

A framework is presented to control phase transformations in colloidal nanocrystal assemblies at liquid-liquid interfaces.
Abstract
Mesostructured matter composed of colloidal nanocrystals in solidified architectures abounds with broadly tunable catalytic, magnetic, optoelectronic, and energy storing properties. Less common are liquid-like assemblies of colloidal nanocrystals in a condensed phase, which may have different energy transduction behaviors owing to their dynamic character. Limiting investigations into dynamic colloidal nanocrystal architectures is the lack of schemes to control or redirect the tendency of the system to solidify. We show how to solidify and subsequently reconfigure colloidal nanocrystal assemblies dimensionally confined to a liquid-liquid interface. Our success in this regard hinged on the development of competitive chemistries anchoring or releasing the nanocrystals to or from the interface. With these chemistries, it was possible to control the kinetic trajectory between quasi–two-dimensional jammed (solid-like) and unjammed (liquid-like) states. In future schemes, it may be possible to leverage this control to direct the formation or destruction of explicit physical pathways for energy carriers to migrate in the system in response to an external field.
INTRODUCTION
Colloidal nanocrystals (NCs) are versatile building blocks for mesostructured matter (1, 2), including colloidal supercrystals (3), superlattices (4, 5), quasi-crystals (6, 7), and NC frameworks (8–11). These multicomponent, architected materials are highly modular, and the tunability of their properties has led to exciting advances in catalysis (12, 13), magnetism (14, 15), energy efficiency (16, 17), and energy conversion (18–24). In principle, the dimensionality of these ordered assemblies [for example, in one dimension (1D), 2D, or 3D] can be controlled at an interface, where crystallization can be spatially directed and where size and shape relationships between components making up the assembly dictate packing arrangements (1, 2, 4, 25). While crystalline, solid-like states of these assemblies have been explored previously (1–11), less is known of the character of dynamic, liquid-like states of colloidal NC assemblies. Furthermore, practical realizations of colloidal NC-based systems with tunable solidification, that is, jamming, from liquid-like states are remarkably scarce due, in part, to the difficulty of controlling the occurrence of jamming. Elucidating design rules to govern the behavior of colloidal NCs in jammed and unjammed states, as well as controlling the interconversion of the system between these states, is key in understanding their potential use as responsive and reconfigurable functional materials. This may also serve to reveal differences between the structural and physical attributes of ordered and glassy colloidal mesostructures, particularly regarding the influence of mesoscopic periodicity or heterogeneities on energy transduction and conversion across multiple length and time scales.
Here, we show that the kinetic trajectory between jammed and unjammed states in 2D assemblies of colloidal NCs at a liquid-liquid interface can be directed in space and time by the competitive binding of small-molecule and polymer ligands at the NC surface. By design, the small-molecule ligand is preferentially soluble in the liquid phase where the NCs are dispersed, and the polymer ligand is selectively partitioned to the opposing phase (Fig. 1). In the absence of the small-molecule ligand, irreversible binding of Lewis base polymer end groups to Lewis acid sites at a “naked” NC surface (that is, an inorganic surface with open metal coordination sites) (26–29) results in the formation of a mesoscopic NC-polymer surfactant (NCPS) that spans the interface between the two fluids (30–32). The number of polymer chains bound to the NC surface is self-regulated and depends on NC size, binding-site density on the NC surface, and polymer molecular weight (33). At steady state, ensembles of NCPSs self-assemble into a monolayer at the interface, minimizing the interfacial energy. The spatial arrangement of NCPSs within the monolayer in this state is dynamic and liquid-like in 2D. When the interface is compressed, NCPSs making up the monolayer jam into a solid-like state in 2D. As is typical for these systems, the structure of this jammed state is stable indefinitely (30, 31), suggesting that the number of NC-NC and NC-polymer contacts is invariant over time. We further show, however, that it is possible to reduce the number of NC-NC and NC-polymer contacts holding the system in the jammed state using a suitably designed small-molecule ligand that reversibly binds to the NC surface, in competition with the polymer ligand. In so doing, the system can be redirected back to its liquid-like state in 2D. Furthermore, by tuning the concentration of the small-molecule ligand, it is possible to control the rate at which these contacts are broken and reformed on the molecular-to-mesoscopic length scales, ultimately dictating the time scale of macroscopic structural reorganization of the entire system.
Fig. 1. Competitive binding by Lewis basic zwitterionic ligands and polymer constituents to Lewis acidic naked NC surfaces dictates the propensity and rate at which NCPSs form at a liquid-liquid interface.
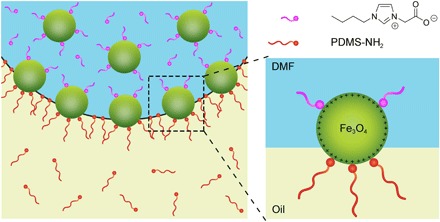
It also dictates important aspects of buckling behavior of the interfacial film when solidified on interfacial compression. At sufficiently high concentrations of the zwitterionic ligand, it becomes possible to reconfigure the solid-like state of a buckled interfacial film of NCPSs back into a liquid-like state (that is, where the wrinkles are no longer observed). The time frame for this dissipative process is also dependent on ligand concentration.
The significance of our work is that molecular cues at interfaces can be exploited in these systems to control the dissipative character of the reconfigurability of NC assemblies between solid- and liquid-like states in 2D. Such control can be used to transiently direct the formation or destruction of physical pathways for energy carriers to migrate between NCs in the assembly. Furthermore, control over the dynamic mechanical properties of the assembly may also be useful in structuring liquids and for the patterning of one fluid phase within another, for example, as are generated using 3D printing, bijel formation, emulsification, and other liquid processing techniques (34–38).
