

Abstract
Purpose of review
The purpose of the review is to highlight developments in autoinflammatory diseases associated with gain-of-function mutations in the gene encoding NLR-family CARD domain-containing protein 4 (NLRC4), the NLRC4-inflammasomopathies.
Recent findings
Three years since the identification of the first autoinflammation with infantile enterocolitis (AIFEC) patients, there is an improved understanding of how the NLRC4 inflammasome and interleukin 18 (IL-18) contribute to gut inflammation in myeloid and also intestinal epithelial cells. This information has opened new therapeutic avenues to treat AIFEC patients with targeted agents like recombinant IL-18 binding protein and anti-interferon-γ antibodies. Additional phenotypes traditionally associated with NLRP3 mutations like familial cold autoinflammatory syndrome and neonatal onset multisystem inflammatory disease (NOMID), have now also been associated with gain-of-function NLRC4 mutations. Finally, NLRC4 somatic mosaicism has now been identified in a NOMID and an AIFEC patient, a finding emphasizing nontraditional modes of inheritance in autoinflammatory diseases.
Summary
The NLRC4 inflammasomopathies comprise a growing autoinflammatory disease category that spans a broad clinical spectrum from cold urticaria to NOMID and the often-fatal disease AIFEC. Rapid case identification with biomarkers like elevated serum IL-18 concentrations and early intervention with targeted immunomodulatory therapies are key strategies to improving outcomes for AIFEC patients.
Keywords: NLRC4, AIFEC, NOMID, FCAS4, IL-18
Introduction
An inflammasome is a pro-inflammatory cytoplasmic structure formed upon detection of exogenous pathogen-associated or danger-associated molecular patterns (PAMPs/DAMPs). The inflammasomopathies describe a group of mechanistically-related diseases, each the result of inappropriate inflammasome activation. Inflammasomopathies can be protean disorders; for instance, activating mutations in the gene encoding NLR family pyrin domain containing 3 (NLRP3) may present with cold urticaria, or progressive hearing loss or neonatal onset multisystem inflammatory disease (NOMID) (1).
In 2014, two groups independently identified gain-of-function mutations in the gene encoding NLR-family CARD domain-containing protein 4 (NLRC4) in four patients displaying recurrent, life-threating episodes of autoinflammation and infantile enterocolitis (AIFEC) (2,3). Since that time, the identification of additional AIFEC patients and their successful treatment with targeted therapies have revealed important insights into AIFEC pathophysiology. Also, new reports have described additional clinical phenotypes associated with novel germline or somatic NLRC4 mutations. Herein, we summarize the growing body of literature describing NLRC4 mutation-associated autoinflammatory diseases, the NLRC4 inflammasomopathies.
NLRC4 inflammasome biology
There are several canonical inflammasomes, and each is organized similarly: cytosolic PAMP/DAMP detectors are linked via an adaptor protein, apoptosis-associated speck-like protein containing a CARD (ASC), to the cleaved, active form of pro-caspase-1 (4). Upon activation, the inflammasome rapidly forms a large wheel-shaped structure (5), exhausting cellular ASC stores. Inflammasome formation initiates pyroptosis, a form of inflammatory cell death (6) and also proteolytically activates pro-interleukin 1 family cytokines (IL-1β and IL-18) into their cleaved, active forms (7–9).
Inflammasome identity and specificity are determined by eponymous detector proteins which include absent in melanoma 2 (AIM2)(10), NLRP3 (11) and NLRC4 (12). Like the NLRP3 inflammasome, which responds to numerous cytosolic DAMPs/PAMPs (13–15), the human NLRC4 inflammasome recognizes at least two bacterial ligands, flagellin and the type three secretion system (T3SS) (16). NLRC4 is distinct from NLRP3 and AIM2 because it does not directly interact with its ligands. Instead, NLRC4 is activated via contact with the sensor protein NLR family of apoptosis inhibitory protein (NAIP), and it is NAIP that physically binds either flagellin or a T3SS (17). This arrangement suggests NLRC4 might be better categorized as a scaffolding protein rather than a PAMP detector, although NLRC4 might also be considered an adaptor. Unlike NLRP3 and AIM2, NLRC4 contains a CARD allowing it to directly contact pro-caspase-1 without ASC (18). Notably, absent ASC, the NLRC4 inflammasome is functionally altered favoring pyroptosis over cytokine production (3,16).
