

Abstract
Background/Aim: According to the reverse Warburg effect, tumor cells may metabolize lactate as an energy source and shuttle L-lactate to neighboring cancer cells, adjacent stroma, and vascular endothelial cells, thus inducing metabolic reprogramming. An increased tumor L-lactate level strictly correlates with increased metastasis, tumor recurrence and a poor outcome. A potent anticancer agent that may act on L-lactate activated cells is 2-metoxyestradiol. Thus, the aim of the study was to evaluate whether a potent anticancer agent, 2-methoxyestradiol, is able to reverse L-lactate-induced metabolic reprogramming in osteosarcoma 143B cells. Materials and Methods: We used flow cytometry in order to determine cell death, autophagy, expression of KI-67, mitochondrial membrane depolarization. We performed cell proliferation assay in order to determine cell viability and cell migration assay to determine invasive potential of osteosarcoma cells. While, CalcuSyn software was used in order to evaluate the interaction between 2-methoxyestradiol and L-lactate. Results: We demonstrated that 2-methoxyestradiol abolished L-lactate-induced migration and proliferation of osteosarcoma cells. Moreover, we observed that this effect was associated with regulation of Ki-67 and induction of autophagy. Conclusion: 2-Methoxyestradiol is a potent anticancer agent also under metabolic reprogramming conditions.
Keywords: 2-Methoxyestradiol, L-lactate, osteosarcoma, tumor microenvironment
Cancer cells are traditionally characterized by increased glucose uptake, high rates of aerobic glycolysis, increased lactic acid production, impaired mitochondrial function and decreased extracellular pH. These characteristics are known as the ‘Warburg effect’ (1-3). The ‘Warburg effect’ is also observed in cancer-associated fibroblasts and mesenchymal stem cells (1-11), which secrete lactate and ketones into the microenvironment of the tumor. Lactic acid exists as L- and D- optical isomers. In mammals including humans, lactate is present almost entirely as L-lactate (12,13). Interestingly, a variety of human cancer cell lines and human tumors, including breast, prostate, head, neck, and osteosarcoma cancers, import these metabolites and deliver them to the mitochondrial TCA cycle, thereby promoting ATP generation and cell proliferation (1-14). According to this ‘reverse Warburg effect’, tumor cells can induce metabolic reprogramming by shuttling lactate as an energy source to neighboring cancer cells, to the adjacent stroma and to vascular endothelial cells (1,14).
Tumors contain both aerobic and hypoxic regions that form a metabolic symbiosis that is crucial for tumor cell survival (15,16). The cancer cells located in the hypoxic regions export lactate, which acidifies the tumor environment; the cells located in the aerobic regions, however, import lactate and utilize it for oxidative phosphorylation (4). Bonuccelli and colleagues (2) suggested that L-lactate metabolism in cancer cells may explain why diabetic patients have an increased risk of cancer development and a tendency towards autophagy/mitophagy in their adipose tissue. An increased tumor L-lactate level strictly correlates with increased metastasis, tumor recurrence and poor outcome (6-9).
L-lactate metabolism and the reverse Warburg effect has been clearly established in osteosarcoma cells. It has been demonstrated that the secretion of lactate and ketones by the mesenchymal stem cells drives mitochondrial biogenesis and increases the mitochondrial activity in osteosarcoma cells (1,3). Mesenchymal stem cells have been demonstrated to feed the osteosarcoma cells by producing and secreting L-lactate (1,3). Co-culture of osteosarcoma cells with mesenchymal stem cells increased the aggressive potential of tumor cells due to the generation of oxidative stress (1).
Here, we set out to determine the anticancer potential of 2-methoxyestradiol (2-ME) (17-30), a natural derivative of 17β-estradiol, in L-lactate-activated osteosarcoma cells. Under the brand name Panzem, 2-ME is currently being evaluated in preclinical studies and for the clinical treatment of numerous types of malignancies including breast, prostate cancer, or osteosarcoma (17-30). Recently, we showed that 2-ME stimulates the nuclear recruitment of neuronal nitric oxide synthase (nNOS) and cellular nitric oxide (NO) generation, which induces DNA damage and cancer cell death (19). We have also determined that 2-ME reverses the metabolic reprogramming that is induced by low concentrations of D-serine and glycine (17).
We demonstrated that 2-ME abolished the L-lactate-induced migration and proliferation of osteosarcoma cells. Both effects were correlated with low Ki-67 levels and high autophagy activities. Moreover, 2-ME effectively induced cell death in L-lactate-activated cells, which also indicated a potent anticancer activity under metabolic reprogramming conditions. Our data indicate that 2-ME acts both directly and indirectly on tumor cells by inducing tumor cell death through effects on the tumor microenvironment.
Materials and Methods
Reagents. 2-methoxyestradiol (No. M6383), L(+)Lactic acid sodium (L7022), the osteosarcoma 143B cell line (No. 91112502), tissue culture media, penicillin streptomycin solution, and fetal bovine sera were obtained from Sigma Aldrich (Warsaw, Poland). The Muse Autophagy LC3-antibody based kit, Ki-67 Proliferation Kit, and Cell Viability Assay were purchased from Merck (Warsaw, Poland).
Cell culture. The 143B osteosarcoma cells were cultured at 37˚C in a humidified atmosphere with 5% CO2. The EMEM (EBSS) medium was supplemented with 2 mM glutamine, 1% Non-Essential Amino Acids (NEAA), and 10% heat-inactivated Fetal Bovine Serum (FBS).
Cell treatment. To evaluate the effect of lactate on tumor, all the analyses were performed using sodium L-lactate (indicated in the text as L-lactate). This is the sodium salt of L(+)-acid lactic, which preserve lactate activity without affecting the pH of media. 143B cells were treated with 2-ME separately and in combination with L-lactate. Initially, the cells were treated for 24 h with 10 mM L-lactate to induce the reverse Warburg effect (1-3). After this, the medium was changed to those containing L-lactate or L-lactate and 2-ME (Figure 1). Cells treated with 2-ME alone were also used in the study (Figure 1). The treatments were administered according to the experimental design. The treatments were performed in EMEM containing 1% charcoal-stripped FBS (Sigma Aldrich, Poland). Charcoal-stripped FBS is used to elucidate the effects of hormones in various in vitro systems. Pyruvate and Lactate-free EMEM medium was chosen for cell culture and treatment due to its low glucose level, in order to avoid the effects of glucose and the Warburg effect in osteosarcoma cells.
Figure 1. Experimental design. Osteosarcoma 143B cells were first treated with L-lactate to induce the reverse Warburg effect. Afterword, the cells were continuously treated with L-lactate and/or 2-ME. Cells treated separately with 2-ME and control cells were also used.
Assessment of cell viability. Osteosarcoma 143B cells were seeded onto six-well plates at a density of 3×105 cells per well. After 24 h, the cells were incubated with 2-ME and/or L-lactate according to the experimental design. The cells were then pelleted and incubated with the Muse Count and Viability Reagent according to the manufacturer’s protocol (Merck Millipore, Warsaw, Poland) (21). After this, the cells were analyzed (5,000 events/sample). The signals were detected using the Muse Cell Analyzer (Merck Millipore, Warsaw, Poland), which is based on flow cytometric technology. The results were then analyzed with the Muse 1.4 analysis software. Each experiment was performed at least three times.
