

Abstract
Objective
To characterize clinically and molecularly an early-onset, variably progressive neurodegenerative disorder characterized by a cerebellar syndrome with severe ataxia, gaze palsy, dyskinesia, dystonia, and cognitive decline affecting 11 individuals from 3 consanguineous families.
Methods
We used whole-exome sequencing (WES) (families 1 and 2) and a combined approach based on homozygosity mapping and WES (family 3). We performed in vitro studies to explore the effect of the nontruncating SQSTM1 mutation on protein function and the effect of impaired SQSTM1 function on autophagy. We analyzed the consequences of sqstm1 down-modulation on the structural integrity of the cerebellum in vivo using zebrafish as a model.
Results
We identified 3 homozygous inactivating variants, including a splice site substitution (c.301+2T>A) causing aberrant transcript processing and accelerated degradation of a resulting protein lacking exon 2, as well as 2 truncating changes (c.875_876insT and c.934_936delinsTGA). We show that loss of SQSTM1 causes impaired production of ubiquitin-positive protein aggregates in response to misfolded protein stress and decelerated autophagic flux. The consequences of sqstm1 down-modulation on the structural integrity of the cerebellum in zebrafish documented a variable but reproducible phenotype characterized by cerebellum anomalies ranging from depletion of axonal connections to complete atrophy. We provide a detailed clinical characterization of the disorder; the natural history is reported for 2 siblings who have been followed up for >20 years.
Conclusions
This study offers an accurate clinical characterization of this recently recognized neurodegenerative disorder caused by biallelic inactivating mutations in SQSTM1 and links this phenotype to defective selective autophagy.
Intracellular clearance of damaged cellular constituents is required for proper cellular function. Autophagy is a major process involved in this control mechanism,1 and its impaired or defective function causes toxic stress, which in turn can drive adverse events, which ultimately may cause cell death.2 Postmitotic neuronal cells are particularly reliant on appropriate autophagic function, and dysfunctional autophagy has been documented to contribute to neurodegenerative disorders.3–5
SQSTM1 (also known as p62) is a multidomain protein involved in a variety of cellular processes, including intracellular signaling, oxidative stress response, and apoptosis.6 In addition, SQSTM1 binds to ubiquitin chains, promoting packaging of ubiquinated cargo by self-oligomerization, and microtubule-associated protein 1A/1B-light chain 3 (LC3), serving as a selective autophagy receptor for degradation of ubiquitinated substrates, and participates in multiple autophagic processes.7 Heterozygous SQSTM1 mutations have been reported in Paget disease of bone (PDB),8,9 amyotrophic lateral sclerosis, and frontotemporal dementia.5,10 More recently, while the present work was underway, biallelic inactivating SQSTM1 mutations were identified to underlie a pediatric neurodegenerative disorder having ataxia, dystonia, and gaze palsy as major features.11
We report 11 affected individuals from 3 consanguineous families carrying homozygous inactivating SQSTM1 mutations. We provide a detailed clinical characterization of this recently recognized neurodegenerative disorder and show that loss of SQSTM1 function causes a slowdown of autophagic flux and impaired production of ubiquitin-positive protein aggregates in response to misfolded protein stress. Finally, we show that suppression of sqstm1 in zebrafish embryos results in a reproducible phenotype affecting the structural integrity of the cerebellum.
Methods
Standard protocol approvals, registrations, and patient consents
Eleven affected individuals from 3 unrelated consanguineous families were included in the study. Approval from the institutional ethical committees of the participating centers was obtained for the genetic studies (Ospedale Pediatrico Bambino Gesù, EC reference 789; Shahid Sadoughi University of Medical Sciences, Institutional Review Board reference 700/D/1109), and written informed consent for the genetic analyses was secured from the guardians of all patients. Affected individuals were examined by experienced neurologists at their primary care centers, and clinical and imaging data were jointly reviewed. Families 2 and 3 were identified through a clinical genetic survey performed on a large cohort of Iranian patients with various pediatric neurologic disorders (≈1,000 families).
