

Abstract
Similar to organophosphate (OP) nerve agents, diisopropylfluorophosphate (DFP) rapidly and irreversibly inhibits acetylcholinesterase, leading to convulsions that can progress to status epilepticus (SE). However, in contrast to the OP nerve agents, the long-term consequences of DFP-induced SE are not well known. Thus, we characterized the spatiotemporal profile of neuropathology during the first 2 months following acute DFP intoxication. Adult, male Sprague Dawley rats administered pyridostigmine bromide (0.1 mg/kg, im) 30 min prior to successive administration of DFP (4 mg/kg, sc), atropine sulfate (2 mg/kg, im), and 2-pralidoxime (25 mg/kg, im), exhibited moderate-to-severe seizure behavior, yet survived until euthanized at 0.5 to 60 days post exposure. Analyses of brains and hearts stained with hematoxylin-eosin, or of brains immunostained for neuronal nuclei (NeuN), glial fibrillary acidic protein (GFAP), or ionized binding adapter molecule 1 (IBA1), revealed progressive neuronal cell death, neuroinflammation, and tissue remodeling across limbic brain regions and the cerebral cortex, with no detectable pathology in the cerebellum or the heart. The lesion type and progression varied according to brain region and time after exposure. Across multiple brain regions, neuronal necrosis peaked after the first week, and neuroinflammation persisted at least 2 months after intoxication. Notably, mineralization was observed at later times in the thalamus, and to a more limited extent, in the hippocampus. Lesion severity was influenced by the initial seizure severity, and spontaneous recurrent seizures were associated with more severe brain damage. These findings parallel descriptions of neuropathology in preclinical models of acute intoxication with OP nerve agents, and other seizurogenic chemicals, suggesting conserved mechanisms of pathology downstream of chemical-induced SE.
Keywords: histopathology, neurodegeneration, neuroinflammation, organophosphate neurotoxicity
The organophosphate (OP) anticholinesterase, diisopropylfluorophosphate (DFP), was originally evaluated as a chemical weapon during World War II, but was subsequently marketed as an insecticide and therapeutic for treating glaucoma (Leopold and Cleveland, 1953; Renshaw, 1946). Recently, however, DFP has been added to the United States Homeland Security list of credible terrorist threat agents (Jessica Cox, Department of Homeland Security, personal communication) due to its high toxicity, ease of access, and similarity to the OP nerve agents, sarin and soman, with respect to both structure and principal mechanism of toxicity. Like the OP nerve agents, DFP rapidly and irreversibly inhibits acetylcholinesterase (Millard et al., 1999), leading to potentially lethal cholinergic crisis and convulsions that can progress to status epilepticus (SE) (Deshpande et al., 2010; Kadar et al., 1992; Kadriu et al., 2009; Rojas et al., 2015). Although the toxicology of OP nerve agents has been extensively studied (de Araujo Furtado et al., 2012; McDonough and Shih, 1997), significantly less is known about the consequences of acute intoxication with DFP. Whether the neuropathology of acute DFP intoxication differs from that of OP nerve agents is not known, but warrants investigation based on evidence that (1) each OP elicits a unique profile of toxic effects, despite a common, primary mechanism of toxicity of acetylcholinesterase inhibition for all OPs (Costa, 2006; Pope, 1999; Rosenbaum and Bird, 2010); (2) inflammatory responses vary between OP nerve agents and OP pesticides (Damodaran and Abou-Donia, 2000; Grauer et al., 2008; Liu et al., 2012); and (3) current therapeutic strategies are not equally effective against different OPs (Eddleston et al., 2008; Rosenbaum and Bird, 2010).
Several preclinical models of acute DFP intoxication have recently been described (Deshpande et al., 2010; Pessah et al., 2016; Pouliot et al., 2016). Characterization of these models has shown that acute intoxication with DFP, at doses that cause SE, triggers a robust neuroinflammatory response (Flannery et al., 2016; Li et al., 2015; Liu et al., 2012; Rojas et al., 2015) and an extensive, delayed neurodegeneration that proceeds via necrotic and apoptotic mechanisms (Kadriu et al., 2009; Li et al., 2011; Rojas et al., 2015). However, little is known about the pathological changes occurring in the brain beyond the first several days following DFP intoxication. The few studies that have examined DFP-induced neuropathology beyond the first week postexposure report persistent neurodegeneration and neuroinflammation for up to 3 weeks following intoxication (Flannery et al., 2016), and the development of deficits in learning and memory evident one to four months post-intoxication (Brewer et al., 2013; Flannery et al., 2016; Rojas et al., 2016; Wright et al., 2010). However, in these studies, neuropathology was only examined in the hippocampus (Flannery et al., 2016; Rojas et al., 2016) and cerebral cortex (Flannery et al., 2016), and neuropathology was not assessed at later time points coinciding with the manifestation of cognitive deficits. Furthermore, the relationship between the duration and severity of seizure behavior and the extent of the ensuing brain injury, whereas well established in OP nerve agent models (Lallement et al., 1994; McDonough et al., 1995), has yet to be defined in the DFP model. Addressing these data gaps is critical to understanding the pathogenesis of clinically relevant cognitive deficits, with the ultimate goal of identifying potential therapeutic targets for protecting the brain against the long-term neurological sequelae of acute intoxication with DFP or other OPs.
Therefore, the purpose of the present study was to fully characterize and map the spatiotemporal progression of persistent neuropathology during the first 2 months following acute DFP intoxication, and to assess the relationship of initial seizure severity to subsequent neuropathology.
MATERIALS AND METHODS
Animals and DFP exposures
Animals were maintained in facilities fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, and all studies were performed with regard for alleviation of pain and suffering according to protocols approved by the UC Davis Institutional Animal Care and Use Committee. Adult, male Sprague Dawley rats (280–320 g; Charles River) were individually housed under controlled environmental conditions (22 ± 20°C, 40%–50% humidity) with a 12 h light/dark cycle and ad libitum access to food and water.
An overview of the DFP exposure paradigm used in these studies is provided in Figure 1A. A total of 41 rats were injected sc with 4 mg/kg DFP (Sigma Chemical Company, St Louis, Missouri) freshly diluted in 300 µl of ice cold PBS. To increase survival, animals received 0.1-mg/kg pyridostigmine bromide (TCI America, Portland, Oregon), im, 30 min prior to DFP administration, and 1 min following DFP, atropine sulfate (Sigma Chemical Company) at 2.0 mg/kg, im, and pralidoxime (2-PAM, Sigma Chemical Company) at 25 mg/kg in saline, im. By blocking peripheral cholinergic toxicity, these rescue compounds significantly reduce mortality. Vehicle controls (n = 10) received 300 µl of PBS in place of DFP, and commensurate administration of pyridostigmine bromide, atropine sulfate and 2-PAM. At 6 h post exposure, DFP-exposed animals were injected sc with 10 ml of 5% dextrose in saline to replace lost fluids and to prevent hypoglycemia. Once returned to their home cages, animals were weighed daily and provided rat chow softened in water for 3–5 days until they were able to locate and consume standard chow and water. At 3, 7, 14, 21, and 28 days post exposure (DPE), animals underwent 1 or 2 rounds of in vivo imaging, with each round requiring 3 hours of isoflurane/O2 (1.0%–2.0% vol/vol) anesthesia per round. Magnetic resonance imaging (MRI) data from these animals are reported in the companion paper by Hobson et al., in this issue of Toxicological Sciences.
FIG. 1.

A, Schematic illustrating the diisopropylfluorophosphate (DFP) exposure paradigm and experimental design. Histopathological examinations were performed in 41 DFP-exposed and 10 saline-exposed rats at varying days post exposure (DPE). B, Scale used to score seizure severity. C, Representative traces of different patterns of seizure behavior during the first four hours following DFP intoxication. SLUD: salivation, lacrimation, urination, defecation; 2-PAM: pralidoxime (2-pyridine aldoxime methyl chloride).
Scoring seizure behavior
Because animals used in this study were also used for MRI, it was not possible to implant electrodes to assess electroencephalographic seizure activity. Thus, seizure behavior was scored using a scale (Figure 1B) developed previously to characterize seizure severity in a preclinical model of acute DFP intoxication (Deshpande et al., 2010). Seizure behavior scores were collected at 5-min intervals from 0- to 120-min post-DFP, and at 20-min intervals from 120 to 240 min post DFP (≥ 16 observations per animal). Previous studies of acute DFP intoxication in rats have shown that a maximal seizure severity score (SSMAX) ≥ 3 corresponds to SE, as determined by electroencephalography (Deshpande et al., 2010). The temporal profile of seizure behavior was found to vary significantly across animals, with equivalent SSMAX scores (Figure 1C). To account for this variation, the seizure severity score was additionally calculated as a time-weighted average of the animal’s individual seizure scores across the 240 min of observation (SSAVR).