Cell proliferation assay. The proliferation assay was performed as previously described (14). Briefly, osteosarcoma cells were seeded in 96-well plates at a density of 8,000 cells/well. The cells were then treated with L-lactate or 2-ME separately and in combination. Twenty microliters of CellTiter 96 AQueous One Solution Reagent was added to each well of the 96-well assay plate containing the samples in 100 μl of culture medium. The plates were incubated at 37˚C for 4 h in a humidified 5% CO2 atmosphere. The absorbance was recorded at 490 nm using a 96-well plate reader. The results are presented as the percentage of that of the control cells (untreated cells). Each experiment was performed at least three times.
CalcuSyn software 2.11 (Biosoft). The analysis of interaction between L-lactate and 2-ME was performed as previously described (14) using a general equation for the dose-effect relationship. This equation shows the relationship between the Dose and the Effect in the simplest possible form:
fa/fu=(D/Dm)m
where:
D the dose of drug
Dm the median-effect dose signifying the potency (see below)
fa the fraction affected by the dose
fu the fraction unaffected, where fu=1-fa
m an exponent signifying the sigmoidicity (shape) of the dose-effect curve (see below)
The median-effect plot: A plot of x=log (D) vs y=log (fa/fu).
Dm value: The median-effect dose or concentration. log Dm is the x-intercept of the median-effect plot.
m value: A measurement of the sigmoidicity of the dose-effect curve; m=1, >1, and <1 indicate hyperbolic, sigmoidal, and negative sigmoidal shapes, respectively. Determined by the slope of the median-effect plot.
r value: The linear correlation coefficient of the median-effect plot.
Combination index (CI): A quantitative measure of the degree of drug interaction in terms of additive effect (CI=1), synergism (CI <1), or antagonism (CI >1) for a given endpoint of the effect measurement.
Cell migration assay kit (BioVendor). The cell migration chips were coated according to the manufacturer’s protocol. Next, the cells were loaded into the pre-filled chips at a density of 9×105 cell/ml in the appropriate medium containing 2-ME, L-lactate, or the combination. The chips were placed in a humid chamber and incubated at 37˚C with 5% CO2. The migration of cells was then observed. The post-migration cell morphology was determined by fixation with 10% formalin and staining the cells with crystal violet. The migration distances were observed using a phase contrast inverted microscope after 0, 6, 12, 24, and 48 h of incubation (magnification ×40, scale bar: 30 μm).
Ki-67 Proliferation assay. Osteosarcoma 143B cells were seeded into 6-well plates at a density of 3×105 cells/dish. After 24 h, the cells were incubated with 2-ME and/or L-lactate according to the experimental design. Afterwards, the cells were fixed, permeabilized and stained with an antibody to Human Ki-67-PE. Afterward, the cells were analyzed (5,000 events/sample), and the signals were detected using the Muse Cell Analyzer (Merck Millipore, Warsaw, Poland). The results were then analyzed with the Muse 1.4 analysis software. Each experiment was performed at least three times.
Mitochondrial membrane depolarization. Osteosarcoma 143B cells were seeded onto 10 cm culture dishes at a density of 2×106 cells/dish. After 24 h, the cells were incubated with 2-ME and/or L-lactate according to the experimental design. The cells were then pelleted and incubated for 20 min with the Muse™ MitoPotential Reagent (21) according to manufacturer’s protocol. After this, the cells were analyzed (5,000 events/sample), and the signals were detected using the Muse Cell Analyzer (Merck Millipore, Poland). The results were then analyzed with the Muse 1.4 analysis software. Each experiment was performed at least three times.
Autophagy LC3–antibody-based detection. Osteosarcoma 143B cells were seeded onto 10-cm culture dishes at a density of 4×104 cells/dish. After 24 h, the cells were incubated with 2-ME and/or L-lactate according to the experimental design. The cells were then pelleted and incubated with antibody detection reagents and stained with Anti-LC3 Alexa Fluor® 555, clone 4E12 according to manufacturer’s protocol. Afterward, the cells were analyzed (5,000 events/sample), and the signals were detected using the Muse Cell Analyzer (Merck Millipore, Poland). The results were then analyzed with the Muse 1.4 analysis software. Each experiment was performed at least three times.
Induction of cell death. The analysis was performed as previously described (19). Osteosarcoma 143B cells were seeded onto six-well plates at a density of 3×105 cells/well. After 24 h, the cells were incubated with 2-ME and/or L-lactate according to the experimental design. The cells were then pelleted and incubated with Annexin V and PI according to manufacturer’s protocol (BD Pharmingen, Poland). Afterwards, the cells (3×104/sample) were analyzed, and the fluorescent signals of the Annexin V conjugate and PI were detected at the fluorescence intensity channels FL1 and FL3 (BD FACScan). The results were then analyzed with Cyflogic software, version 1.2.1. Each experiment was performed at least three times.
Statistical analysis. The results represent the means±SD from at least three independent experiments. All microscopic evaluations were performed using randomized and coded slides. The differences between the control samples and the 2-ME-treated samples were evaluated using one-way analysis of variance (ANOVA) with post hoc testing with a Dunnett’s multiple comparison test or a T test combined with Wilcoxon test. A p-value of less than 0.01 was considered to indicate statistical significance. The data were imported and analyzed with GraphPad Prism (GraphPad Software v.6).
Results
Effect of glucose on treatment with L-lactate. First, to determine whether L-lactate affected the cell viability as a function of low (lg) or high glucose (hg) medium, we observed the impact of a 48 h treatment with 10 mM L-lactate on the viability of the osteosarcoma 143B cells (Figure 2A). As shown in this figure, we did not observe any significant difference in the cell viability between treatments with L-lactate in low glucose and high glucose medium. Thus, for the subsequent studies, we used the low glucose medium to limit the Warburg effect in the osteosarcoma cells.
Figure 2. Effects of L-lactate in low and high glucose medium on osteosarcoma cell viability (A). Effect of 2-ME, L-lactate and combination on proliferation of osteosarcoma 143B cells (B-D). The inhibition of osteosarcoma proliferation was determined using the MTS assay. The values are the means±SE of three independent experiments (N=6 replicate cultures). The absence of an error bar denotes a line thickness greater than the error. **p<0.001, ***p<0.0001, ****p<0.00001 versus control cells (C).

Cytotoxic effects of 2-ME and cytoprotective effect of L-lactate on osteosarcoma 143B cells. Our next goal was to evaluate impact of treatment with L-lactate alone and in combination with 2-ME on proliferation of osteosarcoma 143B cells using the MTS assay. In our experimental model, the osteosarcoma 143B cells were first treated for 24 h with various concentrations of L-lactate to increase the metastatic potential of cells (1,2). Then, to observe the anticancer effect of 2-ME, the media were changed to those containing L-lactate, or L-lactate and 2-ME at various concentrations, and the cells were incubated for 24 h longer. In addition, cells were treated with 2-ME alone for 24 h. The cytotoxicity was then determined, and the EC50 was calculated using CalcuSyn software (Figures 3 and 4).
Consistent with our previously published results (18-20), the EC50 for 2-ME was determined to be in the range of 1.29×10–6 M (Figure 2B). As shown in Figure 2C, L-lactate at the high concentrations of 446 mM and 223 mM decreased the osteosarcoma cell viability with a calculated EC50 value equal to 224.4 mM. In contrast, lower concentrations of L-lactate in the range of 29 mM to 1.7 mM resulted in pro-proliferative effects of the compound (Figure 2B). Importantly, 2-ME abrogated the pro-carcinogenic effect of L-lactate (Figure 2D). On the basis of these results and literature data (1,2), for the next studies we selected 10 mM L-lactate and a high pharmacological concentration of 2-ME: 10 μM.