Molecular analyses
The genomic analyses were carried out by whole-exome sequencing (WES) in families 1 and 2 and by using a combined approach based on homozygosity mapping analysis in 6 affected individuals coupled to WES performed in a single affected member in family 3. Detailed information on genome-wide single nucleotide polymorphism (SNP) genotyping, WES sequencing, and SNP array/WES data analyses is provided in the data available from Dryad (methods, doi.org/10.5061/dryad.r1vp879). Variant validation and segregation analyses were performed by Sanger sequencing.
Biochemical analyses
We obtained primary fibroblasts from the 2 affected individuals (V.5 and V.6, family 1) by dermal skin biopsy and maintained cultures in high-glucose Dulbecco modified Eagle medium with 10% fetal bovine serum and supplements (Euroclone, Pero, Italy). Inhibition of proteasome- and autophagy-mediated SQSTM1 degradation was attained by treating cells with 100 μmol/L MG132 and 200 nmol/L bafilomycin A1 (Sigma-Aldrich, St. Louis, MO), respectively, for 6 hours. In all experiments evaluating autophagy, we starved cells in Earle balanced salt solution (EBSS; Gibco, Thermo Fisher Scientific, Waltham, MA), as specified. For Western blot analyses, cell extracts were separated by 7.5% or 15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to nitrocellulose or polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA). Blots were incubated with mouse monoclonal anti-SQSTM1 (1:200; Santa Cruz Biotechnology, Dallas, TX), rabbit monoclonal anti-LC3 (1:1000; Cell Signaling, Beverly, MA), mouse monoclonal anti-β actin (1:500; Sigma-Aldrich), and anti-rabbit or anti-mouse horseradish peroxidase–coupled secondary antibody (1:3000; Thermo Fisher). Immune complexes were identified with enhanced chemiluminescence (Promega, Madison, WI).
To investigate the effect of loss of SQSTM1 function in the formation of inclusion bodies containing ubiquitinated proteins, we treated primary cells with 5 μg/mL puromycin (2 hours) and analyzed cells by confocal microscopy. We assessed autolysosome function by using a red BODIPY dye-conjugated (DQ) bovine serum albumin (BSA; Invitrogen, Carlsbad, CA)–based assay.12 Control and patient fibroblasts were incubated with the BSA derivative (10 µg/mL) (1 hour, 37°C) in complete culture medium and then in starvation medium (EBSS) (5 and 16 hours). We assessed proteolysis of this BSA derivative by release of fluorescence (590 nm) using confocal microscopy.
We measured mitochondrial ATP content and complex V activity in primary fibroblasts (V.5, family 1) and an age-matched control, as reported.13 To display the mitochondrial network arrangement, cells were fixed and permeabilized with methanol:acetone (2:1) for 10 minutes at room temperature; then a blocking solution (5% BSA in phosphate-buffered saline) was used. The polyclonal rabbit TOMM20 antibody (Santa Cruz Biotechnology) was applied overnight and visualized with Alexa Fluor 647 secondary antibody (Jackson ImmunoResearch, West Grove, PA), both at 1:500 dilution. Images were acquired with a fluorescence inverted microscope (Leica DMi8, Leica Biosystems, Newcastle, UK). An average of 8 image planes were obtained along the z-axis (0.2-μm increments) with LASX 3.0.4 (Leica). We induced mitochondrial depolarization using 20 µmol/L protonophore carbonyl cyanide m-chlorophenyl hydrazine (CCCP) for 3 hours.