RESULTS AND DISCUSSION
Here, we explore the jamming and unjamming of 2D assemblies of colloidal NCs at a liquid-liquid interface. These unusual states of matter form from an ensemble of colloidal NCPSs, consisting here of cationic naked Fe3O4 NCs (6.0 ± 0.8 nm in diameter) to which a self-regulated number of amine-terminated poly(dimethylsiloxane) [PDMS; Mw = 3.0 kg mol−1; 3K-PDMS-NH2] polymers are tethered at the liquid-liquid interface. We introduce cationic and Lewis acidic Fe3O4 NCs to the system as a dispersion in N,N-dimethylformamide(DMF; 0.50 mg ml−1), alongside complementary Lewis basic 3K-PDMS-NH2 polymers (5% w/w) as a solution in PDMS oil (Mw = 2.8 kg mol−1, η = 50 cSt, ρ = 0.980 g ml−1) that contains dodecane (20% w/w) to lower its density (ρ = 0.905 g ml−1). Lowering the density in this manner was required for certain analytical experiments, for example, pendant drop tensiometry; hereafter, we may refer to the nonpolar phase simply as PDMS.
Although Lewis acid–Lewis base interactions govern the affinity of these two components for each other, 3K-PDMS-NH2 polymers do not transfer the NCs from the polar DMF phase into the nonpolar PDMS phase. Instead, the two components form NCPSs that bridge the DMF-PDMS interface, minimizing the interfacial energy per particle. At steady state, a monolayer of these NCPSs forms at the interface, and the NCPSs have translational degrees of freedom along the interface, consistent with a liquid-like state in 2D. We initially performed dynamic surface tension measurements on a system consisting of DMF containing cationic naked Fe3O4 NCs (typically 20 μl) that had been introduced to PDMS containing 3K-PDMS-NH2. Here, the shape of the droplet was monitored using time-resolved optical techniques and subsequently modeled using the Young-Laplace equation, which balances the gravitational force of the droplet against the interfacial tension. These experiments showed that the NCPS monolayer formation occurred over the course of ~1 hour. After monolayer formation reached steady state, the interfacial tension decreased to γpolar/nonpolar = 2.85 mN m−1 (fig. S1). Control measurements of the same system without NCPSs yielded γpolar/nonpolar = 4.65 mN m−1 (fig. S2). In the instance where the 3K-PDMS-NH2 polymer surfactant is used alone, we observed an unusual increase in the interfacial tension as it was recruited to the interface, before a steady-state value of γpolar/nonpolar = 5.25 mN m−1 was reached (fig. S3). This, we argue, arises from a gradual depletion of dodecane relative to PDMS close to the interface as 3K-PDMS-NH2 reaches steady-state occupation. In other words, while the presence of 3K-PDMS-NH2 at the interface decreases the interfacial tension, the exclusion of dodecane at the interface counteracts this. Elsewhere in the system, for example, in the dodecane-enriched bulk, free energy must be gained to compensate for the work being done at the interface. In the context of NCPS formation, then, the rapid increase in the interfacial tension with time is due to polymer recruitment to the interface in the dodecane/PDMS phase, followed by a gradual reduction in interfacial tension over longer periods of time as NCs diffuse to the interface, NCPSs form, and finally a monolayer forms.
The assembly of NCPSs at the interface is irreversible, as evidenced by the wrinkling behavior observed when the volume of the droplet is decreased, which compresses the NCPS assembly at the interface. The NCPSs jam, resulting in a wrinkling of the interfacial film when further compressed. The wrinkles are persistent (Fig. 2), in keeping with the notion that the system is not reconfigurable despite being assembled into a liquid-like state from discrete nanoscale building blocks. Previously, this jamming behavior had only been observed for systems implementing soft or glassy nanoparticles (for example, polymers or silica) dispersed in aqueous media with polymer surfactants dissolved in an oil phase (30–33). Our results highlight that the 2D interfacial jamming phenomenon is general to other multiphasic liquid media and a considerably more diverse set of functional nanomaterials (for example, magnetic, quantum-confined, light-emitting, catalytic) in each phase (26–29).
Fig. 2. Nonreconfigurable, jammed 2D assemblies of colloidal NCPSs.
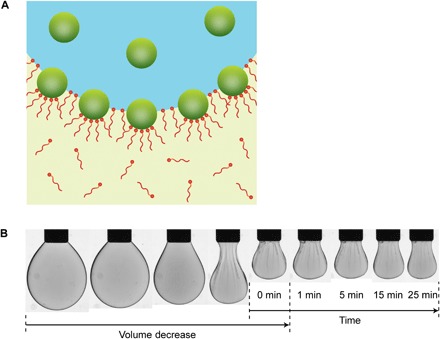
(A) Lewis acid–Lewis base interactions between cationic naked NCs and amine-terminated PDMS polymers allow the formation of NCPSs at the interface between DMF and PDMS oil, which are immiscible. (B) The liquid-like 2D assembly of NCPSs can be solidified by compressing the liquid-liquid interface, shown here as the DMF droplet is retracted into the syringe. This buckled state of the 2D interfacial film exhibits wrinkles, which are persistent indefinitely.