NLRC4 inflammasome biology has primarily been studied in myeloid cells including circulating monocytes and neutrophils, but since NLRC4 detects components of lung and gut-trophic pathogens, its behavior in mucosal tissues is also of vital interest. Recently, a specialized host defense role of the NLRC4 inflammasome was identified in mouse intestinal epithelial cells (IECs). Upon detection of Salmonella within IEC cytoplasm, the NLRC4 inflammasome rapidly forms producing IL-18 and diarrhea-causing eicosanoids (19). Instead of pyroptosis, Salmonella containing IECs undergo IL-18 independent, caspase dependent, non-lytic cell death with subsequent expulsion into the colonic lumen (20). Although this adaptation produces secretory diarrhea, vascular leak and shock, it likely prevents catastrophic, invasive bacterial infections.
NLRC4 inflammasome initiation is exquisitely sensitive; a single ligand-bound NAIP molecule is sufficient to propagate NLRC4 oligomerization (21), yet since systemic inflammation impacts host survival, the process is highly regulated. One level of regulation occurs intrinsically through the autoinhibitory structure of the NLRC4 molecule (22). NLRC4 consists of a caspase activation and recruitment domains (CARD), a ligand binding/NAIP interacting leucine rich repeat (LRR) and a regulatory nucleotide-binding oligomerization domain (NOD) (Fig 1) (23). Within the NOD, helical domain 1 (HD1), winged helix domain (WHD) and helical domain 2 (HD2) form a specialized adenosine diphosphate (ADP) binding pocket that stabilizes NLRC4 in its inactive conformation (12,22,24). Upon LRR detection of ligand-bound NAIP, the NOD undergoes a conformational change that promotes ADP for adenosine triphosphate (ATP) exchange, NLRC4 oligomerization and inflammasome assembly (22). A second regulatory layer controls cytokine production and is inflammasome extrinsic, as production of inactive pro-IL-1 family cytokines requires signals from cell surface PAMP/DAMP receptors including tumor necrosis factor (TNF) receptor 1 and toll-like receptor 4 (TLR4) (25). Physiologically inert, these pro-cytokines accumulate intracellularly until an inflammasome cleaves them into immunologically active compounds.
Figure 1.
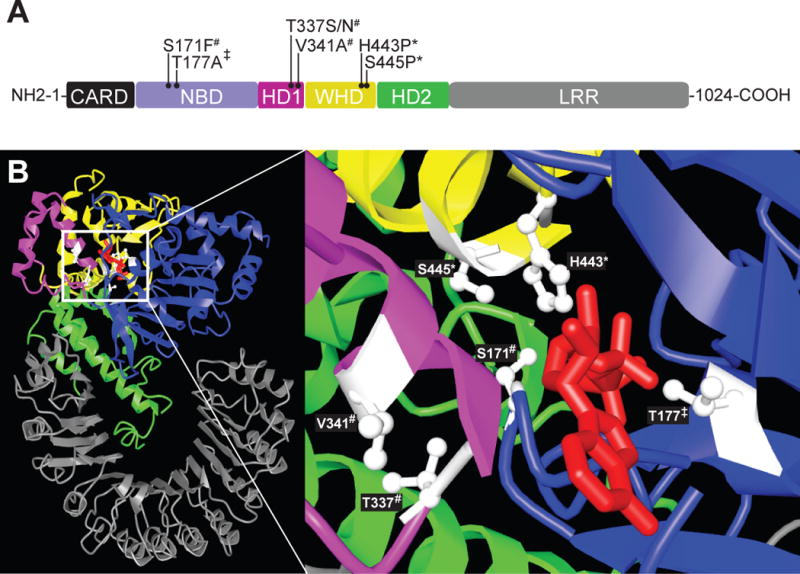
Disease-associated NLRC4 variants cluster near the ADP-binding site. A) The functional domains of NLRC4 are depicted and color coded: CARD domain black; NBD=purple; HD1=pink; WHD=yellow; HD2=green; LRR=gray. Variants associated with predominantly skin phenotypes are marked by *, the NOMID-associated variant is marked by ‡, and AIFEC mutations by #. B) A color coded NLRC4 crystal structure without a CARD is displayed per Ref. 22. ADP (red) is depicted within its binding pocket (inset, right). Disease associated residues (white) are labeled.