Confocal laser scanning microscopy experiments used 20 × 103 cells seeded in 24-well cluster plates on 12-mm coverglasses and cultured in complete medium (48 hours), as described.14 Cells were stained with mouse monoclonal immunoglobulin (Ig) G anti-SQSTM1 (1:5; Santa Cruz), rabbit polyclonal IgG anti-LC3B (1:100; Novus Biologicals, Littleton, CO), mouse polyclonal IgM anti-polyubiquitin (1:20; FK1, Enzo Life Sciences), and goat anti-mouse IgG, goat anti-mouse IgM, or goat anti-rabbit Alexa Fluor 488–, 594–, or 647–conjugated AffiniPure F(ab′)2 secondary antibodies (1:200; Molecular Probes, Eugene, OR). After staining, coverslips were rinsed and mounted on slides with Vectashield mounting medium (Vector Laboratories, Burlingame, CA) containing 1.5 µg/mL DAPI. Observations were performed on a Leica TCS SP2 AOBS apparatus. Image acquisition and processing were performed as reported.14 Signals from different fluorescent probes were taken in sequential scanning mode, and >50 fields (10 cells per field on average) were analyzed for each labeling condition. Representative results are shown.
Zebrafish studies
Two splicing-blocking antisense morpholino oligonucleotides (MOs) targeting sqstm1 exon 2 and exon 4 were obtained from Gene Tools, LLC (Philomath, OR). We injected casper zebrafish clutches from natural pairwise matings with progressively increasing doses of each MO into 1- to 4-cell stage embryos.15 After titration experiments, we used a dose of 12 and 10 ng for the assays for the exon 2 and 4 MOs, respectively. For rescue experiments, wild-type and mutant SQSTM1 constructs were subcloned into the pCS2+ vector, linearized, and in vitro transcribed with the SP6 mMessage mMachine kit (Ambion, Austin, TX). Larvae were phenotyped at 3 days postfertilization. Permeabilized fixed embryos were incubated with anti–α-acetylated tubulin (1:1000; T7451, Sigma-Aldrich) overnight at room temperature. Larvae were stained with Alexa Fluor 488 goat anti-mouse IgG (1:500; A21207, Invitrogen). Dorsal images of the cerebellum were obtained with the Zeiss AxioCam microscope and then qualitatively scored for structural integrity. All experiments were performed with triplicate batches.
Statistical analysis
For ATP content and complex V activity data comparisons, statistical analyses were performed with the Student t test. For the other in vitro and in vivo comparisons, statistical analyses used a χ2 test. In all comparisons, statistical significant differences were assumed with p < 0.05.
Data availability
Any data not published within the article will be shared on request.
Results
Clinical characterization
Family 1
The 2 affected individuals were 41- and 35-year-old male siblings born to healthy consanguineous parents (third cousins) who originated from a small village of central Italy (figure 1A). They shared the same phenotype and exhibited a similar clinical history. The disorder manifested at the age of 6 and 12 years, respectively, and consisted of a slowly progressive syndrome characterized by cerebellar ataxia, severe hypotonia, bilateral dysmetria, intentional tremor, dysarthria, nystagmus in all position of gaze, ophthalmoparesis, which was more pronounced in the vertical gaze, dyskinesia with bilateral choreoathetotic movements, mild cognitive impairment, and hypergonadotropic hypogonadism. A muscle biopsy performed to explore for a possible mitochondrial disorder documented the absence of mitochondrial structural abnormalities in the presence of a mild reduction of mitochondrial respiratory chain enzyme activities of complexes I-II-IV (residual activity 50%–60%). On the basis of the peculiar association of features and progression of disease, the 2 individuals were supposed to have a previously unrecognized neurodegenerative disorder.
Figure 1. Pedigrees of the 3 families included in the study and location of the identified homozygous SQSTM1 mutations.

(A) Family trees. Squares indicate males; circles indicate females. Solid symbols indicate affected individuals; open symbols represent unaffected relatives. (B) Sequence chromatograms showing homozygosity for the SQSTM1 mutations identified in affected members of the 3 families. (C) Functional and structural domains of SQSTM1 and localization of the identified mutations. KIR = Keap1-interacting; LIR = LC3-interaction region; PB1 = Phox 1 and Bem1p; TRAF6 = tumor necrosis factor receptor–associated factor 6; UBA = ubiquitin-associated; ZZ = zinc finger.