Histopathological examination
Brains were harvested at 8 different time point DPE, with at least 1 vehicle (VEH) control per time point (sample sizes at each time point are listed in Figure 1A). Animals were deeply anesthetized with 4% isoflurane in oxygen and subsequently perfused transcardially with cold PBS (pH 7.2) followed by cold 4% paraformaldehyde solution in PBS. After fixation in 4% paraformaldehyde for a week, brains were serially trimmed and placed into 4–6 paraffin-embedded blocks. Five-micron-thick sections were mounted on positive charged adhesion slides, air-dried overnight at 37°C, and subsequently deparaffinized and rehydrated. A subset of sections from each brain was stained with hematoxylin-eosin (H&E). Intact hearts were also harvested from a subset of animals at early and late time points: 0.5 DPE (n = 4 DFP and 1 VEH), 7 DPE (n = 2 DFP), 14 DPE (n = 2 DFP), 21 DPE (n = 1 VEH), and 28 DPE (n = 3 DFP and 2 VEH). All hearts were sagittally trimmed and processed for histopathological examination by H&E staining to investigate cardiac pathology and global ischemia.
Brain sections were also immunostained for NeuN (neuronal nuclei, A60, MAB#377, 1:400; Chemicon International, Temecula, California), GFAP (glial fibrillary acidic protein, Z0334, 1:600; Dako North America, Carpinteria, California), and IBA1 (ionized binding adapter molecular 1, 19-19741, 1:600; Wako, Richmond, Virginia) to assess neuronal cell loss, astrogliosis, and microglial activation, respectively. Endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide in methanol, and sections were subsequently rehydrated through serial dilution of the alcohol with water. Antigen retrieval procedures included either a step of heat-induced epitope retrieval using Dako Target Retrieval Solution (S1699) or, where appropriate, an enzymatic digestion with Dako Proteinase K (S3020), following the manufacturer’s instructions. PBS-Tween 20 (0.02%) was the solvent used for the antibody and blocking solutions, and for all rinses. Sections were blocked in 10% normal horse serum for 20 min. All antigen-antibody complexes were visualized via a 30-min incubation with Dako Envision+ System-horseradish peroxidase–conjugated secondary antibodies (mouse K4001 or rabbit K4003), followed by reaction with Vector NovaRed for peroxidase (SK-4800). Sections were counterstained with Mayer’s hematoxylin. Negative controls were incubated with diluent supplemented with normal serum, rather than the primary antibody. Standardized, positive control tissue was included for each antibody.
Neuropathology associated with DFP intoxication, specifically neuronal necrosis (NN), astrogliosis, microgliosis, and inflammation, was scored by a veterinary neuropathologist blinded to treatment, using a multiparametric scale ranging from 0 to 3, according to the following criteria: (a) acidophilic NN in the prosencephalon: 0, absent; 1, focal and a few neurons; 2, multifocal and > 1 layer of neurons; 3, segmental and > 1 layer of neurons. (b) IBA1 immunoreactivity in the prosencephalon: 0, absent; 1, focal; 2, multifocal; 3, segmental. (c) GFAP immunoreactivity in the prosencephalon: 0, absent; 1, focal; 2, multifocal; 3, segmental. (d) Neuronophagia in the cerebral cortex: 0, absent; 1, focal; 2, multifocal; 3, segmental. (e) Mineralization in thalamic nuclei: 0, absent; 1, multifocal to diffuse; 2, diffuse with neuronal ferrugination (dystrophic mineralization in association with dead neurons); 3, diffuse with ferrugination and spongy change. (f) Astrogliosis in the hippocampal formation: 0, none; 1, focal in one layer; 2, focal in > 1 layer; 3, segmental in at least 2 layers. (g) Spongiosis in the cerebral cortex: 0, absent; 1, focal in white matter; 2, multifocal in white matter; 3, white and gray mater. (h) Inflammatory cells: 0, absent; 1, focal in CC; 2, multifocal in CC; 3, segmental or multifocal in > 1 brain area. Overall lesion severity was evaluated using a total lesion (TL) score, defined as the sum of NN, astrogliosis, microglial activation, neuronophagia, mineralization, and spongiosis scored either within a single brain region (TLregion) or globally across all prosencephalic regions (TLglobal).
Statistical analyses
The quantitative assessment of the severity and spatiotemporal distribution of lesions utilized all animals receiving an SSMAX of 3 or greater (39/41). Means and SDs by DPE were computed. Linear regression was used to assess the association between DPE and the histopathology severity scores; DPE and DPE-squared were included in the models to reflect the quadratic pattern observed in the data. To assess lesion severity across regions, repeated-measures regression models were used. These models included region, DPE, DPE-squared and the interactions between regions and DPE and DPE-squared. Using the estimated curves, the DPE of peak lesion severity was calculated by obtaining the estimated DPE at which the maximum severity occurred both overall and within each region; 95% confidence intervals (CIs) for the DPE of peak severity were computed using Fieller’s method (Fieller, 1954). Spearman correlation coefficients and corresponding 95% CIs were also computed. All analyses were conducted using RStudio, version 0.98.1102.
RESULTS
Seizures and Survival Following Acute DFP Intoxication
DFP (4 mg/kg, sc) produced seizure behavior with a maximal score of ≥ 3 in 39 out of 41 rats within 10 min of subcutaneous injection. Among these 39 animals, the mean maximal seizure score (SSmax) was 4.4 ± 0.7, whereas the mean of the behavioral seizure score averaged across the first 4 hours post exposure (SSAVR) was 2.99 ± 0.42. Among all DFP-exposed rats, 81% survived to 12 hours post exposure, whereas 76% survived until euthanized between 3 and 28 DPE. During handling of animals for husbandry and experimental purposes, behavioral evidence of spontaneous recurrent seizures (SRS), characterized as rearing and falling coincident with rapid, repetitive forelimb and hindlimb clonus that lasted approximately 30 sec, was observed in a subset of DFP animals as early as 7 DPE. Analyses of detailed notes regarding the incidence of SRS behavior during handling of animals 3–4 times per week post-DFP exposure, indicated that those animals observed to exhibit SRS (4/39) had significantly higher SSAVR during the first 4 hours after exposure to DFP, relative to other DFP-exposed animals included in the study (P < .05). This increase in SSAVR was attributed to more frequent scores of 4 and 5 on the scale of seizure behavior severity (Figure 1B).
DFP-Induced Neuronal Necrosis
Of the 39 DFP animals with SSMAX ≥ 3, 37 had lesions in their brains. Lesions were restricted to the allocortex (particularly the piriform and entorhinal cortices, amygdala, and hippocampus), neocortex (cerebral cortex) and thalamus. Lesions in those areas consisted of acidophilic NN (Figure 2A), neuroinflammation (Figure 2B), and thalamic dystrophic mineralization (Figure 2C and D). Notably, none of these lesions were detected in the cerebellum at any time point examined in this study. Hearts were collected from 11 of the DFP animals with brain lesions, and from 4 VEH controls, at multiple times post exposure, ranging from 0.5 to 28 DPE, and H&E stained for histopathological examination. None of the hearts from DFP or VEH animals exhibited lesions or evidence of ischemic damage (data not shown).
FIG. 2.

Types of lesions observed in the brain of rats acutely intoxicated with diisopropylfluorophosphate (DFP). A–D, Representative photomicrographs of hematoxylin-eosin–stained brain sections from the same animal at 28 days post exposure (DPE). A, Neuronal necrosis in the amygdala. B, Reactive astrogliosis in the dentate gyrus of the hippocampus, characterized by hypertrophic astrocytes with long fibrillary hypereosinophilic processes surrounding blood vessels (closed arrowhead). Note that healthy neurons (open arrowheads) are still present in areas of neuronal cell loss and glial scarring. C, D, Mineralization (open arrows) in the thalamus, associated with acute neuronal necrosis and hypertrophic activated astrocytes, characterized by a large cytoplasm, short thick processes and a peripheralized nucleus (closed arrows). Magnification: ×400 (A–C); ×600 (D).
Neuronal necrosis was apparent as early as 12 hours post-DFP exposure, and persisted up to 60 DPE in multiple brain regions (Table 1, Figs. 3–6). Diffuse NN occurred in the different amygdaloid nuclei in 97% [36/37] of rats with brain pathology and in the piriform cortex (Figure 3) in 92% [34/37] of rats with brain pathology. Neuronal necrosis in the piriform cortex frequently affected layers II and III, and involved the adjacent entorhinal cortex deep layers, mostly layers II and III. Dorsolateral and ventromedial thalamic areas were affected in 89% [33/37] of rats with lesions, with consistent NN within the anteromedial, anterioventral, reuniens, and anterolateral thalamic nuclei. In less affected areas, such as the neocortex, NN was restricted to superficial layers I–III in 89% [33/37] of the cases. In the hippocampus, initial necrosis of CA1 pyramidal neurons, followed by necrosis of CA3 pyramidal neurons, occurred in 84% [31/37] of cases (Figure 3), whereas neurons of the polymorphic and the granule-cell layers of the dentate gyrus were necrotic in 73% [27/37] and 35% [13/37] of DFP-intoxicated animals (Figure 3), respectively.
