

Abstract
Hemophilia B, a hereditary bleeding disorder caused by a deficiency of coagulation factor IX (FIX), is an excellent candidate for gene therapy. However, to date, success in hemophilia gene therapy clinical trials has been limited due to failure to achieve or sustain therapeutic levels of factor expression. The phiC31 integrase system efficiently integrates plasmid DNA carrying a transgene and an attB site into a limited number of endogenous pseudo attP sites in mammalian genomes, leading to robust, sustained transgene expression. A strategy utilizing plasmid DNA integrated with phiC31 integrase may offer a facile and safe alternative for sustained human FIX (hFIX) expression. Hydrodynamic tail vein injection was used for delivery of plasmids encoding phiC31 integrase and hFIX to the liver of FIX knockout mice. We demonstrated prolonged therapeutic levels of hFIX in this knockout mouse model of hemophilia B over a 6-month time course when phiC31 integrase was used. Additionally, we observed sustained FIX activity in plasma and phenotypic correction of bleeding after tail clip in phiC31-treated mice. In the livers that received integrase, we also demonstrated prolonged hFIX expression in hepatocytes by immunohistochemistry and documented sequence-specific genomic integration of the hFIX plasmid. These studies suggest the possibility that a similar approach in large animals and humans could lead to a simple and successful gene therapy for hemophilia.
Keywords: hemophilia B, hydrodynamic injection, non-viral, phiC31 integrase
INTRODUCTION
Hemophilia B is a compelling candidate for gene therapy. This X-linked recessive bleeding disorder results from a deficiency of blood coagulation factor IX (FIX) activity and affects an estimated 1 in 30 000 males.1,2 The state-of-the-art therapeutic approach involves frequent infusion of recombinant human factor IX (hFIX) protein. While effective, recombinant factor therapy is intrusive, expensive, and rarely available for patients in less developed countries. There is a need, as well as a strong rationale, for gene therapy. As a well-characterized monogenic disorder, hemophilia B is an appropriate candidate for gene addition therapy. FIX is normally synthesized and secreted by hepatocytes,3 which are accessible by several gene delivery approaches. Furthermore, preclinical studies can be performed in well-established murine4,5 and canine6 models, and primate studies are also possible.7 The severity of the disease is determined by the level of FIX in circulation and is classified as either mild (>5%), moderate (1–5%) or severe (<1%).2 Thus, only 2–3% of normal FIX levels would be considered therapeutic, resulting in marked reduction in spontaneous bleeding symptoms and prevention of most life-threatening episodes.8 Finally, the effects of a successful gene therapy can be easily monitored by obtaining blood samples to measure FIX expression levels and perform clotting assays, as well as by examining changes in bleeding phenotype.
While preclinical gene therapy studies in animal models for hemophilia have been promising, clinical trials have suffered numerous setbacks.9,10 Translating successful preclinical approaches into a long-term cure in human gene therapy trials has been hampered due to challenges associated with limited gene transfer, immunogenicity of viral vectors11 and transience of gene expression using non-viral vectors.12 These limitations highlight the need for an improved vector system that can achieve a sustained therapeutic effect while avoiding toxicity.
The ϕC31 integrase system offers a novel non-viral approach for gene therapy. This integrase is a serine recombinase derived from Streptomyces phage ϕC31 (ref. 13). The enzyme recombines the bacteriophage attP site with the target attB site on the bacterial chromosome, catalyzing precise unidirectional integration. ϕC31 integrase also performs this reaction in mammalian cells.14,15 When used for gene therapy, ϕC31 integrase can recombine the attB site on a plasmid carrying the transgene with sequences that have partial identity to attP called pseudo attP sites in the host genome, resulting in stable integration.15 ϕC31 integrase-mediated integration is sequence-specific and occurs at a relatively small number of genomic locations,16 reducing the risk of insertional mutagenesis. ϕC31 integrase caused no acceleration of tumors in a murine MYC model of liver cancer.17 In addition, the system is not limited by vector size,18 and integration of a plasmid into genomic pseudo attP sites results in robust, prolonged transgene expression (reviewed in ref. 19). Furthermore, the system has been used extensively with safety to create and engineer transgenic organisms.18,19
Plasmids have been effectively introduced into the liver by the high-pressure tail vein injection method.20,21 We previously introduced hFIX in normal mice, as a marker gene to study the potential utility of ϕC31 integrase for long-term liver gene therapy.22 Given the positive results in those experiments, we were interested in advancing the integrase system toward clinical utility by studying hFIX gene therapy in the absence of endogenous FIX, to more closely model the situation in hemophilia. We also wanted to develop plasmids that would be optimally suited for clinical use. The purpose of this study was to evaluate whether the ϕC31 integrase approach with such plasmids could safely and effectively provide long-term therapeutic levels of hFIX that would ameliorate the disease in the FIX knockout (FIX−/−)4 mouse model of hemophilia B. We demonstrated functional activity of the hFIX protein by the one-stage factor IX activity assay (FIX-specific activated partial thromboplastin time, FIX aPTT). Correction of the bleeding phenotype was evidenced by hemostatic protection in the tail-transection bleeding challenge (tail clip). In addition, we assessed the expression of hFIX in liver sections by immunofluorescence and demonstrated ϕC31 integrase-mediated integration of the hFIX plasmid in the mouse genome.
RESULTS
ϕC31 integrase provides prolonged therapeutic levels of hFIX expression in FIX−/−mice
pCS was the plasmid backbone we used for integrase expression in all past studies.14–17,19,22,23 However, the pVax backbone is preferred for clinical studies24 as put forth by the Food and Drug Administration in its document ‘Points to consider on plasmid DNA vaccines for preventive infectious disease indications’ (Docket No. 96N-0400). The pVax plasmid lacks sequences unnecessary for replication in Escherichia coli or expression of the recombinant protein in mammalian cells. In addition, it carries the kanamycin resistance gene in place of ampicillin, because kanamycin is less likely than ampicillin to illicit an allergic response in humans and to avoid dispersal of ampicillin resistance (Figure 1a). Hence, a new plasmid, pVI, expressing wild-type ϕC31 integrase from the pVax backbone, was utilized in these studies. The hFIX expression plasmid, pVFB,23 carries an hFIX mini-gene driven by a hepatocyte-specific human α1 anti-trypsin promoter and the liver-specific apolipoprotein E enhancer.25 This expression cassette has been shown to confer robust, long-term hFIX expression in C57Bl/6 mice.23,25 Even though hFIX is a mammalian gene, some studies have reported that significant increases in hFIX expression can be achieved after codon optimization.26,27 Furthermore, the hFIX gene in the pVFB plasmid did not have a Kozak sequence for translation, suggesting that adding a Kozak sequence upstream of the ATG might increase hFIX expression. Therefore, a codon-optimized hFIX expression cassette with a Kozak sequence was synthesized. The optimized hFIX cassette, including attB, was cloned into the pVax expression plasmid to generate pVB9 (Figure 1b).
