

This work describes the oxidative oxysulfenylation and aminosulfenylation of alkenes using an electrochemical protocol.
Abstract
Difunctionalization of alkenes is a valuable and versatile chemical transformation that could quickly build complex molecules. Extensive efforts have been made, and great achievement, such as Sharpless aminohydroxylation and dihydroxylation, has been reached. However, in marked contrast to the extensive research of aminohydroxylation and dihydroxylation, directly using thiophenols/thiols and O/N-nucleophiles to perform the difunctionalization of alkenes that form the C–S and C–O/N bonds together is still underexplored. The main issue is that thiophenols/thiols are often easily overoxidized to sulfoxides or sulphones under such essential oxidation conditions. We demonstrate an electrochemical oxidative oxysulfenylation and aminosulfenylation of alkenes. A critical feature of this transformation is that neither external chemical oxidants nor metal catalysts are required. This electrochemical oxidative synthetic strategy could also be applied for the hydroxysulfenylation and acyloxysulfenylation of alkenes.
INTRODUCTION
The functionalization of alkenes (1, 2), especially vicinal difunctionalization (2–9), has proved to be one of the most attractive and efficient approaches toward the construction of structurally diverse molecules. As an example, the Sharpless aminohydroxylation and dihydroxylation have been extensively studied and applied in chemical syntheses as shown in Fig. 1A (10, 11). As ubiquitous building blocks for various organic molecules, β-alkoxy and β-amino sulfides showed a wide range of intriguing properties and have been widely applied in organic chemistry (12, 13), biological chemistry (14–17), and especially carbohydrate chemistry (18–20). However, in marked contrast to the extensive research of Sharpless aminohydroxylation and dihydroxylation, oxysulfenylation and aminosulfenylation (ideal strategies for the synthesis of β-alkoxy and β-amino sulfides) remain underexplored, and almost all methods involve presynthesized thiolating agents such as disulfides, sulfonyl hydrazides, sodium sulfinates, and 1-(arylthio)pyrrolidine-2,5-diones, which lead not only to an additional operation step but also to environmental problems (21–26). Undoubtedly, directly using commercially available thiophenols/thiols as thiolating agents to perform oxysulfenylation and aminosulfenylation is a more ideal choice and becomes very attractive (Fig. 1B). However, thiophenols/thiols could easily overoxidize to generate sulfoxides and sulfones, as an oxidant is essential for this transformation (27, 28), making the oxysulfenylation and aminosulfenylation of alkenes become problematic (Fig. 1C).
Fig. 1. Difunctionalization of alkenes.
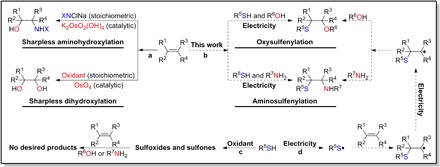
Electrochemical synthesis can directly use an anode to remove electrons to realize the function of oxidants and has become a growing research field in chemical syntheses (29–45). We envisioned that the oxidation and overoxidation of thiophenols/thiols might be controlled in the electrochemical reactions by altering the operating voltage and current. As shown in Fig. 1D, under electrochemical conditions, thiophenols/thiols could be converted to the corresponding thiyl radicals with high efficiency and selectivity. Then, the radical addition of thiyl radicals to alkenes forms a carbon radical, which could be further oxidized and attacked by nucleophiles, providing an opportunity for selective oxysulfenylation and aminosulfenylation of alkenes.
Here, we report a novel and highly selective electrochemical oxidative oxysulfenylation and aminosulfenylation of alkenes directly using thiophenols/thiols as thiolating agents. Specifically, neither external chemical oxidants nor metal catalysts are required in this designed reaction. In terms of substrate scope, various alkenes, thiophenols/thiols, O-nucleophiles, and types of N-nucleophiles were compatible in this transformation, generating the desired products in up to 95% yields. Notably, this electrochemical oxidative synthetic strategy could also be applied for the hydroxysulfenylation and acyloxysulfenylation of alkenes.
