, 1975). But the routine disparity between IC50 values for inhibition of the transport of MPP and the other test substrates (Figs. 6–8) runs counter to this prediction and necessitates that ligand binding to OCT2 can involve interaction with more than one spatially distinct site. Our earlier observation of competitive, noncompetitive, and mixed-type inhibition of OCT2-mediated transport (Harper and Wright, 2013) was also consistent with this conclusion. A closer inspection of the current IC50 data (Figs. 6–8; Supplemental Fig 2) lends credence to the view that ligand binding to OCT2 is not restricted to interaction with a single, spatially restricted site. Consider the inhibition of OCT2-mediated metformin transport displayed by three structurally dissimilar compounds: fluoxetine, quinidine, and trimethoprim. Although their pairwise Tanimoto similarity coefficients are less than 0.5, their measured IC50 values were effectively identical (18.6, 18.6, and 19.8 µM, respectively) (Supplemental Fig. 6; Table 2). These data suggest that the mechanism of stabilization of ligand binding with a site or surface on OCT2 is unlikely to involve discrete points of interaction between substrate/inhibitor and a single, spatially restricted binding site on the transport protein that favors a set of ligand structural features arranged in the same relative orientation. This is the basis of traditional pharmacophores and may explain the failure of our efforts to generate one or more common-feature pharmacophores using the current data set.
Fig. 8.

Pairwise comparisons of measured IC50 values for the inhibition of OCT2-mediated transport of either metformin (x-axes) (A, B, and C) or MPP (y-axes) (D, E, and F) vs. the transport of TEA, NBD-MTMA, and ASP (Table 2). Lines of unity (equal IC50 values for the inhibition of both substrates) are represented by the solid red lines, and dashed lines represent the ratios of IC50 values that differ from one another by 3-fold. Each IC50 value is the mean ± S.E. (n = 2) of values determined in two separate experiments using the experimental design used for the experiments presented in Fig. 6 (also Supplemental Fig. 2).
The differential interaction of MPP with OCT2 is also evident from the impact on transport of single site mutations to OCT2. In the rabbit ortholog of OCT2, although the conversion of glutamate to leucine at position 447 (equivalent to E448 in hOCT2) eliminates the transport of TEA and cimetidine, it has no effect on the transport of MPP (Zhang et al., 2005). The “outlier” status of MPP may also extend to OCT1: the conversion of aspartate to glutamate at position 475 in rat Oct1 (equivalent to D474 in hOCT1) results in a 4- to 14-fold decrease in the apparent Kt values for the transport of TEA, N1-methylnicotinamde, and choline, but has no effect on the Ktapp value for MPP transport (Gorboulev et al., 1999). Both observations are consistent with the view that MPP (and possibly other substrates) may bind to OCTs at multiple sites, one or more of which may exert short distance allosteric effects that contribute to the substrate dependence of ligand interaction with OCTs. Kinetically, this can present as a “mixed-type” interaction that involves simultaneous binding of multiple substrate molecules (or substrate and inhibitor molecules) (Harper and Wright, 2013). Moreover, some ligands exert a biphasic inhibition of OCT transport activity that includes both a high-affinity (IC50 < nanomolar) (partial) inhibition, as well as a lower affinity (generally micromolar to millimolar) inhibition of transport activity, with the lower-affinity interaction being associated with the translocation of substrate (Minuesa et al., 2009; Schophuizen et al., 2013). Lamivudine’s inhibition of MPP transport mediated by OCT1, OCT2, and OCT3 displays such biphasic profiles (Minuesa et al., 2009). Interestingly, the high-affinity IC50 value is markedly elevated (from picomolar to nanomolar) by increasing the concentration of MPP (from nanomolar to micromolar), which suggests that substrate molecules can occupy multiple sites and that at high concentrations (near Kt), substrate occupancy of the high-affinity (allosteric/inhibitory) site limits inhibitor access. The IC50 values we determined did not display biphasic profiles for any of the test inhibitors or substrates, even in those cases (MPP and TEA) where the substrate concentrations were in the nanomolar range (Fig. 6; Supplemental Fig. 2). Nevertheless, the growing body of kinetic and structural evidence underscores the mechanistic complexity of ligand interaction with OCTs.