Figure 1.
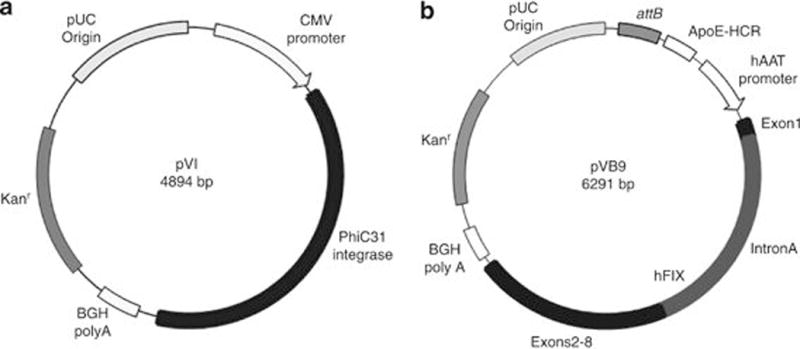
Integrase and hFIX plasmids. (a) pVI, the ϕC31 integrase expression plasmid used in this study. (b) pVB9, liver-specific hFIX expression vector. This plasmid comprises the pVax backbone carrying the kanamycin resistance gene and the ϕC31 attB site. It also carries the apolipoprotein E hepatic control region (ApoE-HCR) enhancer and the human codon-optimized hFIX mini-gene, consisting of Exon 1 separated from Exons 2 to 8 by Intron A, transcribed from the human α1 anti-trypsin (hAAT) promoter.
We evaluated the ability of ϕC31 integrase to provide sustained therapeutic levels of hFIX expression in FIX−/− mice. Hydrodynamic tail vein (HTV) injection was performed on groups of 7–10 mice to achieve efficient delivery of the plasmid DNA to the liver.20,21 A plasmid carrying attB and expressing the codon-optimized hFIX gene, pVB9, was co-injected with either pVI or pVmI. The ‘m’ form of the integrase contains a mutation in the catalytic serine that prevents recombination, so this plasmid constitutes a negative control for integration. Blood was collected and plasma hFIX levels were measured by enzyme-linked immunosorbent assay over a time course. Results of this study are depicted in Figure 2. At the first time point, on day 1, all the experimental animals expressed high levels of hFIX, averaging between 1500 and 3500 ng ml−1. These initial levels ultimately dropped for all groups to different degrees. The hFIX levels in mice that were injected with the ϕC31 integrase (pVI+pVB9) plateaued after 2 weeks, remaining consistent for the remainder of the 6-month study. Animals that received pVI displayed nine- and seven-fold higher sustained expression of hFIX (1022±199 ng ml−1) compared with groups that received either pVB9 alone (114±32 ng ml−1) or the inactive integrase, pVmI (146±42 ng ml−1), respectively. The increased hFIX levels generated by pVI persisted for the 6-month length of the study and represented therapeutic amounts. The Bethesda assay for inhibitors was performed on the pVB9 alone and pVI+pVB9 day 168 mouse plasma samples, with the result that no neutralizing antibodies were detected (data not shown). Antibodies to hFIX and integrase were not expected based on previous experience,22,23,25–27 and as further explained in the Discussion.
Figure 2.
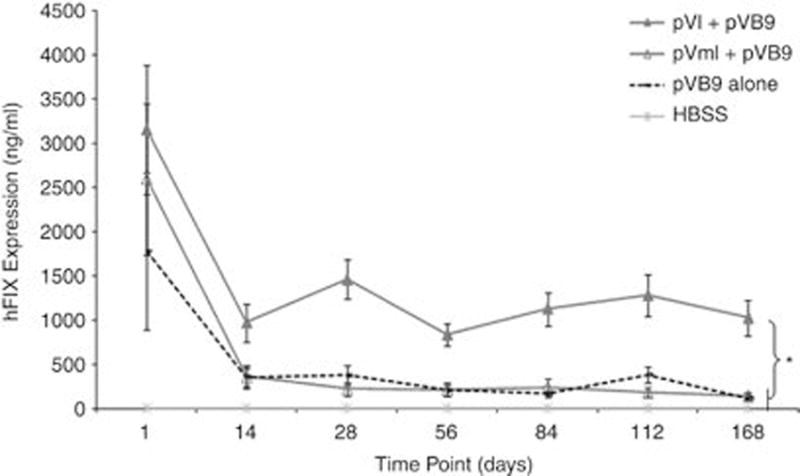
hFIX expression in plasma of FIX−/− mice injected with various plasmids. FIX−/− mice were hydrodynamically injected with pVB9 alone, pVI plus pVB9 or pVmI plus pVB9. Negative control animals were injected only with Hank’s balanced salt solution. Plasma from blood samples was assayed for hFIX expression by enzyme-linked immunosorbent assay at various time points. Asterisk (*) denotes P-value of interest that was <0.05. Values are ± s.e.m.
FIX−/−mice co-injected with ϕC31 integrase have sustained circulating hFIX activity
To quantify the activity of hFIX in the blood, we performed one-stage FIX activity assays (FIX-specific activated partial thromboplastin time, FIX aPTT) on citrated plasma samples obtained from the experimental mice at various time points. As shown in Figure 3a, at day 1, all groups expressed hFIX activities averaging between 20 and 30%. After 2 weeks, the hFIX activities decreased by varying extents in all groups. The hFIX activity initially declined in the pVI+pVB9 group of mice, but then stabilized at ~10% of normal, an amount considered to be sufficient to ameliorate most bleeding symptoms. All treated groups that did not receive wild-type ϕC31 integrase had hFIX activity of less than 3% at the end of the experiment (Figures 3a and b). The percentages of mice expressing hFIX, as well as the levels of hFIX activity, are presented in Table 1.