RESULTS AND DISCUSSION
At the initial stage, 4-chlorothiophenol (1a), styrene (2a), and methanol (3a) were selected as the model substrates to examine our design. The desired β-alkoxy sulfide 4aa was produced in moderate yield in an undivided cell when nBu4NBF4 was used as the electrolyte and CH3CN was used as the solvent under a constant current. This interesting result showed that our design is reasonable. After a series of explorations, the optimal conditions were described as follows: In an undivided cell using a two-electrode system with the graphite rod as the anode, the platinum plate as the cathode, the solution containing nBu4NBF4 as the electrolyte, and CH3CN as the solvent, the reaction was electrolyzed at a constant current of 12 mA for 4 hours at 40°C (for optimization of reaction conditions, see the Supplementary Materials).
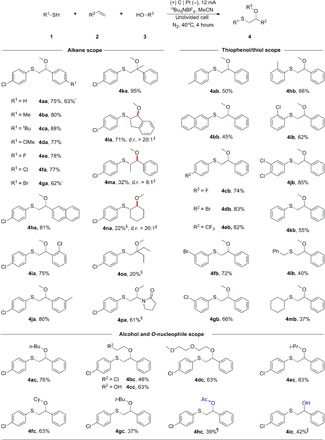
Using the optimized reaction conditions, we investigated a series of alkenes. As shown in Table 1, gratifyingly, a series of styrenes bearing electron-donating groups on the para position of the phenyl ring were compatible with the reaction conditions, providing the desired β-alkoxy sulfides in high yields (Table 1, 4ba to 4da). Similarly, ortho- and meta-substituted styrenes also delivered the corresponding products in good yields (Table 1, 4ia to 4ja). Halo substituents including F, Cl, and Br were tolerated (Table 1, 4ea to 4ga), providing valuable functionality for further transformations. Note that when α-methyl styrene was used, the corresponding β-alkoxy sulfide was obtained in excellent yield (Table 1, 4ka). In addition, indene was also suitable for this transformation, and the desired β-alkoxy sulfide could be obtained in 71% yield with an excellent diastereomeric ratio (Table 1, 4la). In addition to aryl-substituted alkenes, aliphatic alkenes, such as cyclohexene, 2-ethyl-1-butene, and 1-vinyl-2-pyrrolidone, were also suitable for this transformation (Table 1, 4na to 4pa). To evaluate the practicability of this method, we performed the reaction of 4-chlorothiophenol (1a) with styrene (2a) and methanol (3a) on a scale of 5 mmol in an undivided cell. No obvious loss of yield was observed, although the concentration of the electrolyte was reduced from 0.3 to 0.03 M (Table 1, 4aa).
Table 1. Substrate scope for alkene oxysulfenylation.
Reaction conditions: C anode, Pt cathode, constant current = 12 mA, 1 (0.5 mmol), 2 (1.0 mmol), 3 [35 equivalent (equiv.)], nBu4NBF4 (3.0 mmol), MeCN (10 ml), 40°C, 4 hours, isolated yields.
*Gram scale.
†The yield was determined by 1H NMR spectroscopy, with CH2Br2 as the internal standard.
‡Diastereomeric ratio.
§2 (5.0 equiv.).
¶3 (1.0 ml of HOAc).
‖3 (0.7 ml of H2O).
As a follow-up study, the scope with respect to the thiolating agents was also investigated (Table 1). A variety of thiophenols bearing electron-neutral and electron-withdrawing groups gave the desired products in moderate to excellent yields (Table 1, 4ab and 4cb to 4jb). Significantly, the scope of the thiolating agents could be extended to thiols. For example, when benzyl mercaptan and cyclohexyl mercaptan were used as the reaction partners, the corresponding products were isolated in 40 and 37% yields (Table 1, 4lb and 4mb), respectively.