Antagonistic effect between 2-ME and L-lactate calculated by CalcuSyn Software. Subsequently, we used CalcuSyn software to determine the combination index (CI) and evaluate the interaction between 2-ME and L-lactate (Figures 3 and 4). The interaction between compounds is presented in Figure 3 as a Median-effect plot (A), a Dose-effect curve (B), while in Figure 4, an Algebraic estimate (A), and a Fa-CI plot (B) (Methods section). The estimated CI value at EC50 was 24344 whereas the CI value at EC75 was 2, which suggested an antagonism between 2-ME and L-lactate (Figures 3 and 4, Table I).
Figure 3. Antagonistic effects between L-lactate and 2-ME. The data obtained from the MTS assay were analyzed using CalcuSyn software version 2.0 (Biosoft) and presented in the form of a Median-Effect Plot (A) and a Dose-Effect curve (B).
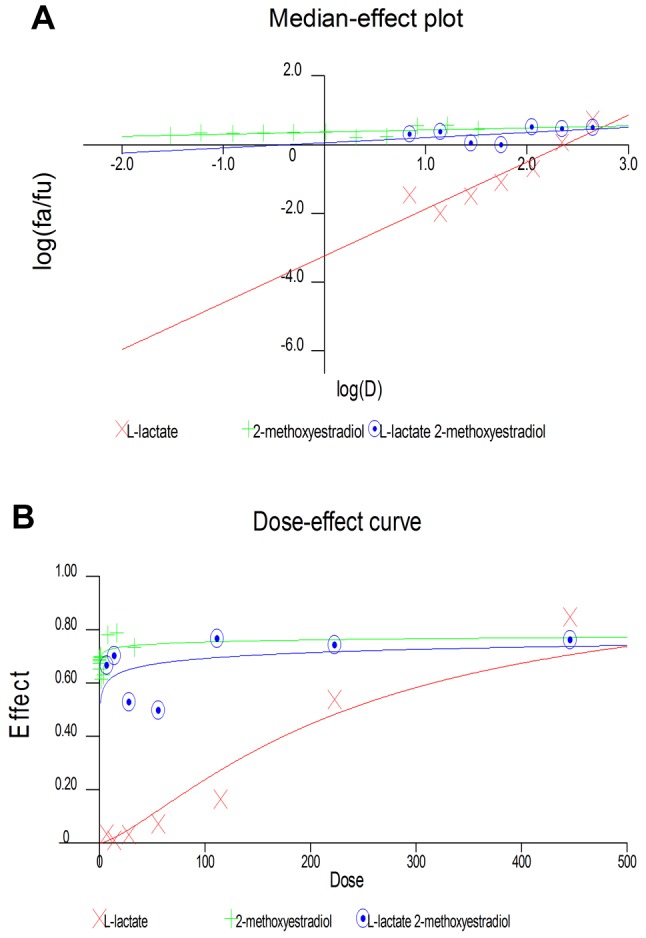
Figure 4. Antagonistic effects between L-lactate and 2-ME. The data obtained from the MTS assay were analyzed using CalcuSyn software version 2.0 (Biosoft) and presented in the form of an algebraic estimate (fractional effect) (A), and a Fa-CI plot (B).
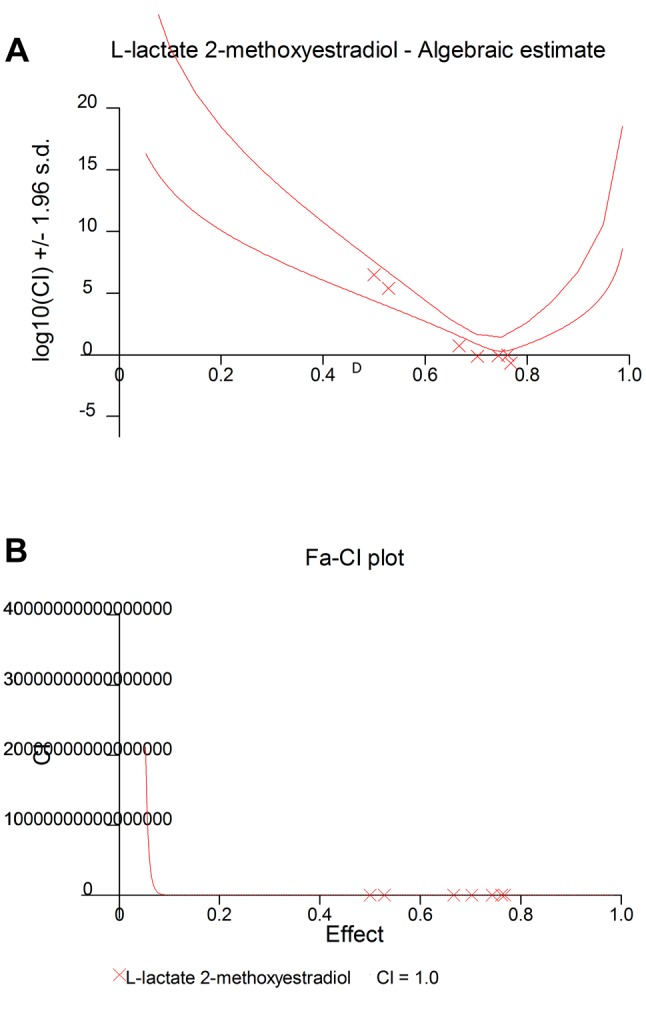
Table I. Antagonistic effects between 2-ME and L-lactate. The data in Table I include the following parameters: Dm value: The median-effect dose or concentration. It is usually represented by the ED50 or IC50. m value: A measurement of the sigmoidicity of the dose-effect curve; m=1, >1, and <1 indicates hyperbolic, sigmoidal, and negative sigmoidal shapes, respectively. Combination index (CI): A quantitative measure of the degree of drug interaction in terms of additive effect (CI=1), synergism (CI <1), or antagonism (CI >1) for a given endpoint of the effect measurement. The analysis was performed using CalcuSyn Software version 2.11 from Biosoft.

Effect of treatment with 2-ME and/or L-lactate on migration of osteosarcoma 143B cells. To determine the migratory potential of osteosarcoma cells that had been treated with 10 mM L-lactate, 10 μM 2-ME, or the combination, we performed a cell migration assay. As demonstrated in Figure 5A, the migration rate of the control cells was determined to be 7.7 μm/h. Moreover, we observed a dramatic increase to a migration rate of 18.6 μm/h for osteosarcoma cells after 48 h of treatment with 10 mM L-lactate (Figure 5A). In contrast, a 48-h treatment with 10 μM 2-ME reduced the migration rate for the osteosarcoma cells to 1.84 μm/h (Figure 5A). Importantly, the pro-migratory effect of L-lactate was significantly reduced to 2.6 μm/h by 10 μM 2-ME (Figure 5A).
Figure 5. Impact of 2-ME and L-lactate separately or in combination on the migratory potential of osteosarcoma cells (A), Ki-67-positive cells (B), and the induction of autophagy (C). The values are the means±SE of three independent experiments (N=6 replicate cultures). The absence of an error bar denotes a line thickness greater than the error. **p<0.001, ***p<0.0001, ****p<0.00001 versus control cells (C).