To impart reconfigurability to the jammed system, we hypothesized that it would be necessary to reduce the number of NC-polymer contacts and, in turn, NC-NC contacts holding the system in the jammed state. We reasoned that this could be achieved using a small molecule that reversibly binds to the NC surface in a manner that was competitive with the binding of 3K-PDMS-NH2. Furthermore, binding of such a small molecule at the NC surface would also influence the energetics of reconfiguring interparticle contacts when in the jammed state. Ultimately, both processes are needed to effectively eject an NC from the interface, which is concomitant with a decrease in interfacial area with each ejection event. If successful, the system could return to its liquid-like state in 2D, with an evolution over time in mesoscale structure dependent on ligand concentration. In other words, this endogenous system input would dictate the rate at which NC-polymer and NC-NC contacts are broken and reformed. It is also possible that the presence of the small-molecule competitive ligand in the system could influence, in a concentration-dependent manner, the dynamics of NCPS formation and subsequent assembly into the 2D liquid-like monolayer at the interface.
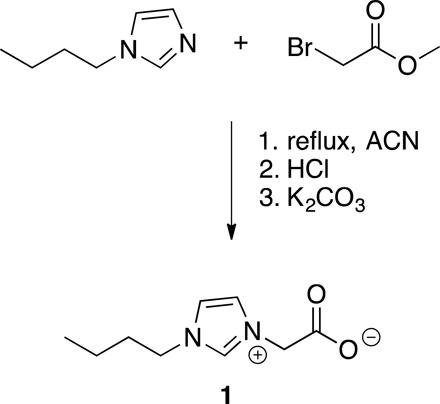
The key to our success in reconfiguring jammed (solid-like) 2D assemblies of colloidal NCs into a liquid-like state was the implementation of a novel zwitterionic ligand—2-(3-butyl-1H-imidazol-3-ium-1-yl)acetate (1)—that reversibly binds to the open metal sites at the naked NC surface. These open metal sites have a Lewis acidic character, while the carboxylate terminus of the zwitterionic ligand is a Lewis base. These zwitterionic ligands selectively partition into the DMF phase and compete with polymer-NC interactions that pin the NCs to the interface as a monolayer of NCPSs. We readily synthesized these ligands in multigram quantities from 1-butylimidazole, which was alkylated with methyl bromoacetate before hydrolyzing the methyl ester under acidic conditions and subsequently neutralizing the HCl (or HBr) adduct to the desired zwitterionic species (Scheme 1). While its structure might suggest interface activity, zwitterionic ligand 1 did not lower the interfacial tension of DMF droplets in PDMS/dodecane mixtures, in the absence or in the presence of 3K-PDMS-NH2 (figs. S4 and S5). The binding of this new ligand motif at naked Fe3O4 NC surfaces was evidenced by dynamic light scattering (DLS), which showed that the NC’s solvodynamic diameter increased from 7.5 to 8.7 nm with ligand 1 present (fig. S6). This ligand-triggered increase in the NC-NC spacing is also integral to the process by which NC-NC contacts are broken when the monolayer of NCPSs is in a jammed state.
Scheme 1. Chemical synthesis of zwitterionic NC ligand 1.
Confirmation that the presence of small-molecule ligand 1 imparts reconfigurability to the system was initially demonstrated by introducing 1 (1.0 mg ml−1) to a dispersion of cationic naked Fe3O4 NCs in DMF (0.50 mg ml−1) and then suspending this mixture as a droplet in a solution of 3K-PDMS-NH2 in PDMS (5% w/w) containing dodecane (20% w/w). In contrast to the rate of monolayer formation observed in the absence of 1, the approach of the system to saturation occurred more rapidly, from 132 min in the absence of 1 to 85 min when 1 was present. Therefore, we reason that ligand 1, when bound to the NC surface, renders the NC more interfacially active due to the presentation of butyl chains at the surface. At steady state, the interfacial tension (γpolar/nonpolar = 2.74 mN m−1) was comparable to that in the absence of ligand 1 (Fig. 3A). Upon compression of the interface by reducing the volume of the droplet, the wrinkling observed was markedly less pronounced than that observed in the absence of 1, suggesting a softer film (that is, lower modulus) (39). More telling with respect to the ability of the jammed assembly to reconfigure, the few instances of wrinkles in this film disappeared over time (~20 min) (Fig. 3B). Here, the disappearance of wrinkles is directly tied to the action of zwitterionic ligand 1 in unjamming NCPSs. Specifically, when the liquid-like assembly of NCPSs is driven to a solid-like jammed state by interfacial compression, the wrinkled film exhibits curvature and, thus, higher interfacial energy and higher Gibbs free energy. The relaxation of the system to a smooth film minimizes both the interfacial area and Gibbs free energy. The decrease in interfacial area requires NCs to be ejected from the interfacial film. This is only possible when the NC-polymer and NC-NC bonds are collectively weakened and ultimately broken, thus necessitating the use of a competitive ligand such as zwitterionic ligand 1.
Fig. 3. Competitive ligand-mediated reconfiguring of jammed 2D assemblies of colloidal NCPSs at a liquid-liquid interface.