To further establish the scope of this transformation, we tested different alcohols (Table 1). Notably, aliphatic alcohols with functional groups such as halogens, alkoxy, and hydroxyl were tolerated in this transformation (Table 1, 4bc to 4dc). Moreover, sterically hindered tertiary alcohols such as tert-butyl alcohol were also suitable (Table 1, 4gc). Note that the other O-nucleophiles, such as acetic acid and water, were also suitable for this transformation, generating the corresponding acyloxysulfenylation and hydroxysulfenylation products in acceptable yields (Table 1, 4hc and 4ic).
Having successfully demonstrated this electrochemical oxidative oxysulfenylation of alkenes, we subsequently investigated alkene aminosulfenylation (Table 2). To our delight, under similar reaction conditions, primary aromatic amines (Table 2, 6aa to 6fa), primary aliphatic amines (Table 2, 6ja to 6la), secondary aromatic amines (Table 2, 6ga), heteroarylamines (Table 2, 6ha), and amides (Table 2, 6ia) all delivered the corresponding products in 30 to 74% yields. These results showed that the aminosulfenylation of alkenes has also a preeminent substrate scope and application potential.
Table 2. Substrate scope for alkene aminosulfenylation.
Reaction conditions: C anode, Pt cathode, constant current = 12 mA, 1 (0.5 mmol), 2 (1.0 mmol), 5 (1.0 mmol), nBu4NBF4 (0.5 mmol), MeCN (10 ml), 40°C, 4 hours, isolated yields.
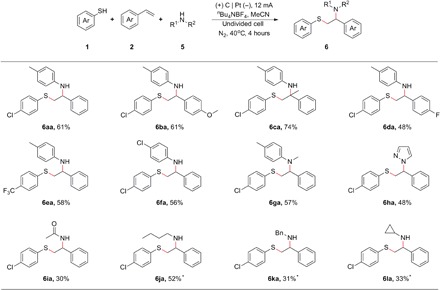
*5 (5.0 mmol).
To gain insights into the transformation mechanism, we carried out control experiments. The radical trapping experiment was performed by using 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) as a radical scavenger. No desired product was found for the standard reaction of 4-chlorothiophenol (1a), styrene (2a), and methanol (3a) (Fig. 2i), while 30% yield of the radical trapping compound 7a was isolated. The reaction of 4-chlorothiophenol (1a) in the absence of styrene (2a) was also investigated under the standard reaction conditions (Fig. 2ii), and bis-(4-chlorophenyl) disulfide 8a (dimers of 1a) was obtained in 78% yield. These results implied that thiyl radicals from thiophenols might be formed under the electrochemical conditions. Furthermore, the reaction of 4-chlorothiophenol (1a), methanol (3a), and radical clock substrate 1-(1-cyclopropylvinyl)benzene (2q) was also examined under the standard conditions, and the reaction afforded the difunctionalization product 4qa in 63% yield; meanwhile, 20% yield of the rearrangement product 9a was isolated. Figure 2iii showed a speculated pathway in which the radical species 2q-A was the key intermediate, and finally, 4qa was obtained by the following oxidation, nucleophilic attack, and deprotonation. In contrast, 9a was obtained via the ring-opening radical clock reaction (46). Another possibility, that 2o-B underwent an SN2′-type reaction, in which MeOH attacks the cyclopropyl group and opens the ring, could not be completely ruled out. Because the alkyl radical 2q-C is much more unstable than 2q-A, 4qa, instead of 9a, was obtained as the major product in this radical clock experiment. Thus, this radical clock experiment also indicated that thiyl radicals might be involved in this transformation.
Fig. 2. Mechanistic studies.