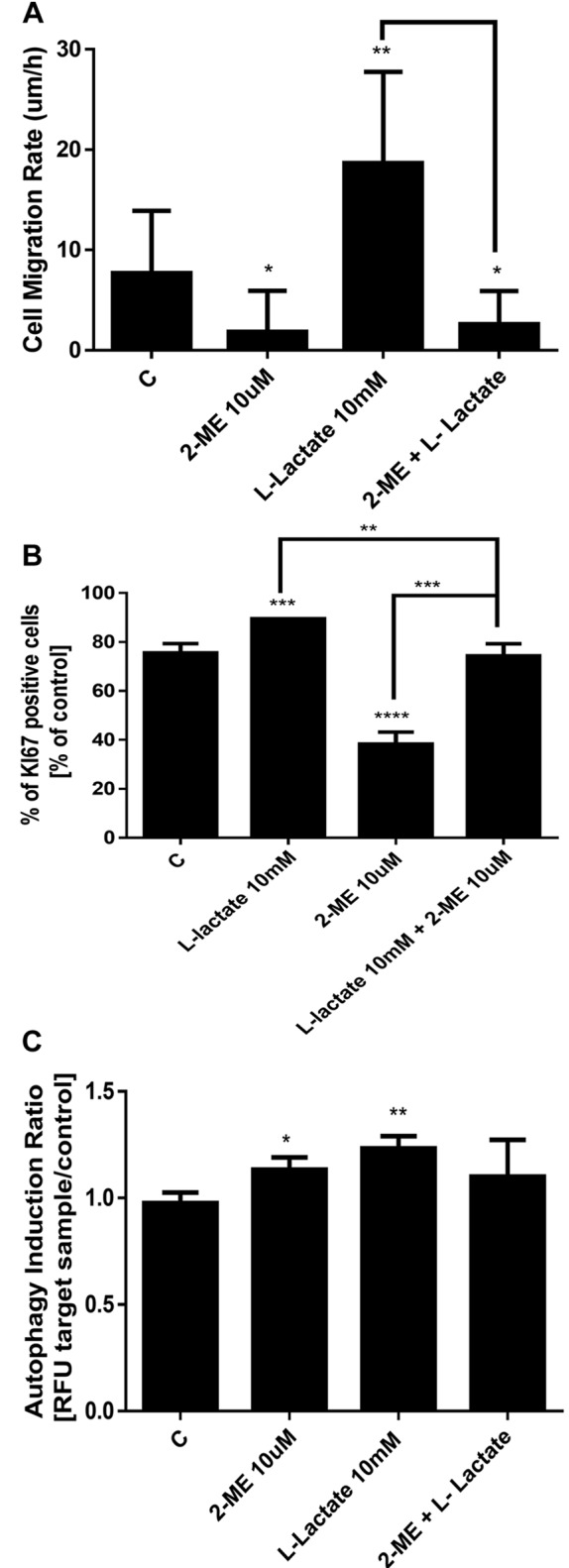
Impact of 2-ME and/or L-lactate on proliferation and expression of Ki-67 of osteosarcoma 143B cells. Because Ki-67 is an important marker for the efficiency of chemotherapy in osteosarcoma cells (31,32), we next measured the expression of Ki-67 after treatment with 2-ME and L-lactate separately and in combination. As in the previous experiments, in our experimental model osteosarcoma 143B cells were first treated for 24 h with 10 mM L-lactate to increase the metastatic potential of the cells (1,2). Subsequently, to observe the anticancer effect of 2-ME in L-lactate-activated cells, the medium was changed to those containing 10 mM L-lactate or the combination of 10 mM L-lactate and 10 μM 2-ME, and the cells were incubated for 24 h longer. In addition, cells were treated for 24 h with 10 μM 2-ME alone. As shown in Figure 5B, a 24-h treatment with 10 μM 2-ME significantly reduced the Ki-67 expression to 38% whereas that of the control cells was 75.5%. In contrast, a 48-h treatment with 10 mM L-lactate increased the Ki-67 expression to 89% (Figure 5B). Notably, the addition of 10 μM 2-ME to the L-lactate-activated cells resulted in a reversal of the L-lactate-induced Ki-67 expression down to 74% (Figure 5B).
Impact of treatment with 2-ME and/or L-lactate on induction of autophagy based on LC3 expression. Ki-67 expression is associated with LC3 expression especially early in the carcinogenesis process (33). The osteosarcoma cells were treated with 10 mM L-lactate alone for 24 h and subsequently treated for 24 h longer with L-lactate or the combination of L-lactate and 10 μM 2-ME. In addition, cells were treated for 24 h with 10 μM 2-ME. The cells were stained with the anti-LC3/Alexa Fluor 555-conjugated antibody and analyzed using the Muse Cell Analyzer (Merck Millipore, Warsaw, Poland). The obtained data are presented as the autophagy induction ratio, which was calculated as the ratio between the target sample fluorescence and that of the control sample. As shown in Figure 5C, a 24-h treatment with 10 μM 2-ME resulted in induction of autophagy in 12% of the cells (Figure 5C). A 48-h treatment with L-lactate resulted in an induction of autophagy in 23% of the cells (Figure 5C). In contrast, treatment with the combination of the agents did not have a significant impact on the induction of autophagy (Figure 5C).
Impact of treatment with 2-ME and/or L-lactate on mitochondrial membrane depolarization. Because one of the mechanisms for the anticancer activity of 2-ME is the induction of reactive nitrogen species (17-21), we next determined whether treatment with 2-ME and/or L-lactate affected the mitochondrial depolarization and further affected the induction of cell death. As in the previous experiments, the osteosarcoma 143B cells were first treated with L-lactate for 24 h, after which the cells were treated for an additional 24 h with 10 μM 2-ME and/or 10 mM L-lactate separately and in combination. As shown in Figure 6A, we observed depolarization of the mitochondrial membrane in 61% of the cells (Figure 6A) which is consistent with previously published data (18,21). L-lactate used alone slightly changed the mitochondrial membrane potential (19% of the total cells were depolarized) compared with control cells (9.7% of total depolarized cells) (Figure 6A). However, after the combined treatment with L-lactate and 2-ME, the mitochondrial membrane depolarization was significantly increased: up to 78% of the cells were depolarized compared to cells treated with L-lactate or 2-ME separately (Figure 6A).
Figure 6. Impact of 2-ME and L-lactate separately or in combination on the mitochondrial membrane depolarization (A), the induction of cell death (B, C). The values are the means±SE of three independent experiments (N=6 replicate cultures). The absence of an error bar denotes a line thickness greater than the error. **p<0.001, ***p<0.0001, ****p<0.00001 versus control cells (C).