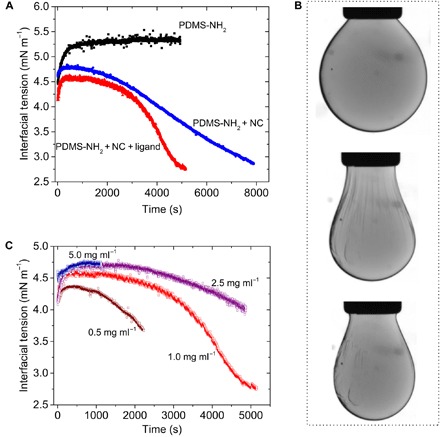
(A) The recruitment of 3K-PDMS-NH2 and naked Fe3O4 NCs to the interface occurs on different time scales and depends on the relative concentration of each. These processes can be observed by monitoring the interfacial tension as a function of time for a pendant droplet of DMF suspended in a bath of PDMS/dodecane for systems configured with either 3K-PDMS-NH2 (black squares), 3K-PDMS-NH2 + Fe3O4 NCs (blue circles), or 3K-PDMS-NH2 + Fe3O4 NCs + competitive ligand 1 (red triangles). [3K-PDMS-NH2] = 5% w/w; [Fe3O4 NCs] = 0.5 mg ml−1; [1] = 1 mg ml−1. (B) The solid-like character of a buckled, 2D film of colloidal NCPSs is evidenced by wrinkling for droplets whose interface has been compressed upon droplet retraction. Only in the instance where competitive ligand 1 is present do the wrinkles in the film return to a smooth state, consistent with a dissipative process whereby the ligands promote the ejection of NCs from the film back into the bulk. Snapshots of such a reconfigurable droplet are shown at steady state, before retraction (top); immediately after droplet retraction (middle), where wrinkles appear; and after 20 min, whereupon the system has relaxed to an unwrinkled state (bottom). (C) Interfacial tension as a function of time for a pendant droplet of DMF suspended in a bath of PDMS/dodecane for systems configured with 3K-PDMS-NH2 (5% w/w), Fe3O4 NCs (0.5 mg ml−1), and varying concentrations of zwitterionic ligand 1 (0.5 to 5.0 mg ml−1).
To better understand the extent to which zwitterionic ligand 1 influences the NCPS monolayer at the DMF-PDMS interface, we varied the concentration of 1 in the DMF phase (0.5 to 5 mg ml−1) and monitored the time evolution of the interfacial tension by pendant drop tensiometry, where a droplet of a cationic Fe3O4 dispersion in DMF (0.50 mg ml−1) was suspended in a solution of 3K-PDMS-NH2 in PDMS (5% w/w) (Fig. 3C). These data demonstrate that the system’s approach to a steady state is strongly dependent on the concentration of zwitterionic ligand 1 in the DMF phase (Fig. 4A). Quantitatively, saturation times varied as follows: 37, 85, 80, and 10 min for ligand concentrations of 0.5, 1.0, 2.5, and 5.0 mg ml−1, respectively. Notable was the return of the system to a fast equilibration regime at the highest concentration of ligand 1 used (that is, 5.0 mg ml−1). This behavior is consistent with our hypothesis that zwitterionic ligand 1 competes with 3K-PDMS-NH2 for binding sites at the NC surface and, in this case, does so effectively at the DMF-PDMS interface despite the dense coverage of the interface by 3K-PDMS-NH2. There are several mechanisms by which this might occur; however, the most likely is that ligand 1 is bound to and in dynamic exchange at the NC surface; the occupation of sites at the NC surface is concentration-dependent, and at high surface coverage, this ligand shell acts as a steric barrier preventing 3K-PDMS-NH2 from accessing the open metal sites (Fig. 4C).
Fig. 4. The time scale of ligand-mediated reconfiguring of jammed, solid-like colloidal NCPS assemblies into a liquid-like state at a liquid-liquid interface depends on ligand concentration.
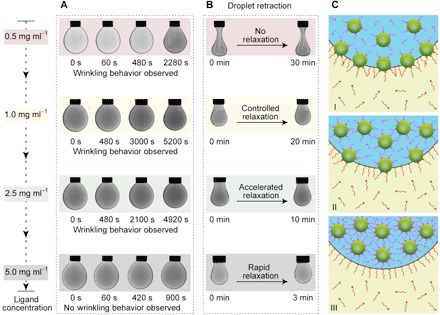
(A) Snapshots of each droplet’s morphology upon NCPS formation at the liquid-liquid interface. (B) Snapshots of each droplet’s morphology after the initial film compression and after the reconfiguring film of NCPSs had reached steady state. (C) Schematic representations of the influence of ligand concentration on interface activity and binding ability of colloidal naked NCs at low (I), moderate (II), and high (III) ligand concentrations.
Having studied NCPS monolayer formation for each of the systems described above, we then sought to understand the response of those systems to compression-induced wrinkle formation. At the lowest concentration of zwitterionic ligand 1 (0.5 mg ml−1) used, we did not observe any relaxation behavior of the assembly of NCPSs at the interface. Instead, we observed a similar wrinkling behavior to the system configured without zwitterionic ligand 1 (Fig. 4B). For higher concentrations of zwitterionic ligand 1 (that is, 1.0–5 mg ml−1), the amplitudes of the wrinkles observed in the compressed film were less and increasingly so at higher ligand concentrations (Fig. 4B). For this range of ligand concentrations, all of the jammed assemblies could relax, that is, the wrinkles eventually disappeared (Fig. 4B). We found that the time required to return to an unjammed state decreased with higher concentrations of zwitterionic ligand 1 in the DMF phase; in the case of a system configured with 5 mg ml−1 of 1 in the DMF phase, only 180 s was required to relax to a steady state with marginal to no wrinkling behavior observed along the way during droplet retraction.