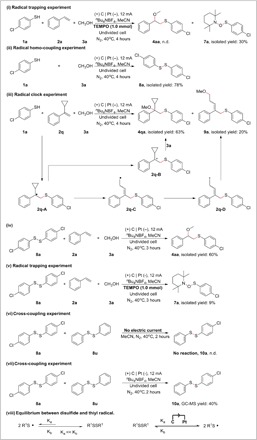
When 4-chlorothiophenol (1a) was replaced by bis-(4-chlorophenyl) disulfide (8a), the reaction gave the desired β-alkoxy sulfide 4aa in 60% yield (Fig. 2iv); when the reaction used TEMPO, no desired product was observed and the radical trapping compound 7a was isolated in 9% yield (Fig. 2v). In addition, the cross-coupling experiment of the mixture of bis-(4-chlorophenyl) disulfide (8a) and phenyl disulfide (8u) was performed. No reaction was observed under the no electric current conditions (Fig. 2vi), while 40% yield of the cross-coupling product 10a was identified under the standard reaction conditions (Fig. 2vii). These results indicate that disulfides could be converted to the corresponding thiyl radicals under the above electrochemical conditions. To rationalize the difunctionalization of oxysulfenylation and aminosulfenylation we described, an equilibrium between disulfides and thiyl radicals promoted by electrodes was proposed (Fig. 2viii). Disulfides could take one electron from the cathode, which led to the formation of disulfide radical anions and then of thiyl radicals and thiolate anions. The thiolate anions could be converted to thiyl radicals at the anode. The undivided electrochemical reaction cell could facilitate the process. To further confirm this speculation, we further performed a sampling experiment, and the results are shown in Fig. 3. In the beginning, both the desired product 4aa and disulfide 8a were accumulated. However, accompanied by the continuous accumulation of 4aa, the highest concentration of 8a was obtained at about 90 min and then gradually went down to zero at the end of this transformation. These results imply that an out cycle of the equilibrium of disulfides and thiyl radicals exists under electrochemical conditions, and thiyl radicals are the key intermediate to react with alkenes. Note that the formation of arylbis(arylthio)sulfonium ions (ArS(ArSSAr))+ from disulfides (ArSSAr) was reported by the electrochemical oxidation in a divided cell at −78°C, and the cationic species could react with alkenes and other nucleophiles in the absence of the electric current (47).
Fig. 3. Sampling experiment.
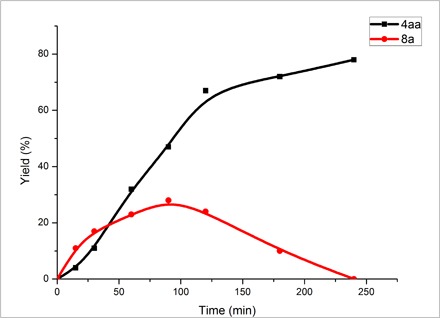
Reaction conditions: C anode, Pt cathode, constant current = 12 mA, 1a (0.5 mmol), 2a (1.0 mmol), 3a (0.7 ml), nBu4NBF4 (3.0 mmol), MeCN (10 ml), 40°C, GC yields.
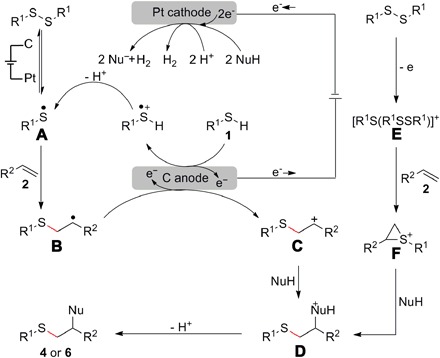
According to the above results, a plausible reaction mechanism is proposed in Fig. 4. First, thiophenols/thiols (1) were converted to thiyl radical A by the electrochemical anodic oxidation and following deprotonation. Subsequently, radical addition of thiyl radical A to alkenes (2) afforded carbon-centered radical B, which could be further oxidized to generate a benzyl cation intermediate C. Finally, C is attacked by the nucleophile (NuH), and the desired product is obtained after deprotonation of intermediate D. Concomitant cathodic reduction of nucleophile or protons leads to the hydrogen evolution. As another possibility, the pathway in which disulfide is oxidized to the corresponding arylbis(arylthio)sulfonium ion E, then nucleophilically attacked by alkene 2 and nucleophile (NuH) to form the desired product, could not be completely ruled out.