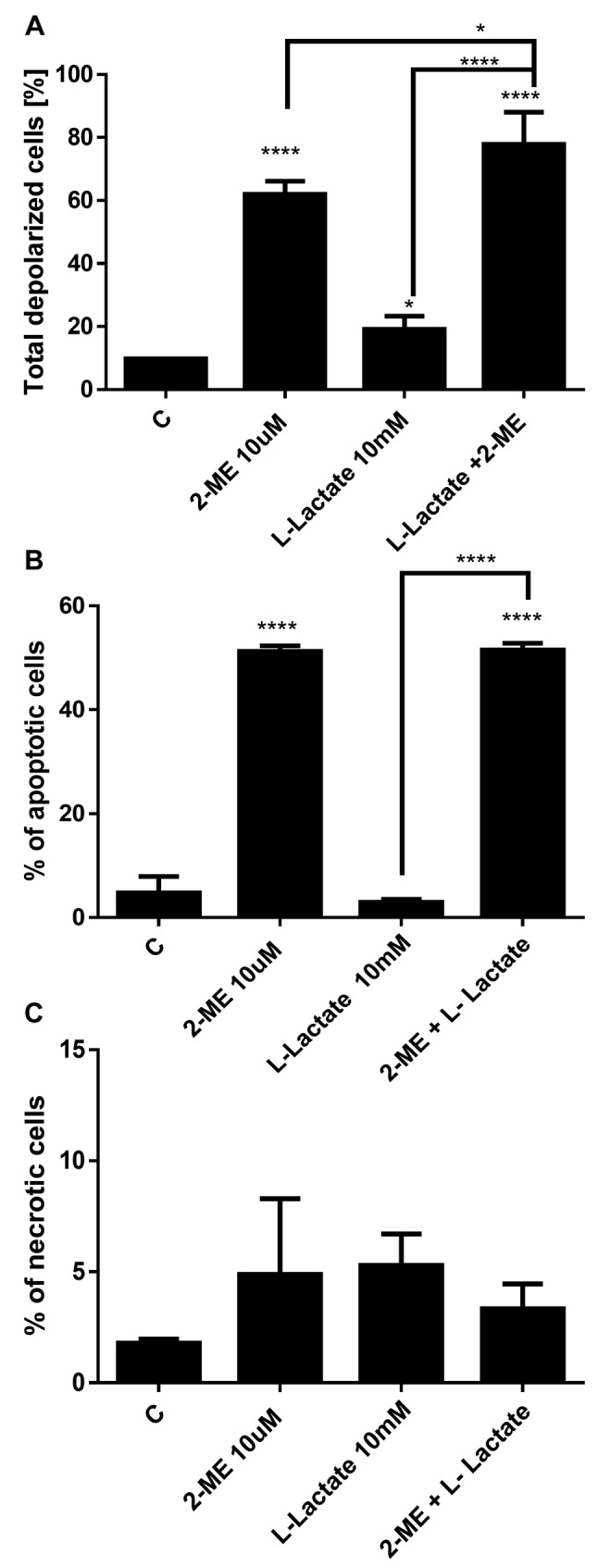
Impact of treatment with 2-ME and/or L-lactate on the induction of cell death. Based on the molecular crosstalk between the induction of autophagy and induction of cell death by 2-ME (34), the data were then compared with the induction of cell death. Consistent with previous studies (18), 10 μM 2-ME increased the number of apoptotic cells up to 51% compared with the control cells (4.7%) (Figure 6B). A 48-h treatment with 10 mM L-lactate did not affect apoptosis in the osteosarcoma cells. Importantly, a 24-h pretreatment and continued treatment of the cells with L-lactate did not affect the pro-apoptotic potential of 2-ME (Figure 5B). We did not observe any impact of the agents used separately or in combination on the induction of necrosis (Figure 6C).
Discussion
Mitochondrial function plays an important role in carcinogenesis and cancer dell death (35). In early studies, cancer cells were thought to metabolize glucose through aerobic glycolysis due to impaired mitochondrial function according to the Warburg theory (36). According to the Warburg effect, cancer cells convert glucose to lactic acid, even in the presence of oxygen. This results in lactate production as an end product of glycolysis. Currently, there is evidence that metabolic symbiosis between hypoxic and aerobic regions exists throughout the cancer microenvironment (37). Hypoxic cancer cells or cancer-associated fibroblasts produce lactate that is taken up by aerobic cells and fuels tumor growth in an effect termed the ‘reversed Warburg effect’ (1-3). This effect has especially been observed in breast cancer cells and osteosarcoma cells (1-3).
Only a few reports have considered the effect of 2-ME on mitochondrial function and the mitochondrial apoptotic pathway (18,21,38-41). An anticancer mechanism of 2-ME is associated with the induction of mitochondrial oxidative stress resulting in the induction of the mitochondrial apoptotic pathway in cancer cells (38,42). It was previously demonstrated that 2-ME decreased mitochondrial membrane potential due to generation of nitro-oxidative stress leading to cancer cell death (17-21,43). However, 2-ME also works as an inhibitor of mitochondrial respiration by inhibiting Complex I (39-41). Importantly, 2-ME-dependent disruption of Complex I was proposed as an alternative mechanism for its proapoptotic effects (39-41). Interestingly, though 2-ME is believed to be nontoxic towards normal cells (44), mitochondrial dysfunction induced by 2-ME was also observed in healthy cells e.g. normal lymphocytes or human embryonic kidney 293 cell line (41,45).
Bonucelli and coworkers demonstrated that L-lactate may play a causative role in breast cancer metastasis (2). Moreover, they observed an association between the altered levels of L-lactate and ketones as the end products of glycolysis in diabetic patients in whom there is an increased incidence of cancer development (2). L-lactate was demonstrated to increase mitochondrial biogenesis and oxidative phosphorylation, which facilitate the migration of cancer cells (1,2). Consistent with these observations, our results confirmed that treatment with 10 mM L-lactate increased the proliferation and migration of osteosarcoma 143B cells. Importantly, the pro-migratory and pro-proliferative effects of L-lactate were reversed by treatment with a pharmacological concentration of 2-ME. The anti-migratory potential of 2-ME has previously been demonstrated in various experimental models (17,23,24). Herein, for the first time, we demonstrated the antagonistic effect between 2-ME and L-lactate. Importantly, 2-ME was able to reverse the pro-proliferative and pro-migratory effects in L-lactate-activated osteosarcoma cells. We demonstrated that the anti-proliferative effect of 2-ME is directly associated with decreases in the expression of Ki-67, which is an important prognostic marker in osteosarcoma (31). An increased level of Ki-67 directly correlates with aggressiveness of tumors and their responses to chemotherapy (31,46). We determined that 2-ME may affect Ki-67 expression in osteosarcoma cells. It has been demonstrated that 2-ME has no antitumor effects on human endometrial carcinoma, in which it did not affect Ki-67 expression (47) whereas 2-ME decreases Ki-67 expression in breast cancer and hepatocellular carcinoma (48,49). Moreover, we demonstrated that treatment with L-lactate results in an increased expression of Ki-67. Previously, the expression of Ki-67 was positively correlated with activity of lactate dehydrogenase (50,51), but not directly correlated with level of L-lactate. Importantly, treatment with 2-ME reversed the L-lactate-induced increase in the expression of Ki-67. The decreased expression of Ki-67 that was induced by 2-ME was directly associated with the decreased mitochondrial membrane potential and induction of apoptosis in osteosarcoma cells. The induction of cell death and tumor growth inhibition that are associated with the abrogated expression of Ki-67 was previously demonstrated (52). The expression of Ki-67 was correlated with the level of LC3, a marker of autophagy (53). Autophagy, for which the formation of double-membrane bound organelles known as autophagosomes is a hallmark, is a lysosome-dependent pathway for protein degradation. Autophagy delivers cytoplasmic material and organelles to the lysosomes for degradation (54). The role of autophagy in carcinogenesis is context-dependent. It was demonstrated that the induction of autophagy is associated with DNA damage (55-57). Autophagy has been shown to regulate some of the DNA repair proteins after DNA damage (55-57). One the other hand, some evidence has demonstrated that some DNA repair molecular have a crucial role in the initiation of autophagy (58,59). The role of oxidative and nitrosative stress in autophagy has also been widely discussed (58). In our previous studies, we determined that one of anticancer mechanisms of actionof 2-ME is the induction of double- and single-strand breaks due to nuclear hijacking of neuronal nitric oxide synthase (18,19). 2-ME was also shown to induce the DNA damage response pathway (19). Moreover, it was suggested that a malfunction of the DNA damage repair system may result in chemoresistance of cancer cells to 2-ME and further progression of the cancer (19). Herein, we observed that the induction of autophagy by 2-ME was strictly correlated with the inhibition of proliferation and migration and the induction of apoptosis in osteosarcoma cells. The correlation of the induction of autophagy by 2-ME with the induction of cell death has previously been demonstrated in various experimental models including osteosarcoma cells (60,61). Moreover, it has been suggested the molecular crosstalk between 2-ME-induced apoptosis and autophagy is associated with its impact on microtubule integrity in cervical adenocarcinoma cells (62). On the other hand, the inhibition of autophagy increased the anticancer potential of 2-ME in chondrosarcoma cells (63). Thus, autophagy could play a dual role in cancer by facilitating either cell death or cell survival. Autophagy is considered to be a tumor-suppressing mechanism in early-stage carcinogenesis and may mediate the therapeutic effect of anticancer agents (54,55). However, autophagy may also act as a pro-survival mechanism to protect cancer cells from various forms of cellular stress (64). Indeed, the inhibition of autophagy was demonstrated to sensitize osteosarcoma cells to chemotherapeutic agents (64). In cancer therapy, adaptive autophagy in cancer cells sustains tumor growth and survival in the face of the toxicity of cancer therapy. We also determined that the L-lactate-induced autophagy in osteosarcoma cells was correlated with its pro-proliferative and pro-migratory effects, whereas the combined treatment with L-lactate and 2-ME resulted in decreased autophagy. This suggested that the autophagy induced by L-lactate leads to resistance of cancer cells to chemotherapy. Importantly, we also observed that the induction of apoptosis was comparable in the cells treated with 2-ME, or the combination of 2-ME and L-lactate.