In conclusion, the differential inhibition of OCT2-mediated transport of six structurally diverse substrate molecules produced by a library of 400–480 compounds confirmed previous reports that the inhibition of OCT2 activity can be influenced by substrate identity. Transport of MPP was least sensitive to inhibition, and the IC50 values determined for 20 structurally distinct compounds against the transport of MPP, metformin, TEA, NBD-MTMA, and ASP were, on average, about 6-fold higher for MPP than for the other test substrates. In contrast, IC50 values for metformin differed on average only by ∼50% from values for the other, non-MPP test substrates. The results support the use of metformin as an OCT2 substrate for assessing clinically relevant interactions with OCT2 (i.e., IC50 values) due to its clinical prevalence and utility for both in vitro and in vivo use. But we also suggest that any of the non-MPP OCT2 substrates studied here can be used to screen NME libraries for the statistical likelihood of adverse drug interactions at the transporter. To that end, well-validated Bayesian models were developed to identify structural elements associated with effective interaction with OCT2.
Acknowledgments
We thank Biovia for providing Discovery Studio to S.E.
Abbreviations
- ASP
4–4-dimethylaminostyryl-N-methylpyridinium
- CHO
Chinese hamster ovary
- DDI
drug-drug interaction
- DMSO
dimethylsulfoxide
- ECFP_6
extended connectivity fingerprint
- hOCT2
human organic cation transporter 2
- Kt
Michaelis-Menten constant
- MATE
multidrug and toxin extruder
- MPP
1-methyl-4-phenylpyridinium
- NBD-MTMA
N,N,N-trimethyl-2-[methyl(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)amino]ethanaminium
- NCC
National Institutes of Health Clinical Collection
- NME
new molecular entity
- OC
organic cation
- OCT
organic cation transporter
- ROC
receiver operating characteristic
- S.A.
specific activity
- WB
Waymouth Buffer
- WT
wild type
Authorship Contributions
Participated in research design: Sandoval, Ekins, Wright.
Conducted experiments: Sandoval.
Performed data analysis: Sandoval, Zorn, Clark, Ekins, Wright.
Wrote or contributed to the writing of the manuscript: Sandoval, Zorn, Clark, Ekins, Wright.
Footnotes
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases [Grant 5R01DK058251]; the National Institutes of Health National Institute of General Medical Sciences [Grant R43GM122196]; the National Heart, Lung, and Blood Institute [Grant 5T32HL07249]; and the National Institute of Environmental Health Sciences [Grant 5P30ES006694].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Aavula BR, Ali MA, Mash EA, Bednarczyk D, Wright SH. (2006) Synthesis and fluorescence of n, n, n -trimethyl-2-[methyl(7-nitrobenzo[ c ][1,2,5]oxadiazol-4-yl)amino]ethanaminium iodide, a pH-insensitive reporter of organic cation transport. Synth Commun 36:701–705. [Google Scholar]
- Belzer M, Morales M, Jagadish B, Mash EA, Wright SH. (2013) Substrate-dependent ligand inhibition of the human organic cation transporter OCT2. J Pharmacol Exp Ther 346:300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentz J, O’Connor MP, Bednarczyk D, Coleman J, Lee C, Palm J, Pak YA, Perloff ES, Reyner E, Balimane P, et al. (2013) Variability in P-glycoprotein inhibitory potency (IC50) using various in vitro experimental systems: implications for universal digoxin drug-drug interaction risk assessment decision criteria. Drug Metab Dispos 41:1347–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budiman T, Bamberg E, Koepsell H, Nagel G. (2000) Mechanism of electrogenic cation transport by the cloned organic cation transporter 2 from rat. J Biol Chem 275:29413–29420. [DOI] [PubMed] [Google Scholar]
- Clark AM, Dole K, Coulon-Spektor A, McNutt A, Grass G, Freundlich JS, Reynolds RC, Ekins S. (2015) Open source Bayesian models. 1. Application to ADME/tox and drug discovery datasets. J Chem Inf Model 55:1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AM, Ekins S. (2015) Open source Bayesian models. 2. Mining a “big dataset” to create and validate models with ChEMBL. J Chem Inf Model 55:1246–1260. [DOI] [PubMed] [Google Scholar]