TABLE 1.
The Spatiotemporal Distribution and Severity of NN in DFP Rats
| DPE | PC | CC | DG | CA | SA | A | T | NN sum |
|---|---|---|---|---|---|---|---|---|
| 0.5 | 0.8 ± 0.3 | 1.1 ± 1.0 | 0.6 ± 0.5 | 0.2 ± 0.2 | 0.3 ± 0.3 | 1.4 ± 0.3 | 1.1 ± 0.5 | 5.0 ± 1.9 |
| 1 | 1.3 ± 0.7 | 1.4 ± 0.8 | 0.7 ± 0.3 | 1.0 ± 0.6 | 0.7 ± 0.3 | 1.2 ± 0.4 | 1.4 ± 0.9 | 6.9 ± 3.1 |
| 3 | 1.8 ± 0.5 | 1.6 ± 0.6 | 0.5 ± 0.6 | 1.9 ± 0.6 | 1.4 ± 0.6 | 2.0 ± 0.0 | 2.5 ± 1.0 | 10.4 ± 2.1 |
| 7 | 2.5 ± 1.2 | 1.2 ± 1.1 | 1.2 ± 1.5 | 1.6 ± 1.4 | 1.3 ± 0.8 | 2.0 ± 0.3 | 2.0 ± 1.1 | 7.8 ± 6.3 |
| 14 | 1.5 ± 1.0 | 1.5 ± 1.2 | 2.0 ± 1.4 | 1.8 ± 1.5 | 1.0 ± 0.8 | 1.6 ± 1.1 | 1.5 ± 1.0 | 9.0 ± 6.3 |
| 21 | 2.0 ± 0.0 | 1.0 ± 0.0 | 0.5 ± 1.0 | 1.0 ± 0.9 | 0.6 ± 0.3 | 2.3 ± 0.5 | 1.9 ± 0.6 | 8.5 ± 0.6 |
| 28 | 2.2 ± 0.8 | 1.3 ± 0.8 | 1.5 ± 1.4 | 1.8 ± 0.8 | 0.9 ± 0.6 | 2.0 ± 0.0 | 2.3 ± 0.5 | 10.3 ± 2.8 |
| 60 | 0.1 ± 0.3 | 0.6 ± 0.9 | 0.1 ± 0.3 | 0.8 ± 0.6 | 0.0 ± 0.0 | 0.3 ± 0.3 | 0.0 ± 0.0 | 1.4 ± 1.1 |
Data presented as mean ± SD (individual animal data can be found in the supplementary file). PC, piriform cortex; CC, cerebral cortex; DG, dentate gyrus; CA, hippocampal cornu ammonis 1 & 3; SA, septal area; A, amygdala, T, thalamus; sum, sum of neuronal necrosis across sub-regions.
FIG. 3.
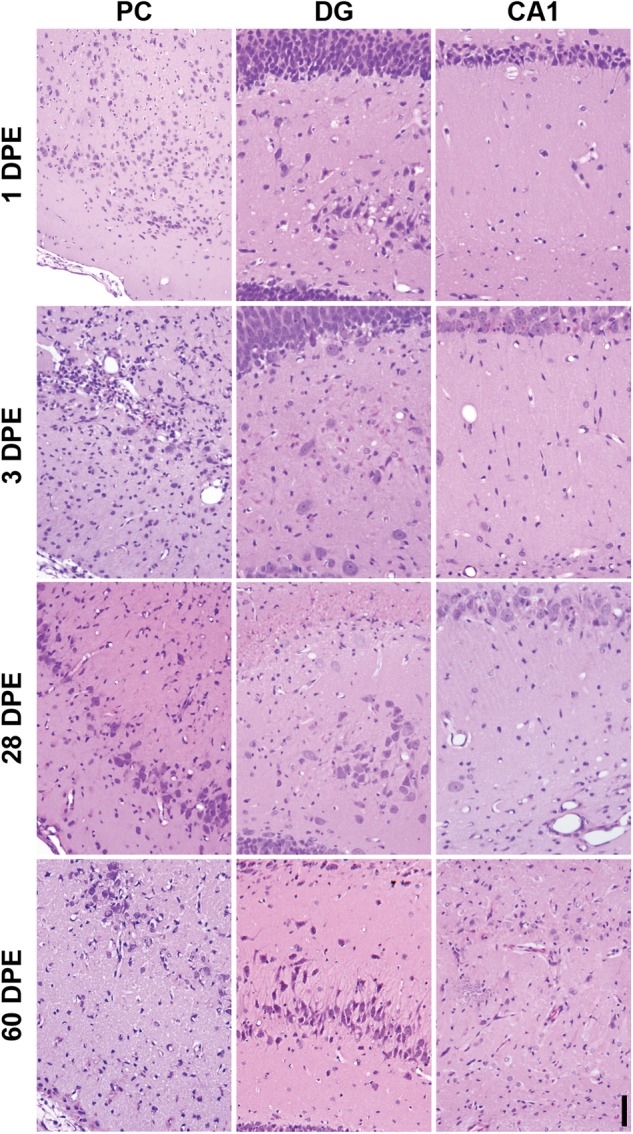
Representative photomicrographs illustrating the differing stages of neuropathology in the piriform cortex (PC), dentate gyrus (DG), and CA1 region of the hippocampus (CA1) in diisopropylfluorophosphate (DFP) rats. All images at × 200 magnification, bar = 50 µm.
FIG. 6.
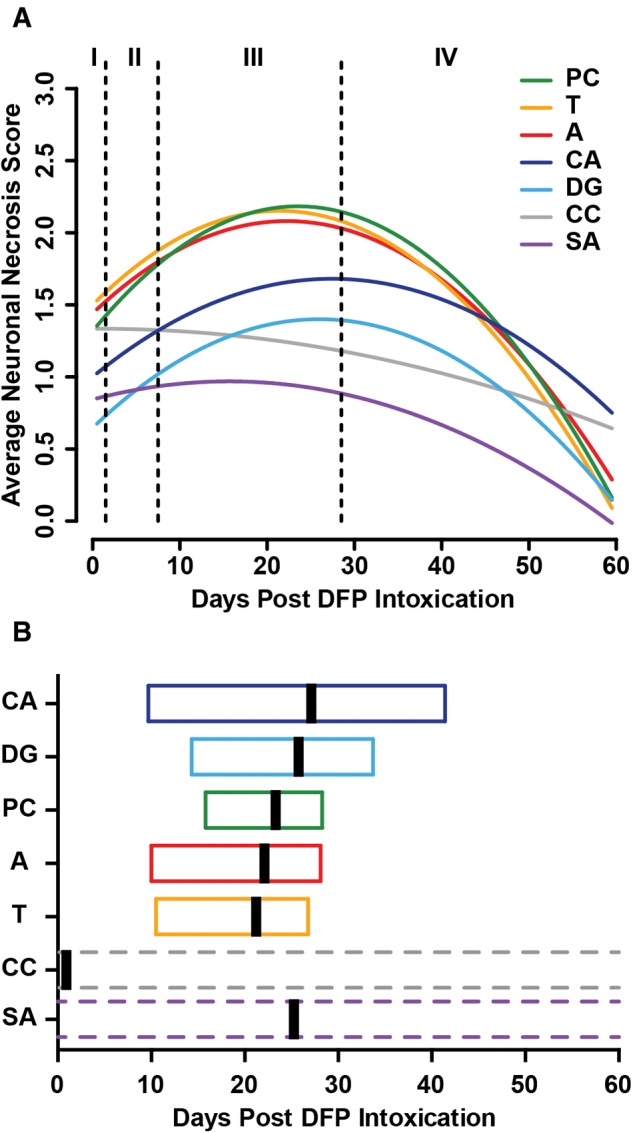
Spatiotemporal modeling of neuronal necrosis following acute diisopropylfluorophosphate (DFP) intoxication. A, Each curve represents a different brain region, as identified by color. Vertical dashed lines identify boundaries between stages of neuropathology, as defined in Figure 3. Each curve is modeled on data from the 39 animals that reached a maximal seizure score of ≥ 3, which is consistent with the induction of SE. B, Box plots indicating estimated global peak severity (vertical black bar) and 95% confidence interval (box) of data in (A). A, amygdala; CA, hippocampus cornu ammonis regions; DG, hippocampus dentate gyrus; PC, piriform cortex; CC, cerebral cortex; SA, septal area; T, thalamus.