Conclusion
All obtained results confirmed that 2-ME effectively induced tumor cell death, but also acted on the tumor micro-environment. We have demonstrated that 2-ME is an efficient anticancer agent in osteosarcoma cells that underwent L-lactate-induced metabolic reprogramming. Due to the obtained data we suggest that L-lactate is not only an intermediate metabolite but also a signaling molecule. Notably, the level of L-lactate may be a marker of the efficiency of anticancer therapies. High Lactate levels are strictly associated with a poor prognosis and increased metastatic potential of the tumors (6,7,12). The poor prognosis associated with an increased level of L-lactate may be strictly associated with its impact on pro-angiogenetic factors and the induction of mitochondrial biogenesis as well as a direct impact on cancer cell migration and proliferation (1-3). Thus, the level of L-lactate may serve as a marker of the efficiency of anticancer therapies. Moreover, regulation of L-lactate level in the tumor microenvironment seems to be a new tool in the arsenal of weapons against cancer.
We are currently evaluating a plausible role for 2-ME in regulation of the mitochondrial biogenesis pathway. Undoubtedly, the role of 2-ME in regulation of bioenergetics and mitochondrial function is very interesting and remains to be elucidated.
Funding
The studies and manuscript publication were supported by the Iuventus Plus Programme from of the Polish Ministry of Science and Higher Education No IP 2015 022074.
Conflicts of Interest
The Authors declare no conflicts of interest.
Acknowledgements
The Authors thank Merck and especially Dr. Barbara Piotrowska for providing the Muse Cell Analyzer for the experiments and for her helpful advice.
References
- 1.Bonuccelli G, Avnet S, Grisendi G, Salerno M, Granchi D, Dominici M, Kusuzaki K, Baldini N. Role of mesenchymal stem cells in osteosarcoma and metabolic reprogramming of tumor cells. Oncotarget. 2014;15(17):7575–7588. doi: 10.18632/oncotarget.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, Frank PG, Flomenberg N, Howell A, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Ketones and lactate "fuel" tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010;9(17):3506–3514. doi: 10.4161/cc.9.17.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sotgia F, Martinez-Outschoorn UE, Lisanti MP. The reverse Warburg effect in osteosarcoma. Oncotarget. 2014;5(18):7982–7983. doi: 10.18632/oncotarget.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Semenza GL. Tumor metabolism: cancer cells give and take lactate. J Clin Invest. 2008;118(12):3835–3837. doi: 10.1172/JCI37373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodwin ML, Gladden LB, Nijsten MW, Jones KB. Lactate and cancer: revisiting the Warburg effect in an era of lactate shuttling. Front Nutr. 2015;1:27. doi: 10.3389/fnut.2014.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brizel DM, Schroeder T, Scher RL, Walenta S, Clough RW, Dewhirst MW, Mueller-Klieser W. Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int J Radiat Oncol Biol Phys. 2001;51(2):349–353. doi: 10.1016/s0360-3016(01)01630-3. [DOI] [PubMed] [Google Scholar]
- 7.Cori CF, Cori HT. The carbohydrate metabolism of tumors II. Changes in the sugar, lactic acid, and CO2-combioning power of blood passing through a tumor. J Biol Chem. 1925;65:397–405. [Google Scholar]
- 8.Warburg O, Winf F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walenta S, Salameh A, Lyng H, Evensen JF, Mitze M, Rofstad EK, Mueller-Klieser W. Correlation of high lactate levels in head and neck tumors with incidence of metastasis. Am J Pathol. 1997;150(2):409–415. [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou W, Liotta LA, Petricoin EF. The Warburg effect and mass spectrometry-based proteomic analysis. Cancer Genomics Proteomics. 2017;14(4):211–218. doi: 10.21873/cgp.20032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou W, Liotta LA, Petricoin EF. Cancer metabolism: what we can learn from proteomic analysis by mass spectrometry. Cancer Genomics Proteomics. 2012;9(6):373–381. [PMC free article] [PubMed] [Google Scholar]
- 12.Romero-Garcia S, Moreno-Altamirano MM, Prado-Garcia H, Sánchez-García FJ. Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front Immunol. 2016;7:52. doi: 10.3389/fimmu.2016.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vassiliou AG, Mastora Z, Jahaj E, Koutsoukou A, Orfanos SE, Kotanidou A. Does serum lactate combined with soluble endothelial selectins at ICU admission predict sepsis development. In Vivo. 2015;29(2):305–308. [PubMed] [Google Scholar]
- 14.Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8(23):3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 15.Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors. a comprehensive review. Front Pharmacol. 2011;2:49. doi: 10.3389/fphar.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Romero-Garcia S, Lopez-Gonzalez JS, Báez-Viveros JL, Aguilar-Cazares D, Prado-Garcia H. Tumor cell metabolism. an integral view. Cancer Biol Ther. 2011;12(11):939–948. doi: 10.4161/cbt.12.11.18140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorska-Ponikowska M, Perricone U, Kuban-Jankowska A, Lo Bosco G, Barone G. 2-methoxyestradiol impacts on amino acids-mediated metabolic reprogramming in osteosarcoma cells by interaction with NMDA receptor. J Cell Physiol. 2017;232(11):3030–3049. doi: 10.1002/jcp.25888. [DOI] [PubMed] [Google Scholar]
- 18.Gorska M, Wyszkowska RM, Kuban-Jankowska A, Wozniak M. Impact of Apparent Antagonism of estrogen receptor β by fulvestrant on anticancer activity of 2-methoxyestradiol. Anticancer Res. 2016;36(5):2217–2226. [PubMed] [Google Scholar]