- Ekins S, Polli JE, Swaan PW, Wright SH. (2012) Computational modeling to accelerate the identification of substrates and inhibitors for transporters that affect drug disposition. Clin Pharmacol Ther 92:661–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Johnson DL, Ekins S, Janiszewski J, Kelly KG, Meyer RD, West M. (2002) Optimizing higher throughput methods to assess drug-drug interactions for CYP1A2, CYP2C9, CYP2C19, CYP2D6, rCYP2D6, and CYP3A4 in vitro using a single point IC(50). J Biomol Screen 7:373–382. [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, et al. International Transporter Consortium (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorboulev V, Volk C, Arndt P, Akhoundova A, Koepsell H. (1999) Selectivity of the polyspecific cation transporter rOCT1 is changed by mutation of aspartate 475 to glutamate. Mol Pharmacol 56:1254–1261. [DOI] [PubMed] [Google Scholar]
- Groves CE, Evans KK, Dantzler WH, Wright SH. (1994) Peritubular organic cation transport in isolated rabbit proximal tubules. Am J Physiol 266:F450–F458. [DOI] [PubMed] [Google Scholar]
- Hacker K, Maas R, Kornhuber J, Fromm MF, Zolk O. (2015) Substrate-dependent inhibition of the human organic cation transporter OCT2: a comparison of metformin with experimental substrates. PLoS One 10:e0136451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenbuch B. (2010) Drug uptake systems in liver and kidney: a historic perspective. Clin Pharmacol Ther 87:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JN, Wright SH. (2013) Multiple mechanisms of ligand interaction with the human organic cation transporter, OCT2. Am J Physiol Renal Physiol 304:F56–F67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillgren KM, Keppler D, Zur AA, Giacomini KM, Stieger B, Cass CE, Zhang L, International Transporter Consortium (2013) Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther 94:52–63. [DOI] [PubMed] [Google Scholar]
- Holohan PD, Ross CR. (1980) Mechanisms of organic cation transport in kidney plasma membrane vesicles: 1. Countertransport studies. J Pharmacol Exp Ther 215:191–197. [PubMed] [Google Scholar]
- Hu SX, Mazur CA, Feenstra KL, Lorenz JK, Merritt DA. (2016) Assessment of inhibition of porcine hepatic cytochrome P450 enzymes by 48 commercial drugs. Vet J 211:26–31. [DOI] [PubMed] [Google Scholar]
- Kido Y, Matsson P, Giacomini KM. (2011) Profiling of a prescription drug library for potential renal drug-drug interactions mediated by the organic cation transporter 2. J Med Chem 54:4548–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen CD, Aleksunes LM. (2010) Xenobiotic, bile acid, and cholesterol transporters: function and regulation. Pharmacol Rev 62:1–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HC, Goldenberg A, Chen Y, Lun C, Wu W, Bush KT, Balac N, Rodriguez P, Abagyan R, Nigam SK. (2016) Molecular properties of drugs interacting with SLC22 transporters OAT1, OAT3, OCT1, and OCT2: a machine-learning approach. J Pharmacol Exp Ther 359:215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Guerrero LJ, Morales MH, Ekins S, Wright SH. (2016) Lack of influence of substrate on ligand interaction with human multidrug and toxin extruder, MATE1. Mol Pharmacol 90:254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minuesa G, Volk C, Molina-Arcas M, Gorboulev V, Erkizia I, Arndt P, Clotet B, Pastor-Anglada M, Koepsell H, Martinez-Picado J. (2009) Transport of lamivudine [(-)-beta-L-2′,3′-dideoxy-3′-thiacytidine] and high-affinity interaction of nucleoside reverse transcriptase inhibitors with human organic cation transporters 1, 2, and 3. J Pharmacol Exp Ther 329:252–261. [DOI] [PubMed] [Google Scholar]
- Neuhoff S, Ungell AL, Zamora I, Artursson P. (2003) pH-dependent bidirectional transport of weakly basic drugs across Caco-2 monolayers: implications for drug-drug interactions. Pharm Res 20:1141–1148. [DOI] [PubMed] [Google Scholar]
- Nies AT, Hofmann U, Resch C, Schaeffeler E, Rius M, Schwab M. (2011a) Proton pump inhibitors inhibit metformin uptake by organic cation transporters (OCTs). PLoS One 6:e22163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nies AT, Koepsell H, Damme K, Schwab M. (2011b) Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol 201:105–167. [DOI] [PubMed] [Google Scholar]