Fig. 4. Proposed mechanism.
CONCLUSION
In summary, we have disclosed efficient and highly selective electrochemical oxidative oxysulfenylation and aminosulfenylation of alkenes directly using thiophenols/thiols as thiolating agents. A series of β-alkoxy and β-amino sulfides were produced in good to excellent yields under electrochemical oxidation conditions. This electrochemical oxidative synthetic strategy can avoid the usage of external oxidants. The anodic oxidation was proved to have certain advantages such as better functional group tolerance and higher atom economy.
MATERIALS AND METHODS
All solvents were purified according to the solvents handbook. Unless otherwise noted, materials were obtained from commercial suppliers and used without further purification. A DJS-292B dual display potentiostat was used for electrolysis. The anodic electrode was a graphite rod (ϕ 6 mm), and the cathodic electrode was a platinum plate (15 mm × 15 mm × 0.3 mm). Glass silica gel plates (0.250 mm) were used for thin-layer chromatography. Flash chromatography columns were packed with 200- to 300-mesh silica gel in petroleum (boiling point, 60° to 90°C). Gas chromatographic (GC) analyses were performed using a Shimadzu GC-2014 GC instrument with a flame ionization detector, and biphenyl was added as an internal standard. GC–mass spectrometry spectra were recorded on a Varian GC MS 3900-2100T or Shimadzu GC MS-2010 mass spectrometer. 1H and 13C nuclear magnetic resonance (NMR) data were recorded with Bruker Advance III (400 MHz) spectrometers with tetramethylsilane as an internal standard.
General procedure for electrochemical oxidative oxysulfenylation of alkenes
In an oven-dried undivided three-necked bottle (25 ml) equipped with a stir bar, thiophenols (0.5 mmol) and nBu4NBF4 (3.0 mmol) were combined and added. The bottle was equipped with the graphite rod (ϕ 6 mm; immersion depth in solution of about 20 mm) as the anode and the platinum plate (15 mm × 15 mm × 0.3 mm) as the cathode and was then charged with nitrogen. Under the protection of N2, alkenes (1.0 mmol), alcohols (35 equiv.), and CH3CN (10 ml) were injected into the tubes via syringes. The reaction mixture was stirred and electrolyzed at a constant current of 12 mA at 40°C for 4 hours (3.7 F). When the reaction was finished, the pure product was obtained by flash column chromatography on silica gel.
General procedure for electrochemical oxidative aminosulfenylation of alkenes
In an oven-dried undivided three-necked bottle (25 ml) equipped with a stir bar, thiophenols (0.5 mmol), amines (1.0 mmol), and nBu4NBF4 (0.5 mmol) were combined and added. The bottle was equipped with the graphite rod (ϕ 6 mm; immersion depth in solution of about 20 mm) as the anode and the platinum plate (15 mm × 15 mm × 0.3 mm) as the cathode and was then charged with nitrogen. Under the protection of N2, alkenes (1.0 mmol) and CH3CN (10 ml) were injected into the tubes via syringes. The reaction mixture was stirred and electrolyzed at a constant current of 12 mA at 40°C for 4 hours (3.7 F). When the reaction was finished, the pure product was obtained by flash column chromatography on silica gel.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Natural Science Foundation of China (21390402 and 21520102003) and the Hubei Province Natural Science Foundation of China (2017CFA010). The Program of Introducing Talents of Discipline to Universities of China (111 Program) is also appreciated. Author contributions: Y.Y. and Y.C. performed and analyzed experiments. A.L., Y.Y., and S.T. conceived the project and designed the experiments. A.L., Y.Y., and Z.H. wrote the manuscript. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaat5312/DC1
Table S1. Optimization of reaction conditions.