Examination of the regional spread of NN as a function of DPE (Figure 4) indicated that NN could be divided into 4 time-dependent stages, as follows: (1) stage I (< 1 DPE), characterized by acute lesions predominantly in the cerebral cortex, piriform cortex, amygdala and thalamus; (2) stage II (3–7 DPE), more severe subacute lesions involving the same brain regions as those listed in stage I with pan-necrosis in the piriform cortex, and additional marked necrosis of hippocampal neurons, particularly in the CA1 and CA3 regions; (3) stage III (14–28 DPE), chronic-active severe lesions with extensive spread, including pan necrosis of the piriform cortex and thalamic nuclei, dystrophic mineralization in the thalamus. During stage III, there was also an increased occurrence of multifocal or segmental necrosis in the dorsal plate of the dentate gyrus (Figure 3) in 8 of the 16 rats, as well as the ventral plate in one rat at 14 DPE; and (4) stage IV (60 DPE), chronic lesions of milder severity compared with stage III with multicentric neuronal cell loss, resulting in distinct brain atrophy (see also Hobson et al., this issue of Toxicological Sciences). This was particularly notable in the neocortex and hippocampal formation, as denoted by cerebrocortical and hippocampal thinning due to moderate to severe neuronal cell loss (Figure 3).
FIG. 4.

Spatiotemporal progression of neuropathology in the brains of rats acutely intoxicated with diisopropylfluorophosphate (DFP). A, Diagrammatic summary of the stages of DFP-induced neuropathology in the rat at bregma -3.14 mm (anatomic outline adapted from Paxinos and Watson 2007) illustrating the spatiotemporal progression of neuronal necrosis and mineralization, assessed by hematoxylin-eosin (H&E) (top, blue shading), microgliosis assessed by IBA1 immunoreactivity (middle, red shading), and astrogliosis assessed by GFAP immunoreactivity (bottom, green shading). Stage I, 12- to 24-hour post exposure corresponding to acute pathology; stage II, 3–7 days post exposure (DPE), subacute pathology; stage III, 14–28 DPE, chronic pathology; stage IV, 60 DPE, resolution phase. B, Representative photomicrographs of DFP-intoxicated (panels ii, iv, and vi) and vehicle control (panels i, iii, and v) animals at 7 DPE illustrating (ii) neuronal necrosis (arrowhead), (iv) microgliosis (arrowhead), and (vi) astrogliosis (arrowhead). Magnification: ×100 (i, ii); ×200 (iii, iv, v, and vi).
Analysis of the temporal progression of global necrosis suggested that mild necrosis was observed during the earliest (stage I, 0.5–1 DPE) and latest (stage IV, 60 DPE) time points; whereas more severe (highest NN scores) and widespread necrosis was observed between 3 and 28 DPE. The severity of NN varied across brain regions. Multifocal or segmental necrosis was consistently observed in the amygdala (24/25), thalamus (22/25), and piriform cortex (22/25) during stages II and III. In contrast, there was significant interanimal variability in the hippocampus, as exemplified by the dentate gyrus, where necrosis was absent in 44% (11/25) of cases, but was multifocal or segmental in 40% (10/25) of cases between 3 and 28 DPE. Interestingly, the 4 animals that exhibited SRS exhibited either multifocal (2/4) or segmental (2/4) NN in the dentate gyrus of the hippocampus during stages II and III.
A linear regression model supported a quadratic association between DPE and global severity of NN, rather than a simple linear increase, with increasing DPE (P < .001; Figure 5). The estimated peak severity of NN was on 23 DPE (95% CI = 12–28). Furthermore, a repeated-measures model of the temporal pattern of regional NN (Figure 6) supported not only that the severity of necrosis was influenced by time (quadratic association), as seen for the global severity, but also that regional differences exist (P = .01). Overall, the amygdala, piriform cortex, and thalamus experienced significantly more severe NN than the cerebral cortex and hippocampus (P < .006, Figure 6). Based on this model, the estimated regional peak severity of NN occurred earliest in the cerebral cortex (0.7 days, 95% CI > window of observation), after 2–3 weeks in the septal area (15.7 days, 95% CI > window of observation), and after > 3 weeks in the thalamus (21.4 days, 95% CI = 10.5, 26.8), amygdala (22.3 days, 95% CI = 10.0, 28.1), piriform cortex (23.5 days, 95% CI = 15.8, 28.3), hippocampal dentate gyrus (26.0 days, 95% CI = 14.3, 33.7), and CA1 (27.4 days, 95% CI = 9.7, 41.4) of the hippocampus (Figure 6B). Neuronal necrosis in the cerebral cortex was estimated to peak within stage I. However, the CI was broader than the study period, which when considered together with histological evidence of NN at 60 DPE, suggests lesions in this brain region developed rapidly and remained relatively steady over time.
FIG. 5.
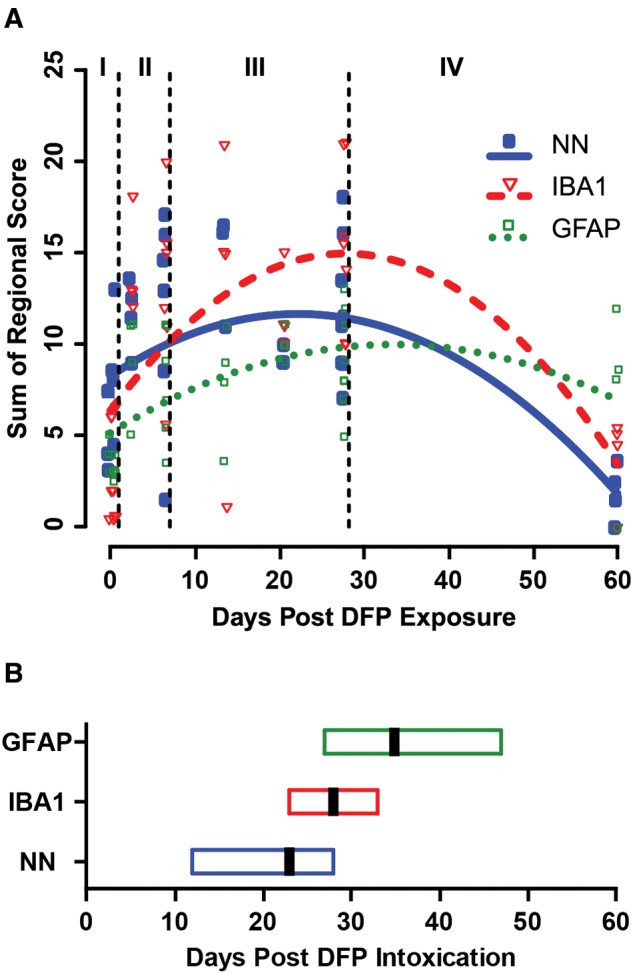
Temporal progression of global neuropathology in the brains of rats acutely intoxicated with diisopropylfluorophosphate (DFP). A, Temporal progression curves of DFP-induced neuropathology, as modeled using scores for global neuronal necrosis and neuroinflammatory responses. Each individual symbol represents data from a single animal. Vertical dashed lines identify boundaries between stages of neuropathology as defined in Figure 3. Each curve is modeled on data from the 39 animals that reached a maximal seizure score of ≥ 3, which is consistent with the induction of status epilepticus. B, Box plots indicating estimated global peak severity (vertical black bar) and 95% confidence interval (box) of data in (A). NN, neuronal necrosis; IBA1, microgliosis; GFAP, reactive astrogliosis.
DFP-Induced Neuroinflammation and Glial Scarring
Additional neuropathology observed in the brains of rats acutely intoxicated with DFP included reduced neuronal cell count, as detected by NeuN immunolabeling (Figure 7A, D, and G), neutrophilic infiltration, histiocytic infiltration, reactive astrogliosis, as detected by GFAP immunoreactivity (Table 2A, Figure 7B, E, and H) and microgliosis, as detected by IBA1 immunoreactivity (Table 2B, Figure 7C, F, and I), with individual neuronophagia of necrotic neurons, spongiosis of the neuropil, and extracellular thalamic mineral accumulation (Table 2C, Figure 2C and D). These lesions varied according to DPE and brain region. However, in all cases, these lesions were colocalized with regions of concurrent NN, as detected by H&E staining, and/or previous neuronal cell loss, as evidenced by decreased NeuN immunoreactivity (Figure 7). Tissue necrosis (malacia) in the olfactory area occasionally progressed into a cyst-like lesion with dilated blood vessels variably apparent between 3 and 28 DPE (Figure 7). Tissue necrosis with inflammation was observed in extra-olfactory tissue in some animals, including the hippocampus at stage III. In all damaged brain regions, astroglial scarring and blood vessel proliferation was evident by stage IV.