There are eleven IL-1 cytokine family members; only two, IL-1β and IL-18, are synthesized as inactive precursors (25). IL-1β mediates the classic signs and symptoms of the febrile response. The IL-1 receptor binds either IL-1β or its endogenously produced competitive inhibitor, IL-1 receptor agonist (IL-1RA) (26,27). Canonically, IL-18 amplifies lymphocyte functions, increasing cytotoxicity through IFNγ production. Circulating IL-18 is sequestered by an endogenous binding protein (IL-18BP), preventing receptor binding (25). The full effects of IL-18 are incompletely understood, but recent work suggests IL-18 equilibrium controls intestinal barrier function in mice and that excessive free IL-18 potentiates colitis (28).
AIFEC
In 2014, two investigative groups simultaneously and independently reported gain-of-function NLRC4 mutations in four patients from two unrelated families (2,3). The disease, called by one group NLRC4 macrophage activation syndrome (NLRC4-MAS) and by the other syndrome of enterocolitis and autoinflammation associated with mutation of NLRC4 (SCAN4), was later designated autoinflammation with infantile enterocolitis (AIFEC) by Online Mendelian Inheritance in Man (OMIM, #616050, *606831) (29). Because the syndromes originally described by each team were remarkably similar and shared a common cause, we will use the umbrella term AIFEC in this review to avoid confusion and build consensus.
AIFEC is a chronic inflammatory disease punctuated by episodes of extreme acuity. All seven reported AIFEC patients have required hospitalizations in intensive care units during inflammatory episodes. Two infantile cases have been fatal. AIFEC flares have been appropriately compared to MAS; both entities share signature IL-1β symptomology (fever, tachycardia) and interferon gamma (IFNγ)-related histopathology (hemophagocytosis). Peripheral blood transcriptional profiles generated from two AIFEC patients in flare suggest MAS-like myeloid cell activation and cytotoxic T-cell dysfunction (2,30). In its most extreme form, an AIFEC flare can be confused with primary hemophagocytic lymphohistiocytosis (HLH), with hypertriglyeridemia, coagulopathy, multi-lineage cytopenias, elevated soluble IL-2 receptor and poor in vitro cytotoxicity (2,3,30,31,32). Importantly, cytotoxic function normalizes in AIFEC patients after flares resolve, indicating intact granule-dependent cytotoxicity machinery. Another feature and useful biomarker distinguishing AIFEC from primary HLH is extremely elevated serum IL-18 concentrations (>104 pg/ml). IL-18 elevations of this degree are described in only a small cadre of inflammatory diseases including AIFEC, x-linked inhibitor of apoptosis (XIAP) deficiency and systemic juvenile idiopathic arthritis/adult-onset Stills disease-related MAS (33,34).
The trigger for AIFEC flares is unknown. At least two older AIFEC patients experienced flares with physical and emotional stressors (3). Another possible trigger could be activation of the mutant NLRC4 inflammasome with flagellin or T3SS expressing bacteria. In vitro, AIFEC macrophages infected with T3SS-expressing Salmonella or Pseudomonas form numerous altered assemblies of inflammasome components that favor pyroptosis over cytokine production (3). Nonetheless, such bacteria have not yet been recovered from flaring AIFEC patients.
One area where MAS and AIFEC do not overlap is gastrointestinal pathology. Diarrhea is an uncommon MAS symptom whereas severe, secretory, neonatal diarrhea is the most common AIFEC presentation. In one case, there is evidence AIFEC enterocolitis began in utero (31). Intestinal biopsies/autopsy specimens from all reported AIFEC patients consistently show a mixed inflammatory infiltrate, villous flattening with tissue edema, epithelial erosions and tissue autolysis (2,3,30,35). Activated macrophages have been visualized in some (31) but not all AIFEC intestinal tissue samples (2,3,30), raising the possibility that mutant NLRC4 in IECs, not myeloid cells, is the primary driver of gut pathology. Due to feeding intolerance, many AIFEC patients temporarily require parenteral nutrition, including the oldest known AIFEC patient, now 46 years old. His course was first described by Harry Shwachman M.D. in a 1973 case series of 16 infants with undifferentiated, protracted diarrhea supported by this nutritive method (36). For this and other surviving AIFEC patients, symptomatic enterocolitis spontaneously normalized after the first year of life even though serum IL-18 concentrations remain persistently elevated into adulthood.