Fig. S1. The experimental setup for electrolysis.
Section S1. Procedure for gram-scale synthesis
Section S2. The cross-coupling experiment
Section S3. Detailed descriptions for products
Section S4. Copies of product NMR spectra
REFERENCES AND NOTES
- 1.Dong K., Fang X., Gülak S., Franke R., Spannenberg A., Neumann H., Jackstell R., Beller M., Highly active and efficient catalysts for alkoxycarbonylation of alkenes. Nat. Commun. 8, 14117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Egami H., Sodeoka M., Trifluoromethylation of alkenes with concomitant introduction of additional functional groups. Angew. Chem. Int. Ed. 53, 8294–8308 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Lin J.-S., Yu P., Huang L., Zhang P., Tan B., Liu X.-Y., Brønsted acid-catalyzed asymmetric hydroamination of alkenes: Synthesis of pyrrolidines bearing a tetrasubstituted carbon stereocenter. Angew. Chem. Int. Ed. 54, 7847–7851 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Bataille C. J. R., Donohoe T. J., Osmium-free direct syn-dihydroxylation of alkenes. Chem. Soc. Rev. 40, 114–128 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Xie Y., Hu J., Xie P., Qian B., Huang H., Palladium-catalyzed difunctionalization of enol ethers to amino acetals with aminals and alcohols. J. Am. Chem. Soc. 135, 18327–18330 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Zhang C., Li Z., Zhu L., Yu L., Wang Z., Li C., Silver-catalyzed radical phosphonofluorination of unactivated alkenes. J. Am. Chem. Soc. 135, 14082–14085 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Courant T., Masson G., Recent progress in visible-light photoredox-catalyzed intermolecular 1, 2-difunctionalization of double bonds via an ATRA-type mechanism. J. Org. Chem. 81, 6945–6952 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Yin G., Mu X., Liu G., Palladium(II)-catalyzed oxidative difunctionalization of alkenes: Bond forming at a high-valent palladium center. Acc. Chem. Res. 49, 2413–2423 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Wang F., Wang D., Wan X., Wu L., Chen P., Liu G., Enantioselective copper-catalyzed intermolecular cyanotrifluoromethylation of alkenes via radical process. J. Am. Chem. Soc. 138, 15547–15550 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Li G., Chang H.-T., Sharpless K. B., Catalytic asymmetric aminohydroxylation (AA) of olefins. Angew. Chem. Int. Ed. Engl. 35, 451–454 (1996). [Google Scholar]
- 11.Jacobsen E. N., Marko I., Mungall W. S., Schroeder G., Sharpless K. B., Asymmetric dihydroxylation via ligand-accelerated catalysis. J. Am. Chem. Soc. 110, 1968–1970 (1988). [Google Scholar]
- 12.Schwartz A., Madan P. B., Mohacsi E., O’Brien J. P., Todaro L. J., Coffen D. L., Enantioselective synthesis of calcium channel blockers of the diltiazem group. J. Org. Chem. 57, 851–856 (1992). [Google Scholar]
- 13.Kesavan V., Bonnet-Delpon D., Bégué J.-P., Fluoro alcohol as reaction medium: One-pot synthesis of β-hydroxy sulfoxides from epoxides. Tetrahedron Lett. 41, 2895–2898 (2000). [Google Scholar]
- 14.Corey E. J., Clark D. A., Goto G., Marfat A., Mioskowski C., Samuelsoon B., Hammarström S., Stereospecific total synthesis of a “slow reacting substance” of anaphylaxis, leukotriene C-1. J. Am. Chem. Soc. 102, 1436–1439 (1980). [Google Scholar]