FIG. 7.

Neuronal cell loss and neuroinflammatory responses within severe lesions in the hippocampus at 28 days post exposure (DPE) and piriform cortex at 3 DPE of rats acutely intoxicated with diisopropylfluorophosphate (DFP). Brain sections from vehicle control (A–C; G–I) or DFP intoxicated (D–F; J–L) rats were immunostained for NeuN (A, D, G, J), GFAP (B, E, H, K), or IBA1 (C, F, I, L) to identify neurons, astrocytes and microglia, respectively. Areas of neuronal necrosis were extensive, and at times, cystic in appearance. NeuN immunohistochemistry (D and J) confirms the extent of neuronal cell damage and the prominent losses of neurons in the dorsal horn of the dentate gyrus and layer II of the piriform cortex. These areas of cystic malacia had large numbers of IBA1 immunoreactive microglial cells and macrophages (F and L). Note the increased GFAP-immunoreactivity (E and K) at the margins of the areas of malacia, in particular, at its most outer margin in the piriform cortex. Magnifications: ×20 (A, D), ×40 (B, C, E–L), ×200 (inset B, E, H, K), and ×400 (inset C, F, I, L).
TABLE 2.
Neuroinflammation and Secondary Pathological Characteristics in DFP Rats
| DPE | PC | CC | HP | A | T | sum |
|---|---|---|---|---|---|---|
| A. Microgliosis (IBA1 Immunoreactivity) | ||||||
| 0.5 | 0.88 ± 0.85 | 0.13 ± 0.25 | 0.5 ± 0.58 | 0.63 ± 0.48 | 0.50 ± 0.58 | 2.6 ± 2.5 |
| 1 | 0.20 ± 0.45 | 0.30 ± 0.45 | 0.25 ± 0.35 | 0.20 ± 0.4 | 0.25 ± 0.45 | 1.2 ± 2.1 |
| 3 | 1.88 ± 1.31 | 1.5 ± 1.29 | 1.44 ± 1.05 | 3.0 ± 0.0 | 3.0 ± 0.0 | 10.8 ± 0.9 |
| 7 | 2.58 ± 1.02 | 1.08 ± 1.16 | 1.63 ± 0.86 | 2.17 ± 0.75 | 2.67 ± 0.82 | 10.1 ± 3.5 |
| 14 | 1.75 ± 1.26 | 1.75 ± 1.26 | 1.63 ± 1.25 | 2.5 ± 1.0 | 2.25 ± 1.5 | 9.9 ± 6.1 |
| 21 | 2.5 ± 0.58 | 1.5 ± 0.58 | 1.13 ± 0.25 | 2.25 ± 0.50 | 2.25 ± 0.50 | 9.6 ± 2.0 |
| 28 | 2.5 ± 0.53 | 1.63 ± 1.03 | 1.81 ± 1.07 | 2.63 ± 0.52 | 2.63 ± 0.52 | 11.2 ± 2.8 |
| 60 | 1.25 ± 0.96 | 1.38 ± 1.38 | 0.06 ± 0.13 | 0.38 ± 0.48 | 0.63 ± 0.48 | 3.7 ± 2.5 |
| B. Reactive Astrogliosis (GFAP Immunoreactivity) | ||||||
| 0.5 | 0.75 ± 0.29 | 0.75 ± 0.29 | 0.75 ± 0.29 | 0.88 ± 0.25 | 0.88 ± 0.25 | 4 ± 0.82 |
| 1 | 0.7 ± 0.27 | 0.6 ± 0.22 | 0.6 ± 0.22 | 0.6 ± 0.22 | 0.6 ± 0.22 | 3.1 ± 0.55 |
| 3 | 1.75 ± 0.5 | 2.25 ± 0.96 | 1.75 ± 0.5 | 1.5 ± 0.58 | 1.75 ± 0.5 | 9 ± 2.83 |
| 7 | 1.83 ± 0.41 | 1.75 ± 1.08 | 1.67 ± 0.82 | 1.42 ± 0.66 | 1.17 ± 0.68 | 7.83 ± 3.04 |
| 14 | 2.0 ± 0.82 | 1.13 ± 0.63 | 1.63 ± 0.75 | 1.38 ± 0.75 | 1.75 ± 0.5 | 7.88 ± 3.17 |
| 21 | 2.5 ± 0.58 | 1.5 ± 0.58 | 2.0 ± 0.0 | 1.75 ± 0.5 | 2.25 ± 0.5 | 10 ± 0.82 |
| 28 | 1.88 ± 0.35 | 1.63 ± 0.74 | 2.13 ± 0.83 | 1.44 ± 0.82 | 2.06 ± 0.78 | 9.13 ± 2.7 |
| 60 | 2.33 ± 1.15 | 2.33 ± 0.58 | 1.17 ± 0.76 | 1.33 ± 0.58 | 2.33 ± 0.58 | 7.13 ± 5.07 |
| C. | Spongiosis | NeuPHG | Mineralization | TL | ||
| DPE | CC | CC | T | All Regions | ||
| 0.5 | 1.5 ± 1.73 | 0 ± 0 | 0 ± 0 | 14.6 ± 1.7 | ||
| 1 | 1.30 ± 0.67 | 0.60 ± 0.55 | 0 ± 0 | 14.7 ± 5.4 | ||
| 3 | 1.63 ± 0.48 | 1.38 ± 0.48 | 0.63 ± 0.75 | 38.3 ± 6.1 | ||
| 7 | 0.85 ± 0.75 | 0.67 ± 0.75 | 0.33 ± 0.82 | 34.6 ± 14.9 | ||
| 14 | 0.88 ± 0.85 | 1.25 ± 0.87 | 1.63 ± 1.18 | 35.5 ± 21.0 | ||
| 21 | 0.63 ± 0.25 | 0.5 ± 0 | 2.13 ± 0.25 | 34.3 ± 2.5 | ||
| 28 | 1.69 ± 1.1 | 1.25 ± 0.53 | 2.38 ± 0.79 | 41.0 ± 11.4 | ||
| 60 | 0.5 ± 0.41 | 2 ± 1.41 | 2.25 ± 1.5 | 17.5 ± 11.8 | ||
Data presented as mean ± SD (individual animal data can be found in the supplementary file). PC, piriform cortex; CC, cerebral cortex; DG, hippocampal dentate gyrus; CA, hippocampal cornu ammonis 1 & 3; SA, septal area; A, amygdala, T, thalamus; HP, hippocampus; NeuPHG, neuronophagia; Total Lesion, sum of neuronal necrosis, microgliosis, astrogliosis, spongiosis, neuronophagia, and mineralization across sub-regions.
There was a significant correlation between neuroinflammation (IBA1 and/or GFAP immunoreactivity) and the severity of regional NN (Spearman correlation; P ≤ .05), with the exception of GFAP in the thalamus (P = .09). The severity of neuroinflammation varied between the stages of neuropathology (Table 2, Figure 3). In stage I, neuroinflammation ranged from minimal to focal perivascular edema with infiltration of blood inflammatory cells, such as neutrophils and macrophages. Reactive astrocytes increased significantly between stage I (0.5 and 1 DPE) and stage II (3–7 DPE) in the piriform cortex (P < .001), cerebral cortex (P < .001), hippocampus (P < .001), amygdala (P = .009), and thalamus (P = .02). In stages II and III, neuroinflammation was characterized by extensive segmental microglial activation and marked astrogliosis surrounding reactive blood vessels in regions of segmental to pan-necrosis (Figure 7), with diffuse increases in ramifications of IBA1 immunoreactive microglial cells (Figure 7F, inset). At stage IV (60 DPE), GFAP+ astrocytes were decreased in number relative to stages II and III, but still increased relative to stage I, whereas IBA1 immunoreactivity had diminished substantially across all brain regions (Figure 3).
Global IBA1 immunoreactivity (IBA1 scores summed across regions) increased with increasing DPE, reaching the estimated peak severity for microgliosis at approximately 28 DPE (95% CI = 23, 33) and declining thereafter (Table 2, Figure 5). Modeling of the temporal profile of regional IBA immunoreactivity suggests that microglial activation peaks first in the hippocampus (26.4 days), followed quickly by the amygdala (27.1 days) and thalamus (27.5 days) and later, in the piriform cortex (30.9 days) and neocortex (36.5 days) (Figure 6). Similarly, global GFAP immunoreactivity (GFAP scores summed across regions) increased with DPE, although the rate of increase lessened as DPE increased (P = .003, Table 2, Figure 5). The estimated peak of GFAP immunoreactivity occurred at 35 DPE (95% CI = 27–47). Modeling of the temporal profile of GFAP immunoreactivity within regions suggests that astrogliosis peaks approximately 1 month post exposure in the hippocampus (31.4 days) and amygdala (34.5 days), followed by the piriform cortex (42.7 days) and thalamus (46.0 days) (Figure 6). Modeling suggests that GFAP immunoreactivity in the neocortex remains relatively unchanged across the study period, which is consistent with the development of persistent segmental astrogliosis observed from 3- to 60-DPE in this brain region.