- Pelis RM, Dangprapai Y, Wunz TM, Wright SH. (2007) Inorganic mercury interacts with cysteine residues (C451 and C474) of hOCT2 to reduce its transport activity. Am J Physiol Renal Physiol 292:F1583–F1591. [DOI] [PubMed] [Google Scholar]
- Schömig E, Lazar A, Gründemann D. (2006) Extraneuronal monoamine transporter and organic cation transporters 1 and 2: a review of transport efficiency. Handb Exp Pharmacol 175:151–180. [DOI] [PubMed] [Google Scholar]
- Schophuizen CM, Wilmer MJ, Jansen J, Gustavsson L, Hilgendorf C, Hoenderop JG, van den Heuvel LP, Masereeuw R. (2013) Cationic uremic toxins affect human renal proximal tubule cell functioning through interaction with the organic cation transporter. Pflugers Arch 465:1701–1714. [DOI] [PubMed] [Google Scholar]
- Segel IH. (1975) Enzyme Kinetics, John Wiley & Sons, New York. [Google Scholar]
- Severance AC, Sandoval PJ, Wright SH. (2017) Correlation between apparent substrate affinity and OCT2 transport turnover. J Pharmacol Exp Ther 362:405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi A, Stockley C, Keal J, Rolan P, Bochner F. (1987) Reduction of metformin renal tubular secretion by cimetidine in man. Br J Clin Pharmacol 23:545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stage TB, Brøsen K, Christensen MM. (2015) A comprehensive review of drug-drug interactions with metformin. Clin Pharmacokinet 54:811–824. [DOI] [PubMed] [Google Scholar]
- Suhre WM, Ekins S, Chang C, Swaan PW, Wright SH. (2005) Molecular determinants of substrate/inhibitor binding to the human and rabbit renal organic cation transporters hOCT2 and rbOCT2. Mol Pharmacol 67:1067–1077. [DOI] [PubMed] [Google Scholar]
- Thévenod F, Ciarimboli G, Leistner M, Wolff NA, Lee WK, Schatz I, Keller T, Al-Monajjed R, Gorboulev V, Koepsell H. (2013) Substrate- and cell contact-dependent inhibitor affinity of human organic cation transporter 2: studies with two classical organic cation substrates and the novel substrate cd2+. Mol Pharm 10:3045–3056. [DOI] [PubMed] [Google Scholar]
- Center for Drug Evaluation and Research. U. S. Food and Drug Administration (2012) Guidance for industry: drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations. Center for Drug Evaluation and Research (CDER), Food and Drug Administration, U.S. Department of Health and Human Services, Silver Spring, MD. [Google Scholar]
- Center for Drug Evaluation and Research. U. S. Food and Drug Administration (2017) In vitro metabolism- and transporter-mediated drug-drug interaction studies: guidance for industry. Center for Drug Evaluation and Research (CDER), Food and Drug Administration, U.S. Department of Health and Human Services, Silver Spring, MD. [Google Scholar]
- Volk C, Gorboulev V, Budiman T, Nagel G, Koepsell H. (2003) Different affinities of inhibitors to the outwardly and inwardly directed substrate binding site of organic cation transporter 2. Mol Pharmacol 64:1037–1047. [DOI] [PubMed] [Google Scholar]
- Wright SH, Dantzler WH. (2004) Molecular and cellular physiology of renal organic cation and anion transport. Physiol Rev 84:987–1049. [DOI] [PubMed] [Google Scholar]
- Xu Y, Liu X, Li S, Zhou N, Gong L, Luo C, Luo X, Zheng M, Jiang H, Chen K. (2013) Combinatorial pharmacophore modeling of organic cation transporter 2 (OCT2) inhibitors: insights into multiple inhibitory mechanisms. Mol Pharm 10:4611–4619. [DOI] [PubMed] [Google Scholar]
- Yin J, Duan H, Wang J. (2016) Impact of substrate-dependent inhibition on renal organic cation transporters hOCT2 and hMATE1/2-K-mediated drug transport and intracellular accumulation. J Pharmacol Exp Ther 359:401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shirahatti NV, Mahadevan D, Wright SH. (2005) A conserved glutamate residue in transmembrane helix 10 influences substrate specificity of rabbit OCT2 (SLC22A2). J Biol Chem 280:34813–34822. [DOI] [PubMed] [Google Scholar]
- Zolk O, Solbach TF, König J, Fromm MF. (2009) Structural determinants of inhibitor interaction with the human organic cation transporter OCT2 (SLC22A2). Naunyn Schmiedebergs Arch Pharmacol 379:337–348. [DOI] [PubMed] [Google Scholar]