- 15.Aveniente M., Pinto E. F., Santos L. S., Rossi-Bergmann B., Barata L. E. S., Structure–activity relationship of antileishmanials neolignan analogues. Bioorg. Med. Chem. 15, 7337–7343 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Rilatt I., Mirabel E., Grand B. L., Perez M., Discovery and SAR of small molecule PAR1 antagonists. Bioorg. Med. Chem. Lett. 20, 903–906 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Souza A. O., Alderete J. B., Minarini P. R. R., Melo P. S., Ferreira I., Barata L. E. S., Silva C. L., Structure activity relationship, acute toxicity and cytotoxicity of antimycobacterial neolignan analogues. J. Pharm. Pharmacol. 63, 936–942 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Kim J.-H., Yang H., Park J., Boons G.-J., A general strategy for stereoselective glycosylations. J. Am. Chem. Soc. 127, 12090–12097 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Adero P. O., Furukawa T., Huang M., Mukherjee D., Retailleau P., Bohé L., Crich D., Cation clock reactions for the determination of relative reaction kinetics in glycosylation reactions: Applications to gluco- and mannopyranosyl sulfoxide and trichloroacetimidate type donors. J. Am. Chem. Soc. 137, 10336–10345 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao X., Feng M., Jiang X., New design of a disulfurating reagent: Facile and straightforward pathway to unsymmetrical disulfanes by copper-catalyzed oxidative cross-coupling. Angew. Chem. Int. Ed. 55, 14121–14125 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Taniguchi N., Copper-catalyzed 1,2-hydroxysulfenylation of alkene using disulfide via cleavage of the S-S bond. J. Org. Chem. 71, 7874–7876 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Gao X., Pan X., Gao J., Jiang H., Yuan G., Li Y., NH4I-mediated three-component coupling reaction: Metal-free synthesis of β-alkoxy methyl sulfides from DMSO, alcohols, and styrenes. Org. Lett. 17, 1038–1041 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Yu J., Gao C., Song Z., Yang H., Fu H., Metal-free oxysulfenylation of alkenes with 1-(arylthio)pyrrolidine-2,5-diones and alcohols. Org. Biomol. Chem. 13, 4846–4850 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Wang D., Zhang R., Ning W., Yan Z., Lin S., Three-component oxysulfenylation reaction: Two simple and convenient approaches to β-alkoxy sulfides. Org. Biomol. Chem. 14, 5136–5140 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Yang F.-L., Wang F.-X., Wang T.-T., Wang Y.-J., Tian S.-K., Iodine-catalyzed three-component oxysulfenylation of alkenes with sulfonyl hydrazides and alcohols. Chem. Commun. 50, 2111–2113 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Sun K., Lv Y., Shi Z., Mu S., Lia C., Wang X., A novel metal-free amidosulfenylation of alkenes leading to β-azolyl sulfides. Org. Biomol. Chem. 15, 5258–5262 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Singh A. K., Chawla R., Keshari T., Yadav V. K., Yadav L. D. S., Aerobic oxysulfonylation of alkenes using thiophenols: An efficient one-pot route to β-ketosulfones. Org. Biomol. Chem. 12, 8550–8554 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Madabhushi S., Jillella R., Sriramoju V., Singh R., Oxyhalogenation of thiols and disulfides into sulfonyl chlorides/bromides using oxone-KX (X = Cl or Br) in water. Green Chem. 16, 3125–3131 (2014). [Google Scholar]
- 29.Tang S., Liu Y., Lei A., Electrochemical oxidative cross-coupling with hydrogen evolution: A green and sustainable way for bond formation. Chem 4, 27–45 (2018). [Google Scholar]
- 30.Yoshida J.-i., Shimizu A., Hayashi R., Electrogenerated cationic reactive intermediates: The pool method and further advances. Chem. Rev. 118, 4702–4730 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Jiang Y., Xu K., Zeng C., Use of electrochemistry in the synthesis of heterocyclic structures. Chem. Rev. 118, 4485–4540 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Yan M., Kawamata Y., Baran P. S., Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev. 117, 13230–13319 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pletcher D., Green R. A., Brown R. C. D., Flow electrolysis cells for the synthetic organic chemistry laboratory. Chem. Rev. 118, 4573–4591 