In addition to enterocolitis and MAS-like episodes, AIFEC patients can develop numerous organ-specific symptoms secondary to lymphohistocytic inflammation. For instance, ephemeral maculopapular and urticarial rashes associated with lymphohistiocytic infiltrates can occur (data not published). Astute clinicians may elect to biopsy and stain skin lesions in suspected AIFEC cases for rapid diagnostic information, as this histologic finding is unusual. Similarly, central nervous system dysfunction (obtundation, seizures) and hepatic dysfunction correlate with activated macrophages in related tissues (3,31).
AIFEC is caused by heterozygous NLRC4 gain-of-function mutations, which exhibit complete penetrance. There are now four published AIFEC-associated NLRC4 mutations: V341A, T337S, T337N and S171F (Table 1 and Fig 1). The V341A variant is described in four individuals from two unrelated pedigrees, while the remaining variants occurred in single cases. Interestingly, the AIFEC patient harboring the S171F variant was mosaic for this allele. Although the variant accounted for only a quarter of NLRC4 peripheral blood transcripts, the patient’s in utero disease onset and fatal outcome suggest it to be highly pathogenic. Amino acid positions 337 and 341 are both located in the autoinhibitory HD-1 subdomain of NOD. NLRC4 crystal structure analysis suggests hydrophobic residues at position 341 are important for closing the “lid” on the ADP binding pocket to prevent ADP/ATP exchange (3,22). Similarly, position 337 may stabilize the pocket’s tertiary structure through interactions with residues 170 and/or 173 (2,22). Presumably, substitution of phenylalanine for serine at position 171 alters these interactions (31).
Table I.
Patients reported with NLRC4 mutation-associated diseases
| variant | number of patients | origin of mutation | age of onset | phenotype | disease-related mortality | reference |
|---|---|---|---|---|---|---|
| p.S171F | 1 | somatic mosaicism | in utero | AIFEC | deceased at 2 months | 31 |
| p.T177A | 1 | somatic mosaicism | birth | NOMID | alive at 19 years | 37 |
| p.T337S | 1 | de novo | 6 months | AIFEC | alive at 7 years | 2 |
| p.T337N | 1 | de novo | 3 weeks | AIFEC | alive at 8 months | 32 |
| p.V341A | 1 | de novo | 6 weeks | AIFEC | alive at 1 year | 30 |
| p.V341A | 3 in 1 family | inherited | 1-2 weeks | AIFEC | deceased at 23 days (proband)/alive at 7 and 46 years | 3 |
| p.H443P | 13 in 1 family | inherited | 2-3 months | FCAS4 | alive into adulthood | 38 |
| p.S445P | 13 in 1 family | inherited | infancy to childhood | FCAS-like | alive into adulthood | 39 |
AIFEC, autoinflammation with infantile enterocolitis; CAPS, cyropyrin-associated periodic syndromes; NOMID, neonatal onset multisystem inflammatory disease; FCAS, familial cold autoinflammatory syndrome
Although infantile diarrhea and MAS-like episodes are the signature features of AIFEC, it is a chronic inflammatory disease. Patients surviving infancy are of short stature and exhibit anemia of chronic disease (3). Between flares, untreated AIFEC patients display moderately elevated acute phase reactants and highly elevated serum IL-18 concentrations (2,3). Amyloidosis, a long-term complication of many untreated autoinflammatory disorders, has not yet been described in AIFEC patients but could be a concern. Longitudinal observation of surviving AIFEC pediatric cases and identification of more adult AIFEC cases may be informative.
Extended NLRC4 phenotypes
Shortly after publication of the original AIFEC cases, 13 members of a Japanese pedigree were reported with a syndrome of neonatal-onset fever, cold-induced urticarial rash and arthralgias (38). A heterozygous NLRC4 H443P variant segregated with disease. Symptoms were sufficiently mild that most affected members did not require treatment. Serum IL-18 concentrations were not assessed, but patient cells exposed to cold in vitro spontaneously produced IL-1β. Given the clear phenotypic similarities to the NLRP3-associated familial cold autoinflammatory syndrome (FCAS1), OMIM designated this disease as FCAS4 (#616115; *606831) (29). In 2017, a second large Dutch kindred was reported with prominent skin manifestations (39). A heterozygous S445P NLRC4 variant segregated with disease. Like FCAS4 patients, these patients developed inflammatory symptoms early in life that did not significantly alter long-term survival; the eldest affected member was age 88 years. Uniquely, in this kindred most patients developed conjunctivitis/uveitis, and many had a nodular, or urticarial rash. Although affected individuals lacked MAS or infantile enterocolitis, two developed intestinal inflammation in adulthood. Functional studies of the S445P variant were not conducted, but many individuals displayed highly elevated serum IL-18 concentrations. Unlike FCAS1 patients whose biopsied skin lesions are characteristically neutrophilic (40), skin infiltrates in Dutch patients’ biopsies were lympho-histiocytic as seen in AIFEC (39).