Loss of Tissue Integrity and Mineralization
A subset of DFP-treated animals exhibited loss of tissue integrity, as characterized by focally extensive pan-necrosis in the piriform cortex as early as 3 DPE (stage II) (Figure 7B), and/or thalamic mineralization initiated by calcium accumulation on lipid cell membranes of necrotic cells. At stages III and IV, animals frequently (18/20) exhibited thick, coarse, extracellular mineralized aggregates within the thalamus, which expanded in size at 28 (Figure 2D) and 60 DPE. Extra-thalamic mineralization was restricted to the hippocampus, and only observed in 2 animals. Thalamic mineralization was highly correlated with DPE (rs = .76, 95% CI = 0.58–0.87, P < .001) and astrogliosis (rs = 0.70, CI = 0.49–0.83, P < .001). Although massive infiltration of IBA1 immunoreactive cells was observed in the thalamus coincident with mineralization, the correlation between thalamic mineralization and microgliosis just missed statistical significance (P = .06). Mineralization in the thalamus was not significantly correlated with initial SSAVR (P = .94) or thalamic NN (P = .74).
Effect of Seizure Severity on Lesion Severity
Average seizure severity scores during the first four hours after DFP injection were positively correlated with the sum of NN across brain regions (Spearman’s ρ = 0.58, P < .001, 95% CI = 0.31–0.75), microgliosis (Spearman’s ρ = 0.51, P < .001, 95% CI = 0.22–0.71), and astrogliosis (Spearman’s ρ = 0.42, P < .008, 95% CI = 0.11–0.65) in the 39 DFP intoxicated animals (Figure 8). The parameter SSAVR was then added to the models used previously to estimate peak regional NN, peak regional microgliosis (IBA1) and peak regional astrogliosis (GFAP), which were modified to consider interactions between seizure severity and DPE or region (when applicable). Overall, an increase in SSAVR was significantly (P < .01) associated with an increase in NN and neuroinflammation in all regions assessed.
FIG. 8.
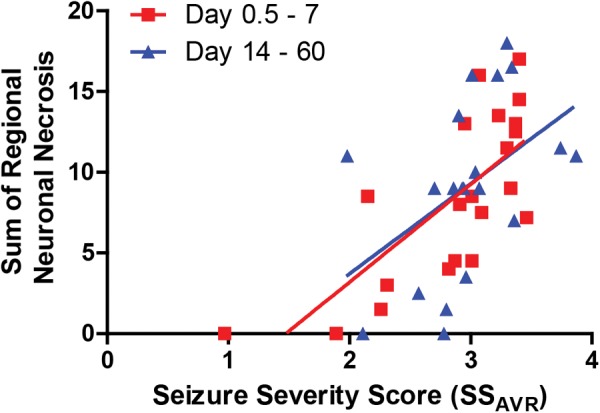
Scatterplot illustrating the relationship between the sum of regional neuronal necrosis and the average seizure severity score (SS AVR) during the first four hours after exposure to diisopropylfluorophosphate (DFP). Each point represents an individual DFP-intoxicated animal. The correlation between seizure severity and neuronal necrosis was stronger during the first week post intoxication (rs = .71 [95% CI = 0.40–0.88], P = .0003) compared with later time points (rs = .55 [95% CI = 0.12–0.80], P < .012), although both were significant.
DISCUSSION
This study shows that acute DFP intoxication causes progressive neuronal cell death, neuroinflammation and tissue remodeling across limbic brain regions and the cerebral cortex, while sparing the cerebellum. Our data confirm previous reports of the neuropathology in the first few days following DFP-induced SE (Deshpande et al., 2010; Flannery et al., 2016; Li et al., 2011; Liu et al., 2012; Rojas et al., 2015) and extend them to demonstrate that across multiple brain regions, NN peaks later than the first week post exposure, and neuroinflammatory responses do not resolve even 2 months after intoxication. Other significant findings from this study include (1) neuropathology varies according to brain region and time after exposure, and is influenced by initial seizure severity; (2) significant neuropathology occurs in the absence of cardiac lesions; and (3) our preliminary evidence suggests that SRS are associated with more severe brain damage.
DFP-induced neuropathology varied significantly between brain regions with respect to lesion composition and progression, suggesting that the pathogenic mechanisms contributing to persistent brain injury may differ between brain regions. In the cerebral cortex, we observed severe NN in stage I that persisted with minimal attenuation out to stage IV. The amygdala, thalamus, and piriform cortex also exhibited severe NN in stage I, but this increased in severity during stages II and III and then resolved by stage IV. In contrast, the hippocampus exhibited minimal neuropathology during stage I that increased in severity during stages II and III, and, by stage IV, had resolved in the dentate gyrus, but persisted in the CA1 and CA3. One possible explanation for why the amygdala and piriform cortex are severely damaged while the hippocampus is relatively spared during the first 24 hours after DFP exposure, is the differential sensitivities of these brain regions to OP-induced seizures. The ventrorostral aspect of the piriform cortex, known as the area tempestas, is a crucial site for seizure initiation following acute OP intoxication (Tang and Loke, 2011). Furthermore, microinjection of the OP nerve agent VX into specific brain regions revealed that of the eleven brain regions tested, the piriform cortex and portions of the amygdala were the most sensitive, whereas the dorsal hippocampus was the least sensitive (McDonough and Shih, 1997). Thus, the piriform cortex and amygdala are direct targets of the acute primary cholinergic insult triggered by acute OP intoxication (McDonough and Shih, 1997).
The minimal NN observed in the hippocampus at stage I relative to other brain regions suggests that the substantial neuronal cell loss and gliosis observed in this brain region beginning in stage II (3 DPE) is mediated by subacute or chronic mechanisms. One mechanism consistent with the delayed increase in hippocampal necrosis is transneuronal degeneration, a phenomenon in which damage to neurons within a circumscribed area leads to propagation of damage along their associated neural circuits (Fornito et al., 2015). There is a well-described neural circuit linking the piriform cortex to the hippocampus via the perforant pathway that runs through the perirhinal and entorhinal cortices (Vismer et al., 2015). It has previously been shown that unilateral transection of the perforant path causes unilateral denervation of the dentate gyrus with delayed neuronal degeneration in the dentate molecular layer at 7 d post-lesioning (Del Turco et al., 2014). In the DFP rat, hippocampal necrosis occurs subsequent to extensive neuronal cell death in the piriform and entorhinal cortices, supporting a model of transneuronal degeneration.
Another potential mechanism contributing to the progressive pathology observed following acute DFP intoxication is neuroinflammation (Banks and Lein, 2012; Chen, 2012; de Araujo Furtado et al., 2012). In experimental models of acute soman intoxication, an early increase in GFAP immunoreactivity, followed by microglial activation, precedes neuronal injury, suggesting that, in this model, an early neuroinflammatory response contributes to neuronal injury (Baille-Le Crom et al., 1995; Zimmer et al., 1997). Consistent with previous studies (Flannery et al., 2016; Kim et al., 1999; Li et al., 2011, 2015; Rojas et al., 2015), we observed that DFP-induced SE also triggers an early and robust neuroinflammatory response that overlaps spatially and temporally with extensive neurodegeneration in multiple brain regions. Although we observed a significant correlation between neuroinflammation and NN, the temporal relationship varied from that reported for sarin. In the DFP rat, the neuroinflammatory responses were minimally apparent during the first 12 hours post-DFP exposure in brain regions with severe NN, while, at later times, global and regional microgliosis and astrogliosis remained elevated even after NN began to subside in corresponding brain regions. Whether these neuroinflammatory responses are a cause or consequence of NN following acute DFP intoxication remains to be determined. However, recent evidence demonstrating that attenuation of neuroinflammation by pharmacological inhibition of the EP2 receptor reduces the severity of neurodegeneration and cognitive impairment in the rat DFP model (Rojas et al., 2015, 2016), supports a model in which neuroinflammation contributes to NN.