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Francke R., Little R. D., Redox catalysis in organic electrosynthesis: Basic principles and recent developments. Chem. Soc. Rev. 43, 2492–2521 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Jutand A., Contribution of electrochemistry to organometallic catalysis. Chem. Rev. 108, 2300–2347 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Sperry J. B., Wright D. L., The application of cathodic reductions and anodic oxidations in the synthesis of complex molecules. Chem. Soc. Rev. 35, 605–621 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Horn E. J., Rosen B. R., Chen Y., Tang J., Chen K., Eastgate M. D., Baran P. S., Scalable and sustainable electrochemical allylic C–H oxidation. Nature 533, 77–81 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Badalyan A., Stahl S. S., Cooperative electrocatalytic alcohol oxidation with electron-proton-transfer mediators. Nature 535, 406–410 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Xiong P., Xu H.-H., Xu H.-C., Metal- and reagent-free intramolecular oxidative amination of tri- and tetrasubstituted alkenes. J. Am. Chem. Soc. 139, 2956–2959 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Wang P., Tang S., Lei A., Electrochemical intramolecular dehydrogenative C–S bond formation for the synthesis of benzothiazoles. Green Chem. 19, 2092–2095 (2017). [Google Scholar]
- 41.Gieshoff T., Kehl A., Schollmeyer D., Moeller K. D., Waldvogel S. R., Insights into the mechanism of anodic N–N bond formation by dehydrogenative coupling. J. Am. Chem. Soc. 139, 12317–12324 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Fu N., Sauer G. S., Saha A., Loo A., Lin S., Metal-catalyzed electrochemical diazidation of alkenes. Science 357, 575–579 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Yang Q.-L., Li Y.-Q., Ma C., Fang P., Zhang X.-J., Mei T.-S., Palladium-catalyzed C(sp3)—H oxygenation via electrochemical oxidation. J. Am. Chem. Soc. 139, 3293–3298 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Ye K.-Y., Pombar G., Fu N., Sauer G. S., Keresztes I., Lin S., Anodically coupled electrolysis for the heterodifunctionalization of alkenes. J. Am. Chem. Soc. 140, 2438–2441 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Fu N., Sauer G. S., Lin S., Electrocatalytic radical dichlorination of alkenes with nucleophilic chlorine sources. J. Am. Chem. Soc. 139, 15548–15553 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Yu X.-Y., Chen J.-R., Wang P.-Z., Yang M.-N., Liang D., Xiao W.-J., A visible-light-driven iminyl radical-mediated C–C single bond cleavage/radical addition cascade of oxime esters. Angew. Chem. Int. Ed. 57, 738–743 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Matsumoto K., Sanada T., Shimazaki H., Shimada K., Hagiwara S., Fujie S., Ashikari Y., Suga S., Kashimura S., Yoshida J.-i., The addition of ArSSAr to alkenes: The implications of a cationic chain mechanism initiated by electrogenerated ArS(ArSSAr)+. Asian J. Org. Chem. 2, 325–329 (2013). [Google Scholar]
- 48.Wang D., Yan Z., Xie Q., Zhang R., Lin S., Wang Y., Three-component difunctionalization of alkenes leading to β-acetamido sulfides and β-acetoxy sulfides. Org. Biomol. Chem. 15, 1998–2002 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Tehri P., Aegurula B., Peddinti R. K., Iodine-catalysed regioselective synthesis of β-hydroxysulfides. Tetrahedron Lett. 58, 2062–2065 (2017). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaat5312/DC1
Table S1. Optimization of reaction conditions.
Fig. S1. The experimental setup for electrolysis.
Section S1. Procedure for gram-scale synthesis
Section S2. The cross-coupling experiment
Section S3. Detailed descriptions for products
Section S4. Copies of product NMR spectra