Both siblings have been treated with coenzyme Q10 (150 mg twice daily) and vitamin E (300 mg/d). Dyskinesia responded dramatically to therapy with l-DOPA (Sinemet, 50 mg 3 times daily), 5-hydroxy tryptophan (50 mg 3 times daily), and trihexyphenidyl (2 mg 3 times daily). Since then, the clinical manifestations have appeared stable for several years. Currently, they are still able to walk with small aid and to manage simple occupational and social tasks, although both individuals manifest a severe cerebellar syndrome, gaze palsy, and mild impairment of ≥1 cognitive domains (video 1). The oldest brother had a Mini-Mental State Examination score of 20, and his executive, mnesic, and attentive functions were impaired at neuropsychological evaluation. The youngest sibling showed a Mini-Mental State Examination score of 24 with selective deficit of executive functions. They do not exhibit pyramidal signs, extrapyramidal symptoms (at the side of involuntary movements), neurosensory impairment, hearing loss, or any other ocular abnormality.
Video of the 2 brothers of family 1 at the ages of 41 and 35 years documenting the cerebellar syndrome. Both siblings exhibit ataxia, hypotonia, incoordination, and gaze palsy.Download Supplementary Video 1 (9.5MB, mp4) via http://dx.doi.org/10.1212/879940_Video_1
Hypergonadotropic hypogonadism with low normal testosterone and small testicles was evident since the first stage of the disease in both patients. There was no other endocrinologic dysfunction or manifestation of bone abnormalities. CSF chemical and immunologic analyses and serum coenzyme Q, lactate, creatine kinase, folate, homocysteine, and vitamins B12 and E were normal, and extensive search for known causes of ataxia was negative. During the 26 years of follow-up, 24-hour Holter EEG, polysomnography, EMG, electroneurography, somatosensory and motor evoked potentials, spine MRI, and respiratory and cardiac evaluation were repeatedly normal. Brain imaging was unrevealing (figure 2, A–F), although it showed a mild, subtle cerebellar atrophy developing over time in the first brother, evident in a comparison of the first (at 12 years old) and last (at 38 years old) MRI examinations, and an enlarged cisterna magna in the second brother. Proton magnetic resonance brain spectroscopy demonstrated a lactate peak in ventricular CSF barely visible in basal ganglia (figure 2, G and H), while choline, creatine, and N-acetylaspartate were normal. Dopamine transporter scan revealed a small bilateral reduction of dopamine transporters uptake in the basal ganglia. Consistent with the cognitive profile of both sibs, 99mTc-HMPAO SPECT showed hypoperfusion of right and left frontal lobes in both patients and right parietal cortex and mild right cerebellum hypoperfusion in the older brother (figure 2, I and J). SPECT examinations displayed the same imaging pattern over a 5-year period.
Figure 2. Brain imaging.

Brain MRI of the affected siblings of family 1: (A–C) F1:V.5 (age 38 years) and (D–F) F1:V.6 (age 31 years). (A, B, D, E) Axial and (C and F) coronal fast spin echo T2 images document the absence of any gross anomaly (enlarged cisterna magna in patient F1:V.6), cortical/cerebellar atrophy, or iron accumulation in basal ganglia. Single-voxel proton magnetic resonance brain spectroscopy using intermediate echo time (144 milliseconds) in patient F1:V.5 documenting an inverted doublet of lactate at 1.33 ppm (G) visible at the level of the ventricular CSF (white arrow) and (H) barely visible in basal ganglia. (I) 99mTc-HMPAO SPECT transaxial images (from the vertex to the cerebellum) in patient F1:V.5 shows mild to moderate reduction in radiotracer uptake (hypoperfusion) in the right parietal cortex and in the right and left frontal cortices, as well as asymmetric radiotracer uptake in the cerebellar hemispheres (right < left). (J) In patient 2, mild reduction in radiotracer uptake (hypoperfusion) is documented in the right and left frontal cortices, but no perfusion asymmetry is seen in the cerebellar hemispheres.
Family history was negative, and there was no record of individuals with a similar condition originating from this village. Neurologic examination and endocrinologic assessment of both parents were normal. None of them showed PDB features. Sequencing of the most common ataxia-associated disease genes and mitochondrial DNA did not provide any insights.