The range of behaviors observed in the tensiometer experiments suggested that the presence of zwitterionic ligand 1 yielded softer NCPS films owing to their dynamic, liquid-like character. To substantiate these differences in mechanical properties for solid- and liquid-like NCPS films at the interface, we conducted in situ atomic force microscopy (AFM) studies to probe directly the interfacial layer (40). Here, a droplet of high-viscosity silicone oil (η = 60,000 cSt) containing 3K-PDMS-NH2 (5% w/w) was placed on a silicon substrate and immersed in a dispersion of naked Fe3O4 NCs in DMF, optionally containing zwitterionic ligand 1 (Fig. 5A). After equilibration, we performed the AFM measurements, including mechanical testing of the NCPS films. Comparing the slopes in the force curves (Fig. 5B), we noted a higher modulus for NCPS films in the absence of zwitterionic ligand 1 than in its presence, when being indented by the AFM cantilever. While the interfacial tension is higher for the NCPS film without ligands (Fig. 3A), the substantially higher stiffness measured by AFM indicates that the jamming of the film is the dominate contributor to the stiffness difference. Furthermore, topography images of the interface show that the interface is rough with NCs only (Fig. 5C), indicating that the films have jammed and wrinkled. After adding zwitterionic ligand 1, the interface is quite smooth and the film is relaxed, consistent with the pendant drop tensiometry measurements.
Fig. 5. Distinguishing the stiffness of NCPS monolayers in solid- and liquid-like states at a liquid-liquid interface using in situ AFM.
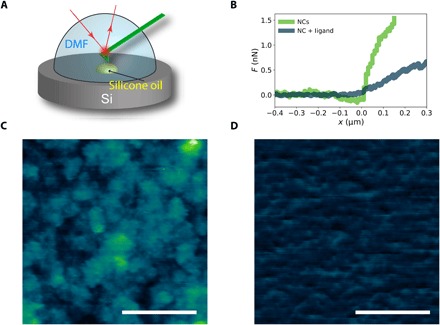
(A) Schematic diagram of the in situ AFM experimental setup. (B) Force (F) curves measured for NCPS films at the DMF–silicone oil interface in the absence and in the presence of zwitterionic ligand 1 confirm that solid-like NCPS films are stiffer than liquid-like films. The distinctive mechanical properties observed for solid- and liquid-like states were concomitant with differentiated morphologies for NCPS films at the DMF–silicone oil interface in the absence (C) and in the presence (D) of zwitterionic ligand 1. Scale bars, 200 nm; z range color scale, 100 nm.
Our use of magnetic Fe3O4 NCs in these 2D interfacial NC assemblies allows us to deform the droplet by applying an external magnetic field. This was demonstrated by placing one or several neodymium magnets in proximity to the 2D assembly of NCPSs (Fig. 6). When a single magnet is applied to the system, the 2D NCPS assembly and the entrapped fluid moved toward the magnet. When two magnets were used, the orientations of the magnets with respect to each other and with respect to the film (and entrapped fluid) determined the response of the system. When the closest two poles of the magnet were of the same polarity, the 2D film did not undergo any significant displacement in space. When the closest poles of the two magnets were of the opposite polarity, the 2D NCPS assembly was displaced in space, and the encapsulated fluid deformed considerably, expanding along the axis of the magnetic field and compressing along the axis normal to the field. The deformation of the droplet by the magnetic field leads to an apparent change in the interfacial tension. For example, at higher concentrations of zwitterionic ligand 1 (2.5 mg ml−1), in the absence of the applied magnetic field, the interfacial tension was measured to be γpolar/nonpolar = 4.0 mN m−1. When the magnetic field was applied, it appeared to increase to γpolar/nonpolar = 5.6 mN m−1. The droplet recovered both its original shape and interfacial tension once the field was removed. We note that in the presence of the magnetic field, the “surface tension,” as shown in Fig. 6, no longer corresponds to the free energy per unit area of the interface but is a parameter describing the shape of the droplet and its deformation by the external field.
Fig. 6. Pendant droplet responses to magnetic fields.
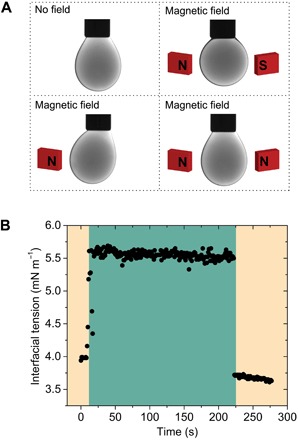
(A) When the pendant droplet was set in the middle of two magnets with the opposite polarity, the 2D NCPS assembly was displaced in space and the encapsulated fluid was expanded along the axis of the magnetic field and was compressed along the axis normal to the field. (B) At 2.5 mg ml−1 concentration of zwitterionic ligand 1, the interfacial tension of the droplet increased from γpolar/nonpolar = 4.0 to 5.6 mN m−1 when the magnetic field was applied. The droplet recovered both its equilibrium shape and interfacial tension once the field was removed.
CONCLUSION
Here, the groundwork has been laid for controlling the solidification and reconfigurability of colloidal NC assemblies that are dimensionally confined to a liquid-liquid interface. The key to our success was the development of dynamic and competitive chemistries, based on Lewis acid–Lewis base pairing, anchoring or releasing the NCs to or from the interface. Our results show that controlling the kinetic trajectory between quasi-2D jammed (solid-like) and unjammed (liquid-like) states is feasible through a simple chemical input consisting of a competitive ligand at a prescribed concentration. With this control, it should be possible to evolve in space and in time explicit physical pathways for energy carriers to migrate in the system between NC components. Upon application of an external field, it may further be possible to direct the energy carriers directionally, suggesting new avenues for manipulating the energy transduction behaviors of quasi-2D quantum materials assembled from interacting NC building blocks.