Following acute DFP intoxication, the thalamus, and to a lesser extent the hippocampus, developed dystrophic mineralization, also known as calcification, which was not observed in other brain regions with severe lesions. Acute OP intoxication triggers excitotoxicity, which increases intracellular calcium to pathological levels (McDonough and Shih, 1997), and neuronal levels of intracellular calcium can remain significantly elevated for up to a month after DFP intoxication (Deshpande et al., 2010). It is hypothesized that in the face of depleted ATP levels, intracellular calcium is aggregated as a defense against calcium-mediated cytotoxicity (Rodriguez et al., 2009). This suggests that the mineralization observed in the thalamus of DFP-intoxicated rats is mediated by calcium dysregulation and energy depletion downstream of excitotoxicity. Progressive neurodegeneration and mineralization are hallmarks of ischemic injury (Ansari et al., 1990; Martin et al., 1998), but whether DFP-induced mineralization is influenced by ischemia, in addition to or independent of seizure-mediated damage, is difficult to determine because both ischemia and seizures converge on similar cellular stress responses (Auer and Siesjo, 1988). No damage was observed in the hearts of DFP intoxicated rats, ruling out global ischemia. However, this does not rule out the potential for local ischemia due to mismatches between the ratio of glucose utilization to oxygen delivery at a regional level in the brain, as has been documented in a rat model of soman exposure (Shih and Scremin, 1992). Interestingly, the regions experiencing relative ischemia in this soman study included the thalamus and hippocampus, but not the piriform cortex or amygdala, which mirrors the distribution of mineralization in our DFP model.
Recent reports indicate that approximately 30% of soman-intoxicated animals develop SRS (de Araujo Furtado et al., 2010). Similarly, we observed that a subset of DFP animals exhibited behavior consistent with SRS. Because we were only able to observe SRS using behavioral criteria, and only during limited time frames when animals were being handled, we cannot rule out the possibility that we did not capture all of the DFP animals with SRS. Given that caveat, DFP animals that were observed to exhibit SRS behavior had more severe neuronal damage than other DFP-intoxicated animals in our study, which is consistent with reports of more severe lesions in soman-intoxicated rats that developed electroencephalographic SRS, compared with those that did not (de Araujo Furtado et al., 2010). In both models, SRS were associated with severe damage in the piriform cortex, thalamus and hippocampus at 2–3 weeks post exposure. It is not clear, however, whether animals with more severe damage in these brain regions are more likely to develop SRS, or whether recurrent seizure activity in the weeks following OP-induced SE further exacerbates existing lesions, promotes new injury or prevents lesion resolution.
Acute DFP intoxication rapidly triggers robust behavioral and electrographic seizure activity that lasts for several hours (Deshpande et al., 2010), raising questions about the relative contribution of seizure activity vs. direct toxic effects of DFP to the resulting neuropathology. SE triggered by kainic acid, lithium, pilocarpine, and flurothyl-induced SE (Gayoso et al., 2003; Ingvar et al., 1988; Wall et al., 2000) has been reported to cause extensive and persistent necrosis and neuroinflammation, as well as progressive expansion of dystrophic mineralization in limbic brain regions. The similar patterns of neuropathology across chemically diverse SE models suggest that DFP-induced neuropathology is primarily dependent on seizure activity. In support of this hypothesis, the time-weighted SSAVR score was highly correlated to neuroinflammation and NN across all brain regions. A potential caveat is that we assessed seizure severity using behavioral criteria. However, the seizure scoring system we used was previously validated against electroencephalography in a comparable DFP rat model (Deshpande et al., 2010). Moreover, the SSAVR captures the magnitude and duration of seizure behavior, and studies of the OP nerve agent soman identified a positive correlation between the magnitude and duration of electroencephalographic seizures and the severity of brain damage (Carpentier et al., 2001; Lallement et al., 1994; McDonough et al., 1995).
In summary, the evolving and region-specific nature of the damage elicited by acute DFP intoxication parallels that observed following SE triggered by OP nerve agents (Collombet, 2011; Kadar et al., 1995; McDonough and Shih, 1997; McLeod et al., 1984; Petras, 1994) and other chemicals (Gayoso et al., 2003; Ingvar et al., 1988; Wall et al., 2000). These findings validate the DFP model as a tool for developing broad-spectrum medical countermeasures against diverse seizure-inducing chemicals, and underscore the need for longitudinal monitoring of pathology and therapeutic response.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Mr Michael Manzer (Histology Laboratory, PMI Department, School of Veterinary Medicine, UC Davis) for his assistance with histological preparation of samples, and Dr Heike Wulff (UC Davis) for providing helpful comments on drafts of the manuscript.
FUNDING
CounterACT Program, National Institutes of Health Office of the Director and the National Institute of Neurological Disorders and Stroke (U54 NS079202 to P.J.L.), the National Institute of General Medical Sciences (T32 GM099608 to B.A.H.), the David and Dana Loury Foundation (predoctoral fellowship to B.A.H.), and the ARCS Foundation (predoctoral fellowship to B.A.H.). The sponsors were not involved in the study design, the collection, analysis and interpretation of data, in the writing of the paper or in the decision to submit the work for publication.
REFERENCES
- Ansari M. Q., Chincanchan C. A., Armstrong D. L. (1990). Brain calcification in hypoxic-ischemic lesions: an autopsy review. Pediatr. Neurol. 6, 94–101. [DOI] [PubMed] [Google Scholar]
- Auer R. N., Siesjo B. K. (1988). Biological differences between ischemia, hypoglycemia, and epilepsy. Ann.Neurol. 24, 699–707. [DOI] [PubMed] [Google Scholar]
- Baille-Le Crom V., Collombet J. M., Carpentier P., Brochier G., Burckhart M. F., Foquin A., Pernot-Marino I., Rondouin G., Lallement G. (1995). Early regional changes of GFAP mRNA in rat hippocampus and dentate gyrus during soman-induced seizures. Neuroreport 7, 365–369. [PubMed] [Google Scholar]
- Banks C. N., Lein P. J. (2012). A review of experimental evidence linking neurotoxic organophosphorus compounds and inflammation. Neurotoxicology 33, 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer K. L., Troendle M. M., Pekman L., Meggs W. J. (2013). Naltrexone prevents delayed encephalopathy in rats poisoned with the sarin analogue diisopropylflurophosphate. Am. J. Emerg. Med. 31, 676–679. [DOI] [PubMed] [Google Scholar]
- Carpentier P., Foquin A., Dorandeu F., Lallement G. (2001). Delta activity as an early indicator for soman-induced brain damage: a review. Neurotoxicology 22, 299–315. [DOI] [PubMed] [Google Scholar]
- Chen Y. (2012). Organophosphate-induced brain damage: mechanisms, neuropsychiatric and neurological consequences, and potential therapeutic strategies. Neurotoxicology 33, 391–400. [DOI] [PubMed] [Google Scholar]
- Collombet J. M. (2011). Nerve agent intoxication: recent neuropathophysiological findings and subsequent impact on medical management prospects. Toxicol. Appl. Pharmacol. 255, 229–241. [DOI] [PubMed] [Google Scholar]
- Costa L. G. (2006). Current issues in organophosphate toxicology. Clin. Chim. Acta 366, 1–13. [DOI] [PubMed] [Google Scholar]
- Damodaran T. V., Abou-Donia M. B. (2000). Alterations in levels of mRNAs coding for glial fibrillary acidic protein (GFAP) and vimentin genes in the central nervous system of hens treated with diisopropyl phosphorofluoridate (DFP). Neurochem. Res. 25, 809–816. [DOI] [PubMed] [Google Scholar]
- de Araujo Furtado M., Lumley L. A., Robison C., Tong L. C., Lichtenstein S., Yourick D. L. (2010). Spontaneous recurrent seizures after status epilepticus induced by soman in Sprague-Dawley rats. Epilepsia 51, 1503–1510. [DOI] [PubMed] [Google Scholar]
- de Araujo Furtado M., Rossetti F., Chanda S., Yourick D. (2012). Exposure to nerve agents: from status epilepticus to neuroinflammation, brain damage, neurogenesis and epilepsy. Neurotoxicology 33, 1476–1490. [DOI] [PubMed] [Google Scholar]