Family 2
This consanguineous Iranian family included 2 affected sibs and 4 healthy siblings (2 sisters and 2 brothers) (figure 1A). Both parents were healthy. The proband is a 33-year-old woman who was born with low birth weight. Her affected brother, 29 years old, was born with normal neonatal period without remarkable complaints. Both affected individuals had mild global delay (e.g., delayed sitting, walking, and speech) compared to the healthy siblings. They presented with poor functional abilities but developed milestone regression between 10 and 12 years of age. At the onset of regression, the older sister began to show progressive limb ataxia and cognitive and language impairment. She developed muscle weakness and atrophy affecting predominantly the upper and lower limbs distally. At the age of 16 years, she lost the ability to walk independently, and currently she is disabled and wheelchair bound. The signs and symptoms, disease course, and age at onset in younger affected brother were similar to those of his afflicted sister, with the severity of the symptoms appearing slightly milder. He has clumsy gait and can walk with help, but he is also wheelchair dependent.
The neurologic assessment also showed that both siblings had cognitive decline, progressive dementia, and additional signs of cerebellar dysfunction, including dysdiadochokinesis, dysmetria, and dysarthria. They both also showed abnormal movements such as dystonia and chorea-athetosis, amyotrophy of the hands, and hyperreflexia. Both patients lost their language fluency and developed similar behavioral and psychiatric manifestations, including mutism, executive dysfunction, and apathy. Additional clinical features in both patients include vertical gaze palsy, abnormal saccadic and pursuit eye movements, oculomotor apraxia and orofacial apraxia, urinary incontinence, and pseudobulbar palsy. EMG and nerve conduction velocities were normal, excluding both a neurogenic and a myogenic process as the cause of the weakness. Brain MRI was not performed, but EEG and hearing and vision assessments carried out in the affected boy were normal.
Family 3
The consanguineous extended Iranian family included 12 children from 3 nuclear families, of which 3 girls and 4 boys between 16 and 39 years of age were affected (figure 1A). All affected individuals had a remarkably similar course with progressive difficulty in standing up from floor, unsteady gait, coordination and balance problems, ataxia, and cognitive decline starting at ≈10 to 11 years of age. The disease progressed until the age of 14 to 15 years, and then the progression decelerated. The last clinical examination revealed ataxia, dysarthria, vertical gaze palsy, and hyperreflexia in all individuals. They showed dysarthria and behavioral abnormalities, including temper and aggression. Hearing and vision were normal. None of them is currently wheelchair bound. Brain imaging, muscle biopsy, and EMG were not available.
Clinical findings for the affected individuals from the 3 families are summarized in the table.
Table.
Clinical and neuroradiologic features of the 11 affected individuals carrying homozygous inactivating mutations in SQSTM1 included in the present study and their frequency in all SQSTM1 mutation–positive individuals reported so far

Molecular analyses
Family 1
WES data annotation predicted 12,420 high-quality nonsynonymous changes and variants affecting splice sites. Among them, 310 clinically associated, private/rare variants were retained. Since the documented consanguinity, data filtering was directed to identify homozygous variants compatible with a recessive inheritance model. After the exclusion of genes with variants predicted as benign by combined annotation-dependent depletion (CADD) and MetaSVM, prioritization and variant validation and cosegregation analysis allowed the identification of a previously unreported homozygous variant affecting the exon 2 splice donor site c.301+2T>A (NM_003900.4) in SQSTM1 as the only excellent candidate underlying the disorder (figure 1B). Reverse transcriptase PCR analysis documented aberrant processing of the SQSTM1 transcript in patients, resulting in skipping of exon 2 (figure 3A) and predicting a shorter protein lacking 34 amino acids of the N-terminal Phox and Bem1p (residues 21–103) domains (figure 1C), which mediate protein self-oligomerization.6
Figure 3. Effect of the c.301+2T>A change on transcript processing and protein stability.