MATERIALS AND METHODS
Synthesis of ligand-coated Fe3O4 NCs
Iron(III) acetylacetonate (0.706 g, 2.00 mmol), 1,2-hexadecanediol (2.584 g, 10.00 mmol), oleic acid (1.904 ml, 6.00 mmol), oleylamine (1.974 ml, 6.00 mmol), and benzyl ether (20 ml) were combined in a 100-ml three-necked flask equipped with an air condenser and an immersed temperature controller. The reaction mixture was degassed under vacuum while stirring, initially at 100°C for 15 min and then at 200°C for 2 hours. The NCs were then grown at a reaction temperature of 287°C for 1 hour. After cooling the reaction mixture to ambient temperature, the NCs were initially isolated by precipitation with ethanol (20 ml) and centrifugation, discarding the supernatant. The precipitate was redispersed into hexanes (5 ml) containing oleylamine (25 μl) and oleic acid (25 μl). Three rounds of precipitation with ethanol and isolation, followed by redispersing with hexanes (2 ml), afforded the desired product. Thermogravimetric analysis showed that the ligand-coated Fe3O4 NCs were 25% organics and that the Fe3O4 concentration in hexanes was 37.5 mg ml−1. The reaction proceeded with an overall yield of 97%.
Synthesis of cationic naked Fe3O4 NCs
In a nitrogen glove box, boron trifluoride etherate (50 μl) was added to DMF (500 μl), followed by hexanes (400 μl) and a dispersion of ligand-coated Fe3O4 NCs in hexanes (100 μl, 37.5 mg ml−1). This biphasic mixture was vortexed for 1 min, after which complete transfer of the NCs from the hexane layer into the DMF layer was observed. Ligand-stripped Fe3O4 NCs were subsequently precipitated from the reaction mixture upon the addition of toluene (3 ml). After centrifugation, the supernatant was discarded, and the pellet was redispersed with DMF. Three rounds of precipitation with hexanes/toluene and redispersion with a minimal amount of DMF were carried out, after which the final dispersion in DMF was obtained (2.5 ml, 1.52 mg ml−1). Naked NC size and size distribution measured by transmission electron microscopy and DLS are shown in fig. S4.
Synthesis of 2-(1-butyl-1H-imidazol-3-ium-3-yl)acetate
A solution of methyl bromoacetate (8.55 g, 55.9 mmol) and 1-butylimidazole (6.94 g, 55.9 mmol) in acetonitrile (50 ml) was stirred at room temperature overnight under a nitrogen atmosphere. The product, methyl 2-(1-butyl-1H-imidazol-3-ium-3-yl)acetate bromide, was isolated as a viscous liquid in quantitative yield after removal of the solvent in vacuum. Acidolysis of the methyl ester was then carried out by dissolving methyl 2-(1-butyl-1H-imidazol-3-ium-3-yl)acetate bromide in concentrated aqueous HCl (37% w/w, 6.109 g) at 100°C for 2 hours. The water was removed in vacuum before the addition of K2CO3 (8.57 g, 62.0 mmol) as a solution in water (40 ml). After 5 min, the water was again removed in vacuum. The crude product was dissolved in dichloromethane (DCM) (40 ml) and further dried over MgSO4. After filtration and concentration, the residue was dissolved in ethanol, and the solution was filtered to remove undissolved solids. The ethanol was removed in vacuum, and the product was isolated as white powder (9.82 g, 96%). 1H NMR (nuclear magnetic resonance) (500 MHz, DMSO-d6): δ 9.16 (s, 1H), 7.71 (s, 1H), 7.63 (s, 1H), 4.51 (s, 2H), 4.19 (t, J = 7.0 Hz, 2H), 1.76 (m, J = 7.2 Hz, 2H), 1.25 (m, 2H), 0.90 (t, J = 7.4 Hz, 3H) parts per million (ppm); 13C NMR (125 MHz, DMSO-d6): δ 166.83, 137.05, 124.13, 121.39, 53.22, 48.72, 31.95, 19.25, 13.77 ppm. High-resolution mass spectrometry (electrospray ionization): m/z (mass/charge ratio) for C9H15O2N2+ calculated 183.1128, found 183.1130. Fourier transform infrared: ν 3379, 3143, 3097, 2960, 2937, 2875, 1616, 1563, 1384, 1307, 1167, 1124, 1033, 983, 917, 790 cm−1.
Supplementary Material
Acknowledgments
Funding: This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division under contract no. DE-AC02-05CH11231 within the Adaptive Interfacial Assemblies Towards Structuring Liquids program (KCTR16). Portions of the work—including NC and zwitterionic ligand synthesis and characterization—were carried out at Molecular Foundry, which is supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. Author contributions: B.A.H. and T.P.R. designed and directed the experiments. Z.Z., Y.J., F.L., and J.F. carried out the pendant drop tensiometry. C.H. and W.F. synthesized and characterized the PDMS-NH2 polymers. Z.Z., E.G., and W.F. synthesized and characterized the zwitterionic ligand. Y.C. and P.D.A. carried out the in situ AFM experiments. Z.Z., E.G., and T.E.W. acquired and analyzed the NC size distributions in the absence or presence of the zwitterionic ligand. Z.Z., C.H., J.F., T.P.R., and B.A.H. wrote the manuscript with contributions from all co-authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from B.A.H. (bahelms@lbl.gov).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaap8045/DC1
Fig. S1. Interfacial tension as a function of time for a system configured with both Fe3O4 NCs and 3K-PDMS-NH2.