Figure 3.
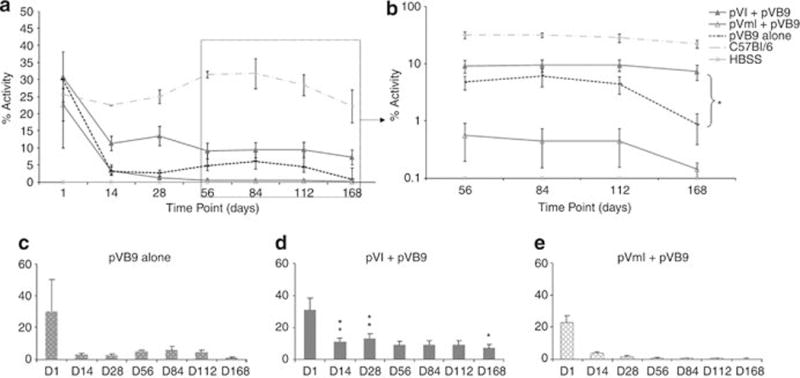
hFIX activity in plasma of FIX−/− mice injected with various plasmids. Animals were hydrodynamically injected with pVB9 alone, pVI and pVB9, pVmI and pVB9, or Hank’s balanced salt solution only. Naive C57Bl/6 mice were used as positive controls. Plasma from blood samples was assayed for the FIX aPTT at various time points. (a) Percentage of hFIX activity is plotted for all the treatment groups throughout the length of the study. (b) Expanded version of (a) showing the later time points only. (c) Percentage of hFIX activity at various time points for pVB9 alone-treated group. (d) Percentage of hFIX activity in the pVI+pVB9 treatment group at various time points. (e) Percentage of hFIX activity in the pVmI+pVB9 group over a time course. Values are ±s.e.m. Asterisks denote values that differ from pVB9 *at P<0.05 or ** at P<0.01 (Student’s t-test).
Table 1.
Frequency of mice that possessed hFIX activity at the last time point (day 168)
| Treatment | Frequency | FIX activity (%) |
|---|---|---|
| pVB9 | 2/6 (33%) | 1.2, 3.0 |
| pVI+pVB9 | 9/9 (100%) | 1.5–17.3 |
| pVml+pVB9 | 1/7 (14%) | 1.0 |
Abbreviations: FIX, factor IX; hFIX, human factor IX.
Compared with the pVB9 alone treatment group (Figure 3c), the pVI+pVB9 group (Figure 3d) showed significantly higher activities at days 14, 28 and 168 (P<0.05 or P<0.01). At the last time point tested (day 168), all the mice in the pVI+pVB9 group displayed hFIX activities of 1.5–17.3%, whereas in the pVB9 group, only 33% maintained low hFIX activities (1.2–3.0%). For the group expressing the inactive version of the integrase (pVmI+pVB9), the treatment had no significant influence on the FIX activity (Figure 3e), compared with the pVB9 alone group. FIX−/− mice treated with Hank’s balanced salt solution only served as negative controls and showed no FIX activity throughout the length of the experiment, whereas wild-type, untreated C57Bl/6 mice served as positive controls and displayed stable FIX activity (22.2–31.7%; Figures 3a and b).
FIX−/− mice co-injected with ϕC31 integrase display hemostatic protection in a bleeding challenge
To assess phenotypic function of hFIX in FIX−/− mice, the tail clip assay was employed at 180 days post-injection. Hemostatic protection was measured by the extent of active bleeding during two 10 min challenges, constituting immediate hemostasis and secondary bleeding. The duration of bleeding through each 10 min challenge was recorded, as well as the amount of blood loss, determined by measuring the amount of hemoglobin in the blood samples collected from the tail. Finally, mice were observed for rebleeding and activity for 4 h after the initial transection.
Consistent with the in vivo hFIX activity results, pVI+pVB9-treated mice had less blood loss than pVmI+pVB9, pVB9 alone and negative control FIX−/− naive mice during secondary bleeding, although there was no significant difference in the pVI+pVB9 and pVB9 alone mice in the first bleeding challenge. Figure 4 indicates the significantly lower optical density values for pVI+pVB9-treated animals, reflecting lower amounts of hemoglobin loss, compared with pVmI+pVB9 and pVB9 alone mice in the second bleeding challenge. There was no significant difference in blood loss between pVI+pVB9 and wild-type C57Bl/6 mice during either the immediate or secondary bleeding challenge.
Figure 4.
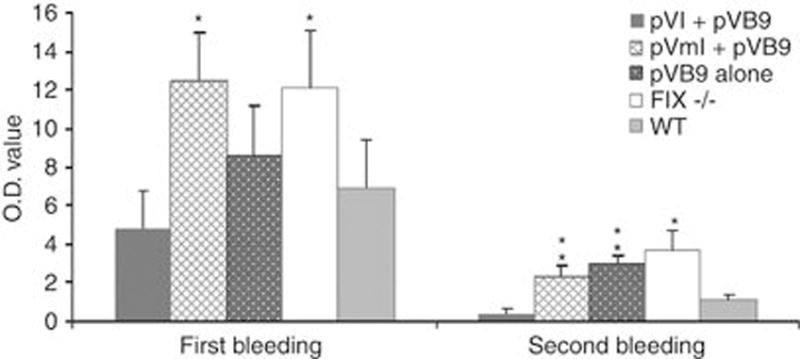
Hemostatic response to tail clips. Hemoglobin measurement indicating total blood loss after tail clips. The collected blood samples were treated with RBC lysis buffer, and then optical density was measured at 575 nm. *Denotes values that differ from pVI+pVB9 at P<0.05 or ** at P<0.01 using the Student’s t-test.