In addition to traditional patterns of disease inheritance, two patients have been described with NLRC4 somatic mosaicism. One, discussed above, presented with prenatal AIFEC (31), the other presented with cardinal NOMID features (fever, rash, inflammatory bone lesions, sensory neural hearing loss and structural brain defects) but he was NLPR3-mutation negative and he displayed chronically elevated serum IL-18 (37). Functional analysis of patient pluripotent stem cell-derived monocytes revealed two distinct populations, one with aberrant and the other with normal IL-1β secretion. The aberrant IL-1β producing cells harbored a novel T177A NLRC4 variant. Notably, this mutation was initially missed on whole exome sequencing analysis.
It is unclear how gain-of-function mutations in the same gene can produce such disparate clinical phenotypes, but a genotype/phenotype relationship may exist. The FCAS phenotype appears to correlate with WHD subdomain variants (H443P and S445P), whereas AIFEC and NOMID associate with HD-1 and NBD subdomain variants (S171F, T177A, T337S, T337N and V341A) (Fig 1). Recent functional work suggests pathologic NLRC4 variants in WHD may differentially promote caspase 8-mediated cell death whereas variants in HD-1 do not (41).
Treatment of NLRC4 inflammasomopathies
Although all NLRC4 inflamasomopathies are categorically autoinflammatory disorders, they manifest across a broad severity spectrum. Without a NLRC4-specific therapy, treating physicians have chosen to target downstream inflammatory mediators based upon their patients’ clinical needs. For instance, many patients with FCAS4 were well controlled with only non-steroidal anti-inflammatory drugs or nothing at all (39). Other non-AIFEC patients were treated with recombinant IL-1RA (anakinra) and although skin manifestations were completely responsive, other disease features were not. Anakinra was also highly effective in treating NOMID-like symptoms due to NLRC4 mutation (37). Used prophylactically, anakinra reduced the severity and frequency of MAS episodes more effectively than low-dose steroids and colchicine in one AIFEC patient with mild gastrointestinal disease (2), but was not efficacious treating another AIFEC patient in flare (30).
The most therapeutically challenging AIFEC manifestations are MAS and enterocolitis. Sadly, NLRC4 mutations were found posthumously in several patients with overwhelming neonatal AIFEC, and their aggressive presentation and rapid progression clouded assessment of treatment efficacy (3,31). MAS episodes were managed successfully with corticosteroids, cyclosporine and IVIg in at least one adult patient prior to his AIFEC diagnosis (3). Notably, this same patient has experienced prolonged periods of treatment-free quiescence between life-threatening MAS flares.
Several agents specifically targeting deranged AIFEC immunologic pathways are under development. For example, we observed dramatic efficacy using recombinant IL-18BP in a critically ill neonatal AIFEC patient whose disease was refractory to combined corticosteroids, cyclosporine, IL-1 inhibition, TNF-inhibition and integrin-inhibition (30). The infant’s clinical improvement corresponded with a precipitous drop in free but not total IL-18 emphasizing the importance of endogenous IL-18BP in humans. This case prompted broader evaluation of IL-18BP in an ongoing clinical trial (NCT03113760). Blockade of IFNγ was also efficacious in one AIFEC flare (32).
A growing literature links the intestinal ecosystem with NLRC4 activation and IL-18 production (19,20,28,42). As such, early gut colonization may promote excessive IL-1 family cytokine production in AIFEC patients. Likewise, spontaneous resolution of AIFEC enterocolitis may coincide with maturation of the gut, mucosal immunity and/or the adoption of a less inflammatory microbiota (42). Without more data, we cannot recommend either intestinal decontamination or fecal transplantation in AIFEC patients. Similarly, because NLRC4 is expressed in both myeloid and intestinal epithelia cells, we would not anticipate hematopoietic stem cell transplantation by itself or intestinal transplantation by itself, would be curative.
Conclusion
The NLRC4 inflammasomopathies comprise a growing category of autoinflammatory diseases that span a broad clinical spectrum from cold urticaria to NOMID and the often-fatal disease AIFEC. Since 2014, 34 patients have been reported with NLRC4 gain-of-function mutation-associated diseases. Of these, most are from two unrelated families with NLRC4 variants that do not appear to confer a significant survival disadvantage.