Fig. S2. Interfacial tension as a function of time for a system without NCPSs.
Fig. S3. Interfacial tension as a function of time for a system configured with only 3K-PDMS-NH2.
Fig. S4. Interfacial tension as a function of time for a system configured with only zwitterionic ligand 1.
Fig. S5. Interfacial tension as a function of time for a system configured with zwitterionic ligand 1 and 3K-PDMS-NH2.
Fig. S6. Zwitterionic ligand binding to naked NC surfaces.
Fig. S7. Steady-state interfacial tension as a function of zwitterionic ligand concentration.
REFERENCES AND NOTES
- 1.Milliron D. J., Buonsanti R., Llordes A., Helms B. A., Constructing functional mesostructured materials from colloidal nanocrystal building blocks. Acc. Chem. Res. 47, 236–246 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Boles M., Engel M., Talapin D. V., Self-assembly of colloidal nanocrystals: From intricate structures to functional materials. Chem. Rev. 116, 11220–11289 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Wang T., LaMontagne D., Lynch J., Zhuang J., Cao Y. C., Colloidal superparticles from nanoparticle assembly. Chem. Soc. Rev. 42, 2804–2823 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Dong A., Chen J., Vora P. M., Kikkawa J. M., Murray C. B., Binary nanocrystal superlattice membranes self-assembled at the liquid–air interface. Nature 466, 474–477 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Paik T., Diroll B. T., Kagan C. R., Murray C. B., Binary and ternary superlattices self-assembled from colloidal nanodisks and nanorods. J. Am. Chem. Soc. 137, 6662–6669 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Talapin D. V., Shevchenko E. V., Bodnarchuk M. I., Ye X., Chen J., Murray C. B., Quasicrystalline order in self-assembled binary nanoparticle superlattices. Nature 461, 964–967 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Ye X., Chen J., Irrgang M. E., Engel M., Dong A., Glotzer S. C., Murray C. B., Quasicrystalline nanocrystal superlattice with partial matching rules. Nat. Mater. 16, 214–219 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Warren S. C., Messina L. C., Slaughter L. S., Kamperman M., Zhou Q., Gruner S. M., DiSalvo F. J., Wiesner U., Ordered mesoporous materials from metal nanoparticle-block copolymer self-assembly. Science 320, 1748–1752 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Buonsanti R., Pick T. E., Krins N., Richardson T. J., Helms B. A., Milliron D. J., Assembly of ligand-stripped nanocrystals into precisely controlled mesoporous architectures. Nano Lett. 12, 3872–3877 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Helms B. A., Williams T. E., Buonsanti R., Milliron D. J., Colloidal nanocrystal frameworks. Adv. Mater. 27, 5820–5829 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Williams T. E., Ushizima D., Zhu C., Anders A., Milliron D. J., Helms B. A., Nearest-neighbour nanocrystal bonding dictates framework stability or collapse in colloidal nanocrystal frameworks. Chem. Commun. 53, 4853–4856 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Kang Y., Ye X., Chen J., Qi L., Diaz R. E., Doan-Nguyen V., Xing G., Kagan C. R., Li J., Gorte R. J., Stach E. A., Murray C. B., Engineering catalytic contacts and thermal stability: Gold/iron oxide binary nanocrystal superlattices for CO oxidation. J. Am. Chem. Soc. 135, 1499–1505 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Kang Y., Ye X., Chen J., Cai Y., Diaz R. E., Adzic R. R., Stach E. A., Murray C. B., Design of Pt–Pd binary superlattices exploiting shape effects and synergistic effects for oxygen reduction reactions. J. Am. Chem. Soc. 135, 42–45 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Dong A., Chen J., Ye X., Kikkawa J. M., Murray C. B., Enhanced thermal stability and magnetic properties in NaCl-Type FePt–MnO binary nanocrystal superlattices. J. Am. Chem. Soc. 133, 13296–13299 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Chen J., Ye X., Oh S. J., Kikkawa J. M., Kagan C. R., Murray C. B., Bistable magnetoresistance switching in exchange-coupled CoFe2O4-Fe3O4 binary nanocrystal superlattices by self-assembly and thermal annealing. ACS Nano 7, 1478–1486 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Williams T. E., Chang C. M., Rosen E. L., Garcia G., Runnerstrom E. L., Williams B. L., Koo B., Buonsanti R., Milliron D. J., Helms B. A., NIR-selective electrochromic heteromaterial frameworks: A platform to understand mesoscale transport phenomena in solid-state electrochemical devices. J. Mater. Chem. C 2, 3328–3335 (2014). [Google Scholar]
- 17.Kim J., Ong G. K., Wang Y., LeBlanc G., Williams T. E., Mattox T. M., Helms B. A., Milliron D. J., Nanocomposite architecture for rapid, spectrally-selective electrochromic modulation of solar transmittance. Nano Lett. 15, 5574–5579 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Urban J. J., Talapin D. V., Shevchenko E. V., Kagan C. R., Murray C. B., Synergism in binary nanocrystal superlattices leads to enhanced p-type conductivity in self-assembled PbTe/Ag2Te thin films. Nat. Mater. 6, 115–121 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Redl F. X., Cho K.-S., Murray C. B., O’Brien S., Three-dimensional binary superlattices of magnetic nanocrystals and semiconductor quantum dots. Nature 423, 968–971 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Ye X., Chen J., Diroll B. T., Murray C. B., Tunable plasmonic coupling in self-assembled binary nanocrystal superlattices studied by correlated optical microspectrophotometry and electron microscopy. Nano Lett. 13, 1291–1297 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Whitham K., Yang J., Savitzky B. H., Kourkoutis L. F., Wise F., Hanrath T., Charge transport and localization in atomically coherent quantum dot solids. Nat. Mater. 15, 557–563 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Wu Y., Li S., Gogotsi N., Zhao T., Fleury B., Kagan C. R., Murray C. B., Baxter J. B., Directional carrier transfer in strongly coupled binary nanocrystal superlattice films formed by assembly and in situ ligand exchange at a liquid-air interface. J. Phys. Chem. C 121, 4146–4157 (2017). [Google Scholar]