As has been previously reported, immediate hemostasis in a tail clip assay may fail to distinguish factor deficiencies.28,29 However, delayed rebleeding is very characteristic of clinical hemophilia, and protection from late hemorrhage is a more specific measure of correction of the hemophilic phenotype.30–32 Indeed, while the initial bleeding time of mice that received the pVI+pVB9 (5.88±1.12 min) was significantly shorter than mice receiving the inactive integrase mutant (pVmI+pVB9, 9.22±0.52 min) and not different from wild-type mice (7.54±1.16 min) or pVB9 alone (7.83±0.78), the secondary bleeding times demonstrated phenotypic differences even more clearly. During the delayed bleeding challenge, the pVI+pVB9 mice bled for 1.9±1.10 min, compared with 7.62±0.86 min for the pVmI+pVB9 mice, 7.38±1.33 min for the pVB9 alone mice and 8.43±0.77 min for untreated FIX−/−mice (P<0.01 for pVI+pVB9 compared with these other three groups). Again, there was no significant difference between pVI+pVB9 and wild-type C57Bl/6 mice in the secondary bleeding time. Table 2 details the observations made for 4 h after the second bleeding challenge. There was no rebleeding observed in any of the seven pVI+pVB9-treated mice, and they continued to remain healthy for months after the bleeding challenges. However, in four out of six pVmI+pVB9 mice, we observed moderate or severe rebleeding. Three out of five pVB9 alone-treated mice demonstrated moderate-to-severe rebleeding, and each appeared to go into shock during the observation, necessitating euthanasia. In the controls, three out of seven FIX−/− naive mice exhibited rebleeding. While no significant rebleeding was observed in wild-type C57Bl/6 mice, three of these female mice went into shock and were euthanized. Female age-matched wild-type C57Bl/6 mice were smaller in size compared with their male counterparts or the FIX−/− mice. This resulted in them having less blood volume, producing distress following blood loss that required euthanasia.
Table 2.
Outcome following second bleeding challenge, beginning at 40 min and continuing until 4 h after the tail transection
| Mouse groups | Delayed rebleeding | Death or distress requiring euthanasia |
|---|---|---|
| pVB9 | 3/5 (60%) | 5/5 (100%) |
| pVI+pVB9 | 0/7 (0%) | 0/7 (0%) |
| pVmI+pVB9 | 4/6 (67%) | 3/6 (50%) |
| FIX−/−(untreated) | 3/7 (43%) | 7/7 (100%) |
| Wild-type C57Bl/6 | 0/7 (0%) | 3/7 (43%)a |
Age-matched wild-type female C57Bl/6 mice were smaller in size and thus had less blood volume. The two bleeding challenges caused distress requiring euthanasia.
ϕC31 integrase mediates sustained hFIX expression and sequence-specific genomic integration in hepatocytes
To analyze the ability of the ϕC31 integrase to provide long-term expression of hFIX and mediate genomic integration in hepatocytes, we employed two approaches. Livers from three mice per group were utilized, killed 200 days after injection. To visualize hFIX expression in hepatocytes, liver sections were prepared for immunofluorescence staining with an antibody specific for hFIX. Representative liver sections are shown in Figure 5a. These sections revealed a significantly higher number of hepatocytes expressing hFIX in livers that received pVB9 plus pVI, compared with the groups that received pVB9 alone, pVB9 plus pVmI or Hank’s balanced salt solution only. Quantitative analysis of the hFIX-positive cells in the pVB9 plus integrase sections revealed that 5–7% of cells expressed hFIX. This figure was in agreement with previous studies23,33 that reported approximately 5–10% of cells expressing hFIX when ϕC31 integrase was co-introduced, compared with ~1% in the absence of ϕC31 integrase, reflecting random integration, after thorough examination of multiple liver sections from each group.
Figure 5.
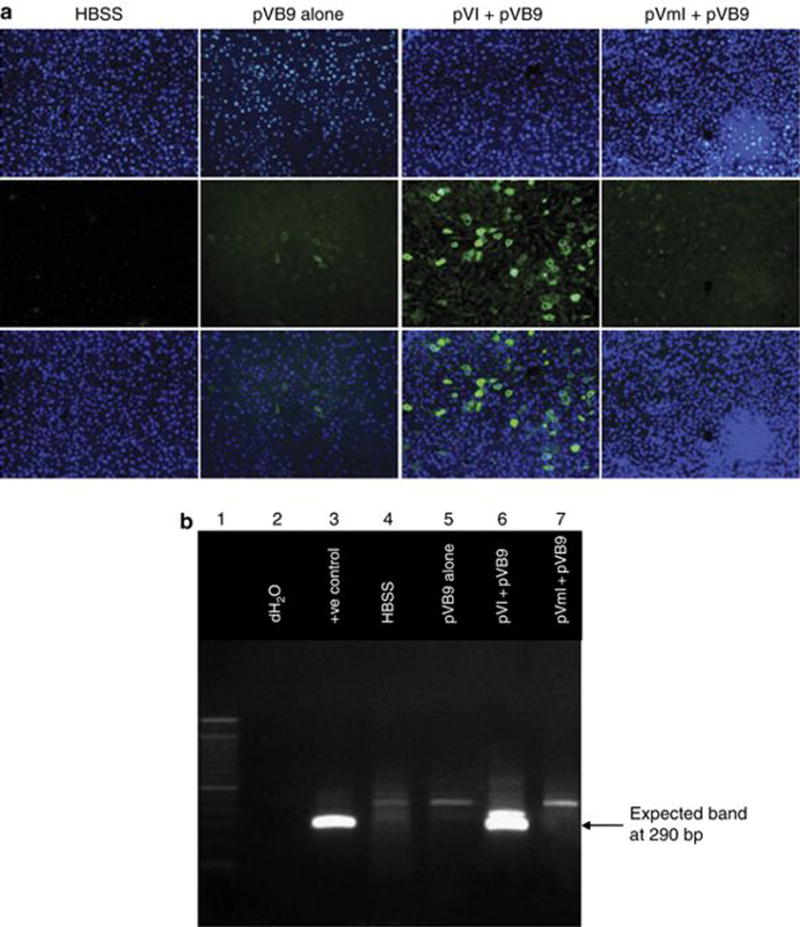
Evidence for prolonged hFIX expression and integration into a genomic pseudo attP site in mouse hepatocytes. (a) Immunofluorescence staining for hFIX in representative FIX−/− mouse liver sections obtained 200 days after HTV injection. The top row shows 4,6-diamidino-2-phenylindole (DAPI)-stained nuclei, the middle row shows staining for hFIX and the bottom row is a merged image of the top and middle rows. Vertical panel 1, Hank’s balanced salt solution (HBSS); panel 2, pVB9 alone; panel 3, pVI+pVB9; and panel 4, pVmI+pVB9. (b) PCR was carried out on DNA extracted from the liver, using primers that detect the junction between the attB site of pVB9 and mpsL1, a preferred pseudo attP site in the mouse genome. Lane 1, DNA ladder; lane 2, no template DNA control; lane 3, liver DNA sample of an injected animal from another experiment, previously shown to have attB-plasmid DNA integrated at mpsL1; lane 4, liver that was injected with HBSS; lane 5, liver that received pVB9 alone; lane 6, liver that received pVI+pVB9; and lane 7, liver that received pVmI+pVB9.