- Del Turco D., Schlaudraff J., Bonin M., Deller T. (2014). Upregulation of APP, ADAM10 and ADAM17 in the denervated mouse dentate gyrus. PLoS One 9, e84962.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande L. S., Carter D. S., Blair R. E., DeLorenzo R. J. (2010). Development of a prolonged calcium plateau in hippocampal neurons in rats surviving status epilepticus induced by the organophosphate diisopropylfluorophosphate. Toxicol. Sci. 116, 623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddleston M., Buckley N. A., Eyer P., Dawson A. H. (2008). Management of acute organophosphorus pesticide poisoning. Lancet 371, 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieller E. C. (1954). Some problems in interval estimation. J. Roy. Stat. Soc. B 16, 175–185. [Google Scholar]
- Flannery B. M., Bruun D. A., Rowland D. J., Banks C. N., Austin A. T., Kukis D. L., Li Y., Ford B. D., Tancredi D. J., Silverman J. L., et al. (2016). Persistent neuroinflammation and cognitive impairment in a rat model of acute diisopropylfluorophosphate intoxication. J Neuroinflammation 13, 267.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornito A., Zalesky A., Breakspear M. (2015). The connectomics of brain disorders. Nat. Rev. Neurosci. 16, 159–172. [DOI] [PubMed] [Google Scholar]
- Gayoso M. J., Al-Majdalawi A., Garrosa M., Calvo B., Diaz-Flores L. (2003). Selective calcification of rat brain lesions caused by systemic administration of kainic acid. Histol. Histopathol . 18, 855–869. [DOI] [PubMed] [Google Scholar]
- Grauer E., Chapman S., Rabinovitz I., Raveh L., Weissman B. A., Kadar T., Allon N. (2008). Single whole-body exposure to sarin vapor in rats: long-term neuronal and behavioral deficits. Toxicol. Appl. Pharmacol. 227, 265–274. [DOI] [PubMed] [Google Scholar]
- Ingvar M., Morgan P. F., Auer R. N. (1988). The nature and timing of excitotoxic neuronal necrosis in the cerebral cortex, hippocampus and thalamus due to flurothyl-induced status epilepticus. Acta Neuropathol. 75, 362–369. [DOI] [PubMed] [Google Scholar]
- Kadar T., Cohen G., Sahar R., Alkalai D., Shapira S. (1992). Long-term study of brain lesions following soman, in comparison to DFP and metrazol poisoning. Hum. Exp. Toxicol. 11, 517–523. [DOI] [PubMed] [Google Scholar]
- Kadar T., Shapira S., Cohen G., Sahar R., Alkalay D., Raveh L. (1995). Sarin-induced neuropathology in rats. Hum. Exp. Toxicol. 14, 252–259. [DOI] [PubMed] [Google Scholar]
- Kadriu B., Guidotti A., Costa E., Auta J. (2009). Imidazenil, a non-sedating anticonvulsant benzodiazepine, is more potent than diazepam in protecting against DFP-induced seizures and neuronal damage. Toxicology 256, 164–174. [DOI] [PubMed] [Google Scholar]
- Kim Y. B., Hur G. H., Shin S., Sok D. E., Kang J. K., Lee Y. S. (1999). Organophosphate-induced brain injuries: delayed apoptosis mediated by nitric oxide. Environ. Toxicol. Pharmacol. 7, 147–152. [DOI] [PubMed] [Google Scholar]
- Lallement G., Pernot-Marino I., Baubichon D., Burckhart M. F., Carpentier P., Blanchet G. (1994). Modulation of soman-induced neuropathology with an anticonvulsant regimen. Neuroreport 5, 2265–2268. [DOI] [PubMed] [Google Scholar]
- Leopold I. H., Cleveland A. F. (1953). Di-isopropyl fluorophosphate, 0.01 percent, in chronic wide-angle glaucoma. Am. J. Ophthalmol. 36, 226–231. [DOI] [PubMed] [Google Scholar]
- Li Y., Lein P. J., Ford G. D., Liu C., Stovall K. C., White T. E., Bruun D. A., Tewolde T., Gates A. S., Distel T. J., et al. (2015). Neuregulin-1 inhibits neuroinflammatory responses in a rat model of organophosphate-nerve agent-induced delayed neuronal injury. J. Neuroinflammation 12, 64.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Lein P. J., Liu C., Bruun D. A., Tewolde T., Ford G., Ford B. D. (2011). Spatiotemporal pattern of neuronal injury induced by DFP in rats: a model for delayed neuronal cell death following acute OP intoxication. Toxicol. Appl. Pharmacol. 253, 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Li Y., Lein P. J., Ford B. D. (2012). Spatiotemporal patterns of GFAP upregulation in rat brain following acute intoxication with diisopropylfluorophosphate (DFP). Curr. Neurobiol. 3, 90–97. [PMC free article] [PubMed] [Google Scholar]
- Martin L. J., Al-Abdulla N. A., Brambrink A. M., Kirsch J. R., Sieber F. E., Portera-Cailliau C. (1998). Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on the contributions of apoptosis and necrosis. Brain Res. Bull. 46, 281–309. [DOI] [PubMed] [Google Scholar]
- McDonough J. H. Jr, Dochterman L. W., Smith C. D., Shih T. M. (1995). Protection against nerve agent-induced neuropathology, but not cardiac pathology, is associated with the anticonvulsant action of drug treatment. Neurotoxicology 16, 123–132. [PubMed] [Google Scholar]
- McDonough J. H. Jr, Shih T. M. (1997). Neuropharmacological mechanisms of nerve agent-induced seizure and neuropathology. Neurosci. Biobehav. Rev. 21, 559–579. [DOI] [PubMed] [Google Scholar]
- McLeod C. G. Jr., Singer A. W., Harrington D. G. (1984). Acute neuropathology in soman poisoned rats. Neurotoxicology 5, 53–57. [PubMed] [Google Scholar]
- Millard C. B., Kryger G., Ordentlich A., Greenblatt H. M., Harel M., Raves M. L., Segall Y., Barak D., Shafferman A., Silman I., et al. (1999). Crystal structures of aged phosphonylated acetylcholinesterase: nerve agent reaction products at the atomic level. Biochemistry 38, 7032–7039. [DOI] [PubMed] [Google Scholar]
- Paxinos G., Watson C. (2007). The Rat Brain in Stereotaxic Coordinates, 6th ed Academic Press/Elsevier, Amsterdam, Boston. [Google Scholar]
- Pessah I. N., Rogawski M. A., Tancredi D. J., Wulff H., Zolkowska D., Bruun D. A., Hammock B. D., Lein P. J. (2016). Models to identify treatments for the acute and persistent effects of seizure-inducing chemical threat agents. Ann. N. Y. Acad. Sci. 1378, 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petras J. M. (1994). Neurology and neuropathology of Soman-induced brain injury: an overview. J. Exp. Anal. Behav. 61, 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope C. N. (1999). Organophosphorus pesticides: do they all have the same mechanism of toxicity? J. Toxicol. Environ. Health B 2, 161–181. [DOI] [PubMed] [Google Scholar]
- Pouliot W., Bealer S. L., Roach B., Dudek F. E. (2016). A rodent model of human organophosphate exposure producing status epilepticus and neuropathology. Neurotoxicology 56, 196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw B. (1946). Chemical warfare agents, and related chemical problems.
- Rodriguez M. J., Pugliese M., Mahy N. (2009). Drug abuse, brain calcification and glutamate-induced neurodegeneration. Curr Drug Abuse Rev 2, 99–112. [DOI] [PubMed] [Google Scholar]
- Rojas A., Ganesh T., Lelutiu N., Gueorguieva P., Dingledine R. (2015). Inhibition of the prostaglandin EP2 receptor is neuroprotective and accelerates functional recovery in a rat model of organophosphorus induced status epilepticus. Neuropharmacology 93, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A., Ganesh T., Manji Z., O'Neill T., Dingledine R. (2016). Inhibition of the prostaglandin E2 receptor EP2 prevents status epilepticus-induced deficits in the novel object recognition task in rats. Neuropharmacology 110, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum C., Bird S. B. (2010). Non-muscarinic therapeutic targets for acute organophosphorus poisoning. J. Med. Toxicol. 6, 408–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih T. M., Scremin O. U. (1992). Cerebral blood flow and metabolism in soman-induced convulsions. Brain Res Bull 28, 735–742. [DOI] [PubMed] [Google Scholar]
- Tang F. R., Kato K., Loke W. K. (2011). Pilocarpine and nerve agent-induced seizures: similarities and differences. In Chemical-Induced Seizures: Mechanisms, Consequences and Treatment, (F.R. Tang, Ed.), pp. 89–90. Bentham Science Publishers, Bentham. ebook, DOI: 10.2174/97816080527451110101.
- Vismer M. S., Forcelli P. A., Skopin M. D., Gale K., Koubeissi M. Z. (2015). The piriform, perirhinal, and entorhinal cortex in seizure generation. Front. Neural Circuits 9, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall C. J., Kendall E. J., Obenaus A. (2000). Rapid alterations in diffusion-weighted images with anatomic correlates in a rodent model of status epilepticus. AJNR Am. J. Neuroradiol. 21, 1841–1852. [PMC free article] [PubMed] [Google Scholar]
- Wright L. K., Liu J., Nallapaneni A., Pope C. N. (2010). Behavioral sequelae following acute diisopropylfluorophosphate intoxication in rats: comparative effects of atropine and cannabinomimetics. Neurotoxicol. Teratol. 32, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer L. A., Ennis M., Shipley M. T. (1997). Soman-induced seizures rapidly activate astrocytes and microglia in discrete brain regions. J. Compar. Neurol. 378, 482–492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.