- 19.Gorska M, Kuban-Jankowska A, Zmijewski M, Marino Gammazza A, Cappello F, Wnuk M, Gorzynik M, Rzeszutek I, Daca A, Lewinska A, Wozniak M. DNA strand breaks induced by nuclear hijacking of neuronal NOS as an anti-cancer effect of 2-methoxyestradiol. Oncotarget. 2015;6(17):15449–15463. doi: 10.18632/oncotarget.3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorska M, Kuban-Jankowska A, Zmijewski MA, Gorzynik M, Szkatula M, Wozniak M. Neuronal nitric oxide synthase induction in the antitumorigenic and neurotoxic effects of 2-methoxyestradiol. Molecules. 2014;28;19(9):13267–13267-. doi: 10.3390/molecules190913267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gorska M, Kuban-Jankowska A, Milczarek R, Wozniak M. Nitro-oxidative stress is involved in anticancer activity of 17β-estradiol derivative in neuroblastoma cells. Anticancer Res. 2016;36(4):1693–1698. [PubMed] [Google Scholar]
- 22.Kumar BS, Raghuvanshi DS, Hasanain M, Alam S, Sarkar J, Mitra K, Khan F, Negi AS. Recent advances in chemistry and pharmacology of 2-methoxyestradiol: An anticancer investigational drug. Steroids. 2016;110:9–34. doi: 10.1016/j.steroids.2016.03.017. [DOI] [PubMed] [Google Scholar]
- 23.Wu SL, Li UJ, Liao K, Shi L, Zhang N, Liu S, Hu YY, Li SL, Wang Y. 2-methoxyestradiol inhibits the proliferation and migration and reduces the radioresistance of nasopharyngeal carcinoma CNE-2 stem cells via NF-ĸB/HIF-1 signaling pathway inactivation and EMT reversal. Oncol Rep. 2016;37(2):793–802. doi: 10.3892/or.2016.5319. [DOI] [PubMed] [Google Scholar]
- 24.Sattler M, Quinnan LR, Pride YB, Gramlich JL, Chu SC, Even GC, Kraeft S, Chen LB, Salgia R. 2-methoxyestradiol alters cell motility, migration, and adhesion. Blood. 2003;102:289–296. doi: 10.1182/blood-2002-03-0729. [DOI] [PubMed] [Google Scholar]
- 25.Fotopoulou C, Baumunk D, Schmidt SC, Schumacher G. Additive growth inhibition after combined treatment of 2-methoxyestradiol and conventional chemotherapeutic agents in human pancreatic cancer cells. Anticancer Res. 2011;30(11):4619–4624. [PubMed] [Google Scholar]
- 26.Foster PA, Newman SP, Leese MP, Bernetiere S, Diolez C, Camara J, Hacher B, Baronnet MM, Ali T, Potter BV, Reed MJ, Purohit A. A new micronized formulation of 2-methoxyestradiol-bis-sulfamate (STX140) is therapeutically potent against breast cancer. Anticancer Res. 2008;28(2A):577–581. [PubMed] [Google Scholar]
- 27.Li L, Heldin NE, Grawé J, Ulmsten U, Fu X. Induction of apoptosis or necrosis in human endometrial carcinoma cells by 2-methoxyestradiol. Anticancer Res. 2004;24(6):3983–3990. [PubMed] [Google Scholar]
- 28.Li L, Bu S, Bäckström T, Landström M, Ulmsten U, Fu X. Induction of apoptosis and G2/M arrest by 2-methoxyestradiol in human cervical cancer HeLaS3 cells. Anticancer Res. 2004;24(2B):873–880. [PubMed] [Google Scholar]
- 29.Banerjeei SK, Zoubine MN, Sarkar DK, Weston AP, Shah JH, Campbell DR. 2-methoxyestradiol blocks estrogen-induced rat pituitary tumor growth and tumor angiogenesis: possible role of vascular endothelial growth factor. Anticancer Res. 2000;20(4):2641–2645. [PubMed] [Google Scholar]
- 30.Fujii H, Honoki K, Tsujiuchi T, Kido A, Yoshitani K, Takakura Y. Growth inhibition and induction of apoptosis by 2-methoxyestradiol in rat osteosarcoma and malignant fibrous histiocytoma cell lines. In Vivo. 2008;22(1):21–25. [PubMed] [Google Scholar]
- 31.Scotlandi K, Serra M, Manara MC, Maurici D, Benini S, Nini G, Campanacci M, Baldini N. Clinical relevance of Ki-67 expression in bone tumors. Cancer. 1995;75(3):806–814. doi: 10.1002/1097-0142(19950201)75:3<806::aid-cncr2820750310>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 32.Robl B, Pauli C, Botter SM, Bode-Lesniewska B, Fuchs B. Prognostic value of tumor suppressors in osteosarcoma before and after neoadjuvant chemotherapy. BMC Cancer. 2015;15:379. doi: 10.1186/s12885-015-1397-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshioka A, Miyata H, Doki Y, Yamasaki M, Sohma I, Gotoh K, Takiguchi S, Fujiwara Y, Uchiyama Y, Monden M. LC3, an autophagosome marker, is highly expressed in gastrointestinal cancers. Int J Oncol. 2008;33(3):461–468. [PubMed] [Google Scholar]
- 34.Theron AE, Nolte EM, Lafanechère L, Joubert AM. Molecular crosstalk between apoptosis and autophagy induced by a novel 2-methoxyestradiol analogue in cervical adenocarcinoma cells. Cancer Cell Int. 2013;13(1):87. doi: 10.1186/1475-2867-13-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanaka T, Kobunai T, Yamamoto Y, Murono K, Otani K, Yasuda K, Nishikawa T, Kiyomatsu T, Kawai K, Hata K, Nozawa H, Ishihara S, Watanabe T. Increased copy number variation of mtDNA in an array-based digital PCR assay predicts ulcerative colitis-associated colorectal cancer. In Vivo. 2017;31(4):713–718. doi: 10.21873/invivo.11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slobodan D. Warburg effect – a consequence or the cause of carcinogenesis. J Cancer. 2016;7(7):817–822. doi: 10.7150/jca.14274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, Wahl ML, Feron O, Dewhirst MW. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118(12):3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee ST, Lee JY, Han CR, Kim YH, Jun do Y, Taub D, Kim YH. Dependency of 2-methoxyestradiol-induced mitochondrial apoptosis on mitotic spindle network impairment and prometa-phase arrest in human Jurkat T cells. Biochem Pharmacol. 2015;94(4):257–269. doi: 10.1016/j.bcp.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 39.Felty Q, Roy D. Estrogen, mitochondria, and growth of cancer and non-cancer cells. J Carcinog. 2005;4(1):1. doi: 10.1186/1477-3163-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chua YS, Chua YL, Hagen T. Structure activity analysis of 2-methoxyestradiol analogues reveals targeting of microtubules as the major mechanism of antiproliferative and proapoptotic activity. Mol Cancer Ther. 2010;9(1):224–235. doi: 10.1158/1535-7163.MCT-09-1003. [DOI] [PubMed] [Google Scholar]
- 41.Hagen T, D'Amico G, Quintero M, Palacios-Callender M, Hollis V, Lam F, Moncada S. Inhibition of mitochondrial respiration by the anticancer agent 2-methoxyestradiol. Biochem Biophys Res Commun. 2004;322(3):923–929. doi: 10.1016/j.bbrc.2004.07.204. [DOI] [PubMed] [Google Scholar]