We also performed polymerase chain reaction (PCR) to investigate whether ϕC31-mediated integration had occurred at a frequently used pseudo attP site in the mouse genome, termed mpsL1 (ref. 22). Previous studies indicated that mpsL1 was by far the most prevalent site for ϕC31-mediated integration in the mouse liver genome, and the only integration site that was universally detected in all treated mice.22,34 Therefore, it represented an excellent marker to detect ϕC31-mediated genomic integration. Genomic DNA was isolated from the same liver samples that were used for immunohistochemistry. A quantity of 200 ng of DNA template was subjected to PCR for analysis of the integration junction between integrated pVB9 and the chromosomal mpsL1 site. Figure 5b is an agarose gel that was run with representative samples from the PCR analysis. The expected PCR band was seen in all three liver samples from the group that was injected with pVB9 plus pVI (lane 6). No bands were visible in livers of animals injected either with pVB9 alone (lane 5) or pVB9 plus pVmI (lane 7). The mpsL1 PCR analysis was consistent with the immunofluorescence and in vivo hFIX expression studies and suggested that wild-type ϕC31 integrase was efficacious in integrating the hFIX plasmid into the mpsL1 site in mouse hepatocytes, contributing to sustained therapeutic levels of hFIX in the blood.
DISCUSSION
Hemophilia B is a monogenic bleeding disorder with considerable potential to be treated by simple gene addition therapy. The goal of this study was to evaluate the efficacy of ϕC31 integrase to mediate integration of the hFIX gene into the genome of hemophilic mice, obtain sustained therapeutic levels of hFIX and ameliorate the disease phenotype. For the disease model, we utilized FIX knockout mice, which exhibit a phenotype similar to hemophilia B in patients.4 FIX−/− mice bred on a C57Bl/6 background do not develop antibodies that would effect clearance of FIX or neutralization of transgenic FIX pro-coagulant function by ‘inhibitor antibodies’,35 making them a good model system for hemophilia B gene therapy.
When we administered hFIX-carrying plasmid with wild-type ϕC31 integrase by hydrodynamic tail vein injection, the hemophilic mice yielded substantially higher levels of hFIX (Figure 2). Furthermore, these mice continued to express hFIX in the clinically relevant range for 24 weeks, the length of the study. The 1022 ng ml−1 of hFIX expressed by pVI+pVB9-treated mice is a level that would provide significant therapeutic correction for patients with hemophilia B, as greater than 250 ng ml−1 (~5% of normal circulating levels) results in a mild hemophilia B phenotype and an absence of spontaneous hemorrhages.4,36
The prolonged high levels of expression of hFIX that we obtained using ϕC31 integrase indicated that neutralizing antibodies that would have led to the elimination of the xenoprotein were not developed. The immunologic tolerance of hFIX observed in these experiments likely stems from several factors, including (1) the FIX transgene, which is poorly immunogenic due to its shared structural homology with other circulating and tissue-specific vitamin K-dependent proteins; (2) the liver-specific transcriptional regulatory elements, ApoE-HCR enhancer and hAAT promoter, used in our hFIX expression cassette minimized protein expression in antigen-presenting cells;27 (3) the site of expression of hFIX from the liver, which is a relatively well-tolerated route;27,28 and (4) the relatively immunotolerant strain background of C57Bl/6 mice (MHC H-2b).35 The latter two reasons also likely limited immune response to integrase, in addition to the limited period of integrase expression, short half-life of the integrase protein and rapid integration of the hFIX plasmid in the liver.33
The FIX aPTT assay carried out to test the biological activity of hFIX in hemophilic mice additionally revealed correction to mild hemophilia levels in pVI+pVB9-treated animals (Figure 3b), and was supported by their protection from severe bleeding in the hemostatic challenge. As expected, the pVI+pVB9-treated mice had short clotting times, protection from delayed rebleeding and less hemoglobin loss, as determined by the tail clip assay (Figure 4; Table 2).
Extended expression of hFIX in hepatocytes was detected in the livers of mice treated with pVB9 plus pVI (Figure 5a). By counting hFIX-positive cells in the liver sections, approximately 5–7% of hepatocytes were determined to express hFIX in these livers. Most of these cells presumably expressed integrase at one time, which suggests that there was no substantial immune response against the wild-type integrase. Sustained, high levels of hFIX expression appeared to require ϕC31 integrase-mediated integration of pVB9 into the mouse genome. Genomic integration into a frequently used pseudo attP site, called mpsL1, in the mouse liver was demonstrated by PCR analysis (Figure 5b). Positive results for integration at mpsL1 do not exclude the possibility that integration also occurred at other sites in the hepatocyte genome. Several other such sites have been detected in previous studies.22,34 Integration at the mpsL1 site appears to be safe, as it does not lie within a known coding sequence.22 The higher levels of hFIX expression seen when integrase was used, versus the pVB9 plasmid alone, indicate that, even though the hFIX expression cassette has been optimized for long-term expression,25 chromosomal integration is still advantageous for prolonged expression, versus extrachromosal plasmids. It is likely that most of the hFIX expression resulted from sequence-specific integration of the hFIX-expressing plasmid into the mouse genome and not from non-integrated episomal plasmid. A recent study from our group analyzed persistence of the hFIX and ϕC31 integrase plasmids in the mouse liver by Southern blot analysis and plasmid rescue in E. coli. This study revealed that most of the hFIX and ϕC31 integrase plasmid DNA was lost by 24 h after hydrodynamic injection.33
Our finding that wild-type ϕC31 integrase can mediate stable, therapeutic levels of hFIX expression that ameliorate the bleeding diathesis provides a novel potential strategy for development of a gene therapy treatment for hemophilia B patients. Furthermore, use of a plasmid rather than a viral system may allow repeated delivery of the hFIX transgene, a potentially important advantage. To minimize the development of hFIX antibodies that can be associated with life-threatening hypersensitivity reaction, patients enrolled in a Phase I clinical trial of FIX gene therapy should have missense mutations and a prolonged uncomplicated history of FIX protein therapy, excluding patients with complete gene deletions. Careful characterization of immunologic responses (or immunologic non-responsiveness) following integrase-mediated hepatic FIX expression in Phase I human application would be warranted to inform the safety of extending the therapy to individuals with null mutations. An essential element of a plasmid-mediated gene therapy approach is an effective and clinically acceptable method for delivery of plasmid DNA to the target tissue. The success of the hydrodynamic method for delivery of plasmid DNA to the liver in rodents has led several groups to explore translation of this method to large animals. In these strategies, systemic injection is replaced with liver-specific, catheter-mediated delivery that can be achieved by interventional radiology.37–40 Because a high percentage of older patients with severe hemophilia have been exposed to hepatitis C and other viruses, DNA delivery to the liver already at risk for inflammation would need to be carried out cautiously, with hemostatic support to counter mechanical stress at the time of DNA delivery. The combination of such delivery methods with ϕC31 integrase for permanent placement of the hFIX gene in the hepatocytes of patients may lead to new options for treatment of hemophilia B.