There are seven published AIFEC patients; two were fatal cases and all experienced significant morbidity due, in part, to diagnostic delay. Even now that it has been established as a distinct clinical entity, AIFEC continues to pose a diagnostic challenge because most cases are sporadic. Accordingly, early recognition of telltale AIFEC symptoms and a rapid diagnosis, using disease biomarkers like serum IL-18 concentrations, characteristic skin biopsy findings and ultimately gene sequencing, will be paramount to improving disease outcomes. Likewise, the timely implementation of anti-inflammatory therapies, either already approved (anakinra, steroids, IVIg, cyclosporine) or available through ongoing clinical trials (recombinant IL-18BP; NCT03113760 or anti-IFNγ monoclonal antibodies; NCT02069899), will continue to be a key determinate of survival.
Key points.
The NLRC4 inflammasomopathies are caused by gain-of-function mutations in the gene encoding the innate immune protein NLRC4. Mutant NLRC4 promotes spontaneous formation of the NLRC4 inflammasome, production of IL1 family cytokines (IL-1β and IL-18) and inflammatory cell death.
Three NLRC4 inflammasomopathy phenotypes have been described: AIFEC, NOMID and FCAS4. AIFEC is associated with very early onset enterocolitis, macrophage activation syndrome and neonatal death.
Rapid case identification using disease biomarkers like elevated serum IL-18 concentrations, characteristic skin biopsy findings and ultimately diagnostic gene sequencing, combined with timely application of immune modulatory therapy are key strategies to improving AIFEC patient outcomes.
Acknowledgments
None
Financial support
The authors would like to thank their funding sources including the Jeffrey Modell Foundation (N.R.) and the RK Mellon Foundation (S.W.C).
Footnotes
Conflicts of interest
S.W.C. is a paid consultant for AB2Bio, Ltd and T.P.V. attended their clinical trial investigator meeting. For the remaining author, no conflicts were declared.
References and recommended reading
* of special interest
** of outstanding interest
- 1.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle–Wells syndrome. Nat Genet. 2001 Nov;29(3):301. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. 2014 Oct;46(10):1140–6. doi: 10.1038/ng.3089. One of two initial descriptions of AIFEC. The report details the genetic cause, molecular mechanism, clinical manifestations, and biologic signature of a patient with recurrent MAS and mild enterocolitis. It also describes the patient’s clinical response to anakinra. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Romberg N, Al Moussawi K, Nelson-Williams C, Stiegler AL, Loring E, Choi M, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet. 2014 Oct;46(10):1135–9. doi: 10.1038/ng.3066. One of two initial descriptions of AIFEC. The report details the genetic cause, molecular mechanism, clinical manifestations, and biologic signature of three patients with MAS, two of these patients experienced severe infantile enterocolitis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407–20. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 5.Cheng J, Waite AL, Tkaczyk ER, Ke K, Richards N, Hunt AJ, et al. Kinetic properties of ASC protein aggregation in epithelial cells. J Cell Physiol. 2010 Mar;222(3):738–47. doi: 10.1002/jcp.22005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol. 2000 Oct;38(1):31–40. doi: 10.1046/j.1365-2958.2000.02103.x. [DOI] [PubMed] [Google Scholar]
- 7.Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. 1997 Apr 10;386(6625):619–23. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- 8.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell. 1995 Feb 10;80(3):401–11. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 9.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995 Mar 31;267(5206):2000–3. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 10.Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009 Mar 26;458(7237):509–13. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004 Mar;20(3):319–25. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 12.Poyet JL, Srinivasula SM, Tnani M, Razmara M, Fernandes-Alnemri T, Alnemri ES. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J Biol Chem. 2001 Jul 27;276(30):28309–13. doi: 10.1074/jbc.C100250200. [DOI] [PubMed] [Google Scholar]