- 42.Ting CM, Lee YM, Wong CK, Wong AS, Lung HL, Lung ML, Lo KW, Wong RN, Mak NK. 2-Methoxyestradiol induces endoreduplication through the induction of mitochondrial oxidative stress and the activation of MAPK signaling pathways. Biochem Pharmacol. 2010;79(6):825–841. doi: 10.1016/j.bcp.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 43.Chang I, Majid S, Saini S, Zaman MS, Yamamura S, Chiyomaru T, Shahryari V, Fukuhara S, Deng G, Dahiya R, Tanaka Y. Hrk mediates 2-methoxyestradiol-induced mitochondrial apoptotic signaling in prostate cancer cells. Mol Cancer Ther. 2013;12(6):1049–1059. doi: 10.1158/1535-7163.MCT-12-1187. [DOI] [PubMed] [Google Scholar]
- 44.Gorska M, Kuban-Jankowska A, Slawek J, Wozniak M. New insight into 2-methoxyestradiol- a possible physiological link between neurodegeneration and cancer cell death. Curr Med Chem. 2016;23(15):1513–1527. doi: 10.2174/0929867323666160316123443. [DOI] [PubMed] [Google Scholar]
- 45.Georgieva E, Zhelev Z, Aoki I, Bakalova R, Higashi T. Detection of redox imbalance in normal lymphocytes with induced mitochondrial dysfunction – EPR study. Anticancer Res. 2016;36(10):5273–5279. doi: 10.21873/anticanres.11098. [DOI] [PubMed] [Google Scholar]
- 46.Jong R, Davis AM, Mendes MG, Wunder JS, Bell RS, Kandel R. Proliferative activity (Ki-67 expression) and outcome in high grade osteosarcoma: A study of 27 cases. Sarcoma. 2000;4(1-2):47–55. doi: 10.1155/S1357714X00000086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li L, Yu F, Wu X, Cheng J, Ulmsten U, Fu X. Effects of 2-methoxyestradiol on endometrial carcinoma xenografts. J Cancer Res Clin Oncol. 2007;133(5):315–320. doi: 10.1007/s00432-006-0173-x. [DOI] [PubMed] [Google Scholar]
- 48.Du B, Wang SY, Shi XF, Zhang CF, Zhang ZZ. The effect of 2-methoxyestradiol liposome on growth inhibition, angiogenesis and expression of VEGF and Ki-67 in mice bearing H22 hepatocellular carcinoma. Tumori. 2011;97(5):660–665. doi: 10.1177/030089161109700520. [DOI] [PubMed] [Google Scholar]
- 49.Azab SS, Salama SA, Hassan MH, Khalifa AE, El-Demerdash E, Fouad H, Al-Hendy A, Abdel-Naim AB. 2-methoxyestradiol reverses doxorubicin resistance in human breast tumor xenograft. Cancer Chemother Pharmacol. 2008;62(5):893–902. doi: 10.1007/s00280-008-0679-9. [DOI] [PubMed] [Google Scholar]
- 50.Himani B, Meera S, Abhimanyu S, Usha R. Ki-67 Immunostaining and its correlation with microvessel density in patients with mutiple myeloma. Asian Pac J Cancer Prev. 2016;17(5):2559–2564. [PubMed] [Google Scholar]
- 51.Alexandrakis MG, Passam FH, Kyriakou DS, Dambaki K, Niniraki M, Stathopoulos E. Ki-67 proliferation index: correlation with prognostic parameters and outcome in multiple myeloma. Am J Clin Oncol. 2004;27(1):8–13. doi: 10.1097/01.coc.0000045810.91816.41. [DOI] [PubMed] [Google Scholar]
- 52.Kausch I, Jiang H, Ewerdwalbesloh N, Doehn C, Krüger S, Sczakiel G, Jocham D. Inhibition of Ki-67 in a renal cell carcinoma severe combined immunodeficiency disease mouse model is associated with induction of apoptosis and tumour growth inhibition. BJU Int. 2005;95(3):416–420. doi: 10.1111/j.1464-410X.2005.05312.x. [DOI] [PubMed] [Google Scholar]
- 53.Wu WK, Coffelt SB, Cho CH, Wang XJ, Lee CW, Chan FK, Yu J, Sung JJ. The autophagic paradox in cancer therapy. Oncogene. 2012;31(8):939–953. doi: 10.1038/onc.2011.295. [DOI] [PubMed] [Google Scholar]
- 54.Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793(4):664–7344. doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 55.Zhang D, Tang B, Xie X, Xiao XF, Yang SM, Zhang JW. The interplay between DNA repair and autophagy in cancer therapy. Cancer Biol Ther. 2015;16(7):1005–1013. doi: 10.1080/15384047.2015.1046022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eliopoulos AG, Havaki S, Gorgoulis VG. DNA damage response and autophagy: a meaningful partnership. Front Genet. 2016;7:204. doi: 10.3389/fgene.2016.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eapen VV, Waterman DP, Bernard A, Schiffmann N, Sayas E, Kamber R, Lemos B, Memisoglu G, Ang J, Mazella A, Chuartzman SG, Loewith RJ, Schuldiner M, Denic V, Klionsky DJ, Haber JE. A pathway of targeted autophagy is induced by DNA damage in budding yeast. Proc Natl Acad Sci USA. 2017;114(7):E1158–E1167. doi: 10.1073/pnas.1614364114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22(3):377–388. doi: 10.1038/cdd.2014.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793(4):664–673. doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 60.Bravo D, Shogren KL, Zuo D, Wagner ER, Sarkar G, Yaszemski MJ, Maran A. 2-methoxyestradiol-mediated Induction of Frzb contributes to cell death and autophagy in MG63 osteosarcoma cells. J Cell Biochem. 2017;118(6):1497–1504. doi: 10.1002/jcb.25809. [DOI] [PubMed] [Google Scholar]
- 61.Visagie MH, Joubert AM. 2-methoxyestradiol-bissulphamate refrains from inducing apoptosis and autophagy in a non-tumorigenic breast cell line. Cancer Cell Int. 2012;12(1):37. doi: 10.1186/1475-2867-12-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Theron AE1, Nolte EM, Lafanechère L, Joubert AM. Molecular crosstalk between apoptosis and autophagy induced by a novel 2-methoxyestradiol analogue in cervical adenocarcinoma cells. Cancer Cell Int. 2013;13(1):87. doi: 10.1186/1475-2867-13-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reumann S, Shogren KL, Yaszemski MJ, Maran A. Inhibition of autophagy increases 2-methoxyestradiol-induced cytotoxicity in SW1353 chondrosarcoma cells. J Cell Biochem. 2016;117(3):751–759. doi: 10.1002/jcb.25360. [DOI] [PubMed] [Google Scholar]
- 64.Shimizu T, Sugihara E, Yamaguchi-Iwai S, Tamaki S, Koyama Y, Kamel W, Ueki A, Ishikawa T, Chiyoda T, Osuka S, Onishi N, Ikeda H, Kamei J8, Matsuo K, Fukuchi Y, Nagai T, Toguchida J, Toyama Y, Muto A, Saya H. IGF2 preserves osteosarcoma cell survival by creating an autophagic state of dormancy that protects cells against chemotherapeutic stress. Cancer Res. 2014;74(22):6531–6541. doi: 10.1158/0008-5472.CAN-14-0914. [DOI] [PubMed] [Google Scholar]