MATERIALS AND METHODS
Plasmids
A codon-optimized hFIX cassette including attB was synthesized by GENEART (Burlingame, CA, USA). This cassette was then cloned into the SpeI/XhoI sites of pVax1 (Invitrogen, Carlsbad, CA, USA) to generate pVB9. To create pVI and pVmI, pCSI14 and pCSmI22 were digested with KpnI/BamHI to release the wild-type or the inactive version of the integrase genes, respectively. In parallel, pVax1 was digested with KpnI/BamHI, and the linearized vector was dephosphorylated with shrimp alkaline phosphatase. Excised fragments containing wild-type or inactive integrase were inserted into the linearized, dephosphorylated pVax backbone. Sequences of the constructs were confirmed by diagnostic digests followed by sequencing by Elim Biopharmaceuticals (Hayward, CA, USA).
Mice
Mice were housed in the Research Animal Facility at Stanford University. C57Bl/6 mice were purchased from Charles Rivers Laboratories (Wilmington, MA, USA). Two mating pairs of FIX−/− mice were obtained from Dr Mark Kay (Stanford University, CA, USA) and bred. Animals were fed water and chow ad libitum. Eight- to ten-week-old C57Bl/6 mice or six- to eight-week-old FIX−/− mice were used for the studies. Experimental protocols were approved by the Administrative Panel on Laboratory Animal Care (A-PLAC) at Stanford University.
Hydrodynamic tail vein injection in mice
Plasmid DNA used for HTV delivery into mouse liver was prepared using a Qiagen EndoFree Plasmid Maxi kit (Qiagen, Valencia, CA, USA), except that the DNA pellet was dissolved in Hank’s balanced salt solution (Invitrogen). The DNA solution to be injected was prepared by diluting 20 μg of each plasmid into 1.8 ml of Hank’s balanced salt solution per injection. The animals were placed under a heat lamp until their tail veins were clearly dilated and then the DNA solution was injected within 5–7 s using a 3-ml Luer-Lok syringe (Becton Dickinson, Franklin Lakes, NJ, USA) and a 27-G butterfly needle (Becton Dickinson).
Blood collection and storage
Whole blood was drawn from the retro-orbital plexus of experimental mice over a time course. Immediately, the blood samples were diluted into a final concentration of 0.38% tri-sodium citrate (Vacuette, Greiner Bio-One, Monroe, NC, USA), followed by centrifugation at 2500 r.p.m. for 15–20 min. The citrated plasma was separated and stored at −80 °C for future analysis.
Enzyme-linked immunosorbent assay for hFIX
For quantification of hFIX expression, citrated plasma samples from experimental mice were used. A sandwich enzyme-linked immunosorbent assay was performed as described previously.23
FIX activity and Bethesda inhibitor assays
In order to quantify the activity of hFIX in blood, citrated plasma samples from experimental mice were used. The hFIX-specific, one-stage clotting assay (FIX aPTT) was performed using a STart 4 Coagulation Analyzer (Diagnostica Stago, Asnieres, France) as described previously.5 The human Bethesda inhibitor antibody assay, based on this FIX aPTT, was performed as previously described.5
Mouse tail bleeding and rebleeding
The method of tail bleeding was that of Gui et al.,30 with a few modifications. Briefly, mice were anesthetized with ketamine xylazine and maintained at 37 °C on a warming pad. The tail was transected at a cross-sectional diameter of 1.5 mm, immersed in 14 ml of 37 °C saline, and time to cessation of bleeding was recorded. After 10 min, bleeding was stopped by applying pressure to the tip of the tail, and the mice were left without manipulation for 20 min. Thirty minutes after the transection, the clot was stripped, and the tail was placed in 14 ml of 37 °C saline for an additional 10 min, and secondary bleeding time was recorded. Mice were observed following the second bleeding challenge, beginning at minute 40 and continuing until 4 h after the tail transection. Although death was not designed as an end point for these studies, mice too weak to drink water, feed or ambulate in the cage were euthanized to minimize distress. The collected blood in saline was centrifuged to collect erythrocytes and re-suspended in 5 ml RBC lysis buffer (Sigma, St Louis, MO, USA). The blood loss during the initial hemorrhage and the secondary rebleeding challenge was quantified by measuring optical density of the hemoglobin at 575 nm.
Immunofluorescence on mouse livers
FIX−/− experimental mice were killed, and their livers were harvested 200 days post-HTV injection. Liver tissues were paraffin embedded and sectioned by Histo-tec Laboratories (Hayward, CA, USA). The staining procedure has been described.23 Images of stained sections were taken on an Axioshop 2 Plus microscope with an AxioCam MRc camera (Zeiss, Thornwood, NY, USA).