- 13.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006 Mar 9;440(7081):228–32. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 14.Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006 Mar 9;440(7081):237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 15.Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A. 2008 Jul 1;105(26):9035–40. doi: 10.1073/pnas.0803933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. 2007 Dec 24;204(13):3235–45. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011 Aug 28;477(7366):592–5. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004 Jul 8;430(6996):213–8. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 19**.Rauch I, Deets KA, Ji DX, von Moltke J, Tenthorey JL, Lee AY, et al. NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and -8. Immunity. 2017 Apr 18;46(4):649–59. doi: 10.1016/j.immuni.2017.03.016. This report emphasizes a unique role for the NLRC4 inflammasome in intestinal epithelial cells, both for preventing invasive bacterial infections and for driving intestinal and systemic immunopathology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sellin ME, Müller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A, et al. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe. 2014 Aug 13;16(2):237–48. doi: 10.1016/j.chom.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Hu Z, Zhou Q, Zhang C, Fan S, Cheng W, Zhao Y, et al. Structural and biochemical basis for induced self-propagation of NLRC4. Science. 2015 Oct 23;350(6259):399–404. doi: 10.1126/science.aac5489. [DOI] [PubMed] [Google Scholar]
- 22.Hu Z, Yan C, Liu P, Huang Z, Ma R, Zhang C, et al. Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science. 2013 Jul 12;341(6142):172–5. doi: 10.1126/science.1236381. NLR-proteins have been notoriously resistant to structural analysis. These authors solved the murine Nlrc4 crystal structure and relate the molecule’s structural elements to its ATPase activity and method of autoinhibition. [DOI] [PubMed] [Google Scholar]
- 23.Damiano JS, Stehlik C, Pio F, Godzik A, Reed JC. CLAN, a novel human CED-4-like gene. Genomics. 2001 Jul;75(1–3):77–83. doi: 10.1006/geno.2001.6579. [DOI] [PubMed] [Google Scholar]
- 24.Danot O, Marquenet E, Vidal-Ingigliardi D, Richet E. Wheel of Life, Wheel of Death: A Mechanistic Insight into Signaling by STAND Proteins. Struct Lond Engl 1993. 2009 Feb 13;17(2):172–82. doi: 10.1016/j.str.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 25.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 26.Carter DB, Deibel MR, Dunn CJ, Tomich CS, Laborde AL, Slightom JL, et al. Purification, cloning, expression and biological characterization of an interleukin-1 receptor antagonist protein. Nature. 1990 Apr 12;344(6267):633–8. doi: 10.1038/344633a0. [DOI] [PubMed] [Google Scholar]
- 27.Sims JE, Acres RB, Grubin CE, McMahan CJ, Wignall JM, March CJ, et al. Cloning the interleukin 1 receptor from human T cells. Proc Natl Acad Sci U S A. 1989 Nov;86(22):8946–50. doi: 10.1073/pnas.86.22.8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28**.Nowarski R, Jackson R, Gagliani N, de Zoete MR, Palm NW, Bailis W, et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell. 2015 Dec 3;163(6):1444–56. doi: 10.1016/j.cell.2015.10.072. This report establishes the role of free IL-18 and IL-18BP deficiency in murine intestinal inflammation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University; Baltimore, MD: MIM Number: 616050: 05/16/2017: World Wide Web URL: https://omim.org/ [Google Scholar]
- 30**.Canna SW, Girard C, Malle L, deJesus A, Romberg N, Kelsen J, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol. 2017 May 1;139(5):1698–701. doi: 10.1016/j.jaci.2016.10.022. This report is the first description of recombinant IL-18BP as a treatment for AIFEC, highlighting the utility of free IL-18 and differences between intestinal and systemic IL-18 regulation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang J, Alfano DN, Squires JE, Riley MM, Parks WT, Kofler J, et al. Novel NLRC4 Mutation Causes a Syndrome of Perinatal Autoinflammation With Hemophagocytic Lymphohistiocytosis, Hepatosplenomegaly, Fetal Thrombotic Vasculopathy, and Congenital Anemia and Ascites. Pediatr Dev Pathol Off J Soc Pediatr Pathol Paediatr Pathol Soc. 2017 Jan 1; doi: 10.1177/1093526616686890. This report describes a fatal AIFEC case, an infant with somatic mosaicism for a novel NLRC4 variant. The authors report systemic inflammation likely commenced in utero. [DOI] [PubMed] [Google Scholar]