- 23.Bigioni T. P., Lin X.-M., Nguyen T. T., Corwin E. I., Witten T. A., Jaeger H. M., Kinetically driven self assembly of highly ordered nanoparticle monolayers. Nat. Mater. 5, 265–270 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Korgel B. A., Nanocrystal superlattices: Assembly at liquid interfaces. Nat. Mater. 9, 701–703 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Choi J. J., Bian K., Baumgardner W. J., Smilgies D.-M., Hanrath T., Interface-induced nucleation, orientational alignment and symmetry transformations in nanocube superlattices. Nano Lett. 12, 4791–4798 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Dong A., Ye X., Chen J., Kang Y., Gordon T., Kikkawa J. M., Murray C. B., A generalized ligand-exchange strategy enabling sequential surface functionalization of colloidal nanocrystals. J. Am. Chem. Soc. 133, 998–1006 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Rosen E. L., Buonsanti R., Llordes A., Sawvel A. M., Milliron D. J., Helms B. A., Exceptionally mild reactive stripping of native ligands from nanocrystal surfaces by using Meerwein’s salt. Angew. Chem. Int. Ed. 51, 684–689 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Rosen E. L., Sawvel A. M., Milliron D. J., Helms B. A., Influence of surface composition on electronic transport through naked nanocrystal networks. Chem. Mater. 26, 2214–2217 (2014). [Google Scholar]
- 29.Doris S. E., Lynch J. J., Li C., Wills A. W., Urban J. J., Helms B. A., Mechanistic insight into the formation of cationic naked nanocrystals generated under equilibrium control. J. Am. Chem. Soc. 136, 15702–15710 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Cui M., Emrick T., Russell T. P., Stabilizing liquid drops in nonequilibrium shapes by the interfacial jamming of nanoparticles. Science 342, 460–463 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Huang C., Sun Z., Cui M., Liu F., Helms B. A., Russell T. P., Structured liquids with pH-triggered reconfigurability. Adv. Mater. 28, 6612–6618 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Toor A., Helms B. A., Russell T. P., Effect of nanoparticle surfactants on the breakup of free-falling water jets during continuous processing of reconfigurable structured liquid droplets. Nano Lett. 17, 3119–3125 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Huang C., Cui M., Sun Z., Liu F., Helms B. A., Russell T. P., Self-regulated nanoparticle assembly at liquid/liquid interfaces: A route to adaptive structuring of liquids. Langmuir 33, 7994–8001 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Whitby C. P., Fornasiero D., Ralston J., Liggieri L., Ravera F., Properties of fatty amine–silica nanoparticle interfacial layers at the hexane–water interface. J. Phys. Chem. C 116, 3050–3058 (2012). [Google Scholar]
- 35.Vashisth C., Whitby C. P., Fornasiero D., Ralston J., Interfacial displacement of nanoparticles by surfactants molecules in emulsions. J. Colloid Interface Sci. 349, 537–543 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Dommersnes P., Rozynek Z., Mikkelsen A., Castberg R., Kjerstad K., Hersvik K., Fossum J. O., Active structuring of colloidal armour on liquid drops. Nat. Commun. 4, 2066 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cates M. E., Clegg P. S., Bijels: A new class of soft materials. Soft Matter 4, 2132–2138 (2008). [Google Scholar]
- 38.Forth J., Liu X., Hasnain J., Toor A., Miszta K., Shi S., Geissler P. L., Emrick T., Helms B. A., Russell T. P., Reconfigurable printed liquids. Adv. Mater. 30, 1707603 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Knoche S., Vella D., Aumaitre E., Degen P., Rehage H., Cicuta P., Kierfeld J., Elastometry of deflated capsules: Elastic moduli from shape and wrinkle analysis. Langmuir 29, 12463–12471 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Chai Y., Lukito A., Jiang Y., Ashby P. D., Russell T. P., Fine-tuning nanoparticle packing at water–oil interfaces using ionic strength. Nano Lett. 17, 6453–6457 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaap8045/DC1
Fig. S1. Interfacial tension as a function of time for a system configured with both Fe3O4 NCs and 3K-PDMS-NH2.
Fig. S2. Interfacial tension as a function of time for a system without NCPSs.
Fig. S3. Interfacial tension as a function of time for a system configured with only 3K-PDMS-NH2.
Fig. S4. Interfacial tension as a function of time for a system configured with only zwitterionic ligand 1.
Fig. S5. Interfacial tension as a function of time for a system configured with zwitterionic ligand 1 and 3K-PDMS-NH2.
Fig. S6. Zwitterionic ligand binding to naked NC surfaces.
Fig. S7. Steady-state interfacial tension as a function of zwitterionic ligand concentration.