PCR on liver genomic DNA
For integration site analysis, experimental animals were killed and the livers collected at 200 days after HTV delivery. Genomic DNA was isolated using the DNeasy Blood and Tissue Kit (Qiagen) based on the manufacturer’s protocol. Integration of the donor plasmid pVB9 into the mouse genome at the mpsL1 pseudo site was detected by PCR analysis using primers and conditions that have been described.23 The PCR product was electrophoresed on a 1% agarose gel under conditions that would display a band of the expected size.
Statistical analysis
Data were analyzed using the Microsoft Excel program. The ‘Student’s t-test assuming unequal variances’ was used to analyze significant differences between groups. P-value of <0.05 was considered to be statistically significant.
Acknowledgments
We would like to thank Kristen Newburn for assistance with blood draws and Dr Mark A Kay for providing mating pairs of FIX−/− mice. We are also very grateful to Alfonso Farruggio for help with plasmid constructions. This work was supported in part by NIH grant HL68012 to MPC. LEW was funded by PHS grant CA09302, awarded by the National Cancer Institute, DHHS. GH and PEM received support from the Hemophilia and Thrombosis Research Society and from Homecare for the Cure.
Footnotes
CONFLICT OF INTEREST
MPC is an inventor on Stanford-owned patents covering ϕC31 integrase.
References
- 1.Tuddenham E, Cooper D. The von Willebrand factor and von Willebrand disease. In: Tuddenham E, Cooper D, editors. The Molecular Genetics of Haemostasis and Its Inherited Disorders. Oxford University Press; Oxford: 1994. pp. 374–401. [Google Scholar]
- 2.Soucie JM, Evatt B, Jackson D. Occurrence of hemophilia in the United States. The Hemophilia Surveillance System Project Investigators. Am J Hematol. 1998;59:288–294. doi: 10.1002/(sici)1096-8652(199812)59:4<288::aid-ajh4>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 3.Lin Y, Chang L, Solovey A, Healey JF, Lollar P, Hebbel RP. Use of blood outgrowth endothelial cells for gene therapy for hemophilia A. Blood. 2002;99:457–462. doi: 10.1182/blood.v99.2.457. [DOI] [PubMed] [Google Scholar]
- 4.Lin HF, Maeda N, Smithies O, Straight DL, Stafford DW. A coagulation factor IX-deficient mouse model for human hemophilia B. Blood. 1997;90:3962–3966. [PubMed] [Google Scholar]
- 5.Jin DY, Zhang TP, Gui T, Stafford DW, Monahan PE. Creation of a mouse expressing defective human factor IX. Blood. 2004;104:1733–1739. doi: 10.1182/blood-2004-01-0138. [DOI] [PubMed] [Google Scholar]
- 6.Evans JP, Brinkhous KM, Brayer GD, Reisner HM, High KA. Canine hemophilia B resulting from a point mutation with unusual consequences. Proc Natl Acad Sci USA. 1989;86:10095–10099. doi: 10.1073/pnas.86.24.10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lozier JN, Metzger ME, Donahue RE, Morgan RA. The rhesus macaque as an animal model for hemophilia B gene therapy. Blood. 1999;93:1875–1881. [PubMed] [Google Scholar]
- 8.Herzog RW, Yang EY, Couto LB, Hagstrom JN, Elwell D, Fields PA, et al. Long-term correction of canine hemophilia B be gene transfer of blood coagulation factor IX mediated by adeno-associated viral vector. Nat Med. 1999;5:56–63. doi: 10.1038/4743. [DOI] [PubMed] [Google Scholar]
- 9.Murphy SL, High KA. Gene therapy for hemophilia. Br J Haematol. 2008;140:479–487. doi: 10.1111/j.1365-2141.2007.06942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Viiala NO, Larsen SR, Rasko JE. Gene therapy for hemophilia: clinical trials and technical tribulations. Semin Thromb Hemost. 2009;35:81–92. doi: 10.1055/s-0029-1214151. [DOI] [PubMed] [Google Scholar]
- 11.Mingozzi F, High KA. Immune responses to AAV in clinical trials. Curr Gene Ther. 2007;7:316–324. doi: 10.2174/156652307782151425. [DOI] [PubMed] [Google Scholar]
- 12.Roth DA, Tawa NE, Jr, O’Brien JM, Treco DA, Selden RF. Nonviral transfer of the gene encoding coagulation factor VIII in patients with severe hemophilia A. N Engl J Med. 2001;344:1735–1742. doi: 10.1056/NEJM200106073442301. [DOI] [PubMed] [Google Scholar]
- 13.Thorpe HM, Smith MC. In vitro site-specific integration of bacteriophage DNA catalyzed by a recombinase of the resolvase/invertase family. Proc Natl Acad Sci USA. 1998;95:5505–5510. doi: 10.1073/pnas.95.10.5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Groth AC, Olivares EC, Thyagarajan B, Calos MP. A phage integrase directs efficient site-specific integration in human cells. Proc Natl Acad Sci USA. 2000;97:5995–6000. doi: 10.1073/pnas.090527097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thyagarajan B, Olivares EC, Hollis RP, Ginsburg DS, Calos MP. Site-specific genomic integration in mammalian cells mediated by phage ϕC31 integrase. Mol Cell Biol. 2001;21:3926–3934. doi: 10.1128/MCB.21.12.3926-3934.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chalberg TW, Portlock JL, Olivares EC, Thyagarajan B, Kirby PJ, Hillman RT, et al. Integration specificity of phage ϕC31 integrase in the human genome. J Mol Biol. 2006;357:28–48. doi: 10.1016/j.jmb.2005.11.098. [DOI] [PubMed] [Google Scholar]
- 17.Woodard LE, Keravala A, Jung WE, Wapinski O, Yang Q, Felsher DW, et al. Evaluating the cancer risk of hydrodynamic injection and phiC31 integrase in a mouse model of MYC-induced hepatocellular carcinoma. PLoS ONE. 2010;5:e11367. doi: 10.1371/journal.pone.0011367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venken K, He Y, Hoskins RA, Bellen HJ. P[acman]: A BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science. 2006;314:1747–1751. doi: 10.1126/science.1134426. [DOI] [PubMed] [Google Scholar]