- 32*.Bracaglia C, Gatto A, Pardeo M, Lapeyre G, Ferlin W, Nelson R, et al. Anti interferon-gamma (IFNγ) monoclonal antibody treatment in a patient carrying an NLRC4 mutation and severe hemophagocytic lymphohistiocytosis. Pediatr Rheumatol. 2015 Sep 28;13(1):O68. This abstract describes AIFEC in an infant with a novel NLRC4 variant. It also is the first description of treating AIFEC with anti-IFNγ monoclonal antibodies. [Google Scholar]
- 33.Maeno N, Takei S, Nomura Y, Imanaka H, Hokonohara M, Miyata K. Highly elevated serum levels of interleukin-18 in systemic juvenile idiopathic arthritis but not in other juvenile idiopathic arthritis subtypes or in Kawasaki disease: Comment on the article by Kawashima et al. Arthritis Rheum. 2002 Sep 1;46(9):2539–41. doi: 10.1002/art.10389. [DOI] [PubMed] [Google Scholar]
- 34.Wada T, Kanegane H, Ohta K, Katoh F, Imamura T, Nakazawa Y, et al. Sustained elevation of serum interleukin-18 and its association with hemophagocytic lymphohistiocytosis in XIAP deficiency. Cytokine. 2014 Jan;65(1):74–8. doi: 10.1016/j.cyto.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 35.Shwachman H, Lloyd-Still JD, Khaw K-T, Antonowicz I. Protracted Diarrhea of Infancy Treated by Intravenous Alimentation: II. Studies of Small Intestinal Biopsy Results. Am J Dis Child. 1973 Mar 1;125(3):365–8. doi: 10.1001/archpedi.1973.04160030039007. One of two publications describing a case series of infants with protracted diarrhea of infancy. One of the infants is the oldest identified patient with AIFEC. This report focuses on intestinal biopsy findings. [DOI] [PubMed] [Google Scholar]
- 36.Lloyd-Still JD, Shwachman H, Filler RM. Protracted Diarrhea of Infancy Treated by Intravenous Alimentation: I. Clinical Studies of 16 Infants. Am J Dis Child. 1973 Mar 1;125(3):358–64. doi: 10.1001/archpedi.1973.04160030032006. The other aforementioned report. [DOI] [PubMed] [Google Scholar]
- 37.Kawasaki Y, Oda H, Ito J, Niwa A, Tanaka T, Hijikata A, et al. Identification of a High-Frequency Somatic NLRC4 Mutation as a Cause of Autoinflammation by Pluripotent Cell–Based Phenotype Dissection. Arthritis Rheumatol. 2017 Feb 1;69(2):447–59. doi: 10.1002/art.39960. [DOI] [PubMed] [Google Scholar]
- 38.Kitamura A, Sasaki Y, Abe T, Kano H, Yasutomo K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med. 2014 Nov 17;211(12):2385–96. doi: 10.1084/jem.20141091. This report first describes FCAS4, its genetic cause, molecular mechanism, clinical manifestations, and biologic signature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39*.Volker-Touw CML, de Koning HD, Giltay JC, de Kovel CGF, van Kempen TS, Oberndorff KMEJ, et al. Erythematous nodes, urticarial rash and arthralgias in a large pedigree with NLRC4-related autoinflammatory disease, expansion of the phenotype. Br J Dermatol. 2017 Jan;176(1):244–8. doi: 10.1111/bjd.14757. This report describes a large and phenotypically diverse Dutch kindred including novel features like urticarial and nodular rashes, conjunctivitis, arthritis and IBD. Skin biopsies displayed a lymphohistiocytic infiltrate distinct from FCAS1 and similar to AIFEC. [DOI] [PubMed] [Google Scholar]
- 40.Kolivras A, Theunis A, Ferster A, Lipsker D, Sass U, Dussart A, et al. Cryopyrin-associated periodic syndrome: an autoinflammatory disease manifested as neutrophilic urticarial dermatosis with additional perieccrine involvement. J Cutan Pathol. 2011 Feb;38(2):202–8. doi: 10.1111/j.1600-0560.2010.01638.x. [DOI] [PubMed] [Google Scholar]
- 41.Raghawan AK, Sripada A, Gopinath G, Pushpanjali P, Kumar Y, Radha V, et al. A disease-associated mutant of NLRC4 shows enhanced interaction with SUG1 leading to constitutive FADD dependent caspase-8 activation and cell death. J Biol Chem. 2016 Dec 14; doi: 10.1074/jbc.M116.763979. jbc.M116.763979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chudnovskiy A, Mortha A, Kana V, Kennard A, Ramirez JD, Rahman A, et al. Host-Protozoan Interactions Protect from Mucosal Infections through Activation of the Inflammasome. Cell. 2016 Oct 6;167(2):444–456.e14. doi: 10.1016/j.cell.2016.08.076. [DOI] [PMC free article] [PubMed] [Google Scholar]