- 19.Calos MP. The phiC31 integrase system for gene therapy. Curr Gene Ther. 2006;6:633–645. doi: 10.2174/156652306779010642. [DOI] [PubMed] [Google Scholar]
- 20.Liu F, Song YK, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Therapy. 1999;6:1258–1266. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 21.Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum Gene Ther. 1999;10:1735–1737. doi: 10.1089/10430349950017734. [DOI] [PubMed] [Google Scholar]
- 22.Olivares EC, Hollis RP, Chalberg TW, Meuse L, Kay MA, Calos MP. Site-specific genomic integration produces therapeutic factor IX levels in mice. Nat Biotechnol. 2002;20:1124–1128. doi: 10.1038/nbt753. [DOI] [PubMed] [Google Scholar]
- 23.Keravala A, Lee S, Thyagarajan B, Olivares EC, Gabrovsky VE, Woodard LE, et al. Mutational derivatives of phiC31 integrase with increased efficiency and specificity. Mol Ther. 2009;17:112–120. doi: 10.1038/mt.2008.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma CH, Zhang Y, Wang XY, Gao LF, Liu H, Guo C, et al. Human endostatin gene transfer, either naked or with liposome, has the same inhibitory effect on growth of mouse liver tumor cells in vivo. World J Gastroenterol. 2004;10:2874–2877. doi: 10.3748/wjg.v10.i19.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miao CH, Ohashi K, Patijn GA, Meuse L, Ye X, Thompson AR, et al. Inclusion of the hepatic locus control region, an intron, and untranslated region increases and stabilizes hepatic factor IX gene expression in vivo but not in vitro. Mol Ther. 2000;1:522–532. doi: 10.1006/mthe.2000.0075. [DOI] [PubMed] [Google Scholar]
- 26.Nathwani AC, Gray JT, Ng CY, Zhou J, Spence Y, Waddington SN, et al. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood. 2006;107:2653–2661. doi: 10.1182/blood-2005-10-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Z, Sun J, Zhang T, Yin C, Yin F, Van Dyke T, et al. Optimization of self-complementary AAV vectors for liver-directed expression results in sustained correction of hemophilia B at low vector dose. Mol Ther. 2008;16:280–289. doi: 10.1038/sj.mt.6300355. [DOI] [PubMed] [Google Scholar]
- 28.Dejana E, Callioni A, Quintana A, de Gaetano G. Bleeding time in laboratory animals. II—a comparison of different assay conditions in rats. Thromb Res. 1979;15:191–197. doi: 10.1016/0049-3848(79)90064-1. [DOI] [PubMed] [Google Scholar]
- 29.Broze GJ, Jr, Yin ZF, Lasky N. A tail vein bleeding time model and delayed bleeding in hemophiliac mice. Thromb Haemost. 2001;85:747–748. [PubMed] [Google Scholar]
- 30.Gui T, Reheman A, Ni H, Gross PL, Yin F, Monroe D, et al. Abnormal hemostasis in a knock-in mouse carrying a variant of factor IX with impaired binding to collagen type IV. J Thromb Haemost. 2009;7:1843–1851. doi: 10.1111/j.1538-7836.2009.03545.x. [DOI] [PubMed] [Google Scholar]
- 31.Borchgrevink CF, Waaler BA. The secondary bleeding time; a new method for the differentiation of hemorrhagic diseases. Acta Med Scand. 1958;162:361–374. doi: 10.1111/j.0954-6820.1958.tb01782.x. [DOI] [PubMed] [Google Scholar]
- 32.Tomokiyo K, Nakatomi Y, Araki T, Teshima K, Nakano H, Nakagaki T, et al. A novel therapeutic approach combining human plasma-derived Factors VIIa and X for haemophiliacs with inhibitors: evidence of a higher thrombin generation rate in vitro and more sustained haemostatic activity in vivo than obtained with Factor VIIa alone. Vox San. 2003;85:290–299. doi: 10.1111/j.0042-9007.2003.00365.x. [DOI] [PubMed] [Google Scholar]
- 33.Chavez CL, Keravala AK, Woodard LE, Hillman RT, Stowe TR, Chu J, et al. Kinetics and longevity of phiC31 integrase in mouse liver and cultured cells. Hum Gene Ther. 2010;21:1287–1297. doi: 10.1089/hum.2010.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Held PK, Olivares EC, Aguilar CP, Finegold M, Calos MP, Grompe M. In vivo correction of murine hereditary tyrosinemia type I by phiC31 integrase-mediated gene delivery. Mol Ther. 2005;11:399–408. doi: 10.1016/j.ymthe.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 35.Kung SH, Hagstrom JN, Cass D, Tai SJ, Lin HF, Stafford DW, et al. Human factor IX corrects the bleeding diathesis of mice with hemophilia B. Blood. 1998;91:784–790. [PubMed] [Google Scholar]
- 36.Hedner U, Davie EW. Introduction to homeostasis and the vitamin K dependent coagulation factors. In: Scriver C, Beaudet AL, Sly WS, Valle D, editors. The Metabolic Basis of Inherited Diseases. Vol. 2. McGraw Hill; New York: 1989. pp. 2107–2134. [Google Scholar]
- 37.Alino SF, Herrero MJ, Noguera I, Dasi F, Sanchez M. Pig liver gene therapy by noninvasive interventionist catheterism. Gene Therapy. 2007;14:334–343. doi: 10.1038/sj.gt.3302873. [DOI] [PubMed] [Google Scholar]
- 38.Khorsandi SE, Bachellier P, Weber JC, Greget M, Jaeck D, Zacharoulis D, et al. Minimally invasive and selective hydrodynamic gene therapy of liver segments in the pig and human. Cancer Gene Ther. 2008;15:225–230. doi: 10.1038/sj.cgt.7701119. [DOI] [PubMed] [Google Scholar]
- 39.Suda T, Kamimura K, Kubota T, Tamura Y, Igarashi M, Kawai H, et al. Progress toward liver-based gene therapy. Hepatol Res. 2009;39:325–340. doi: 10.1111/j.1872-034X.2008.00479.x. [DOI] [PubMed] [Google Scholar]
- 40.Kanimura K, Suda T, Xu W, Zhang G, Liu D. Image-guided, lobe-specific hydrodynamic gene delivery to swine liver. Mol Ther. 2009;17:491–499. doi: 10.1038/mt.2008.294. [DOI] [PMC free article] [PubMed] [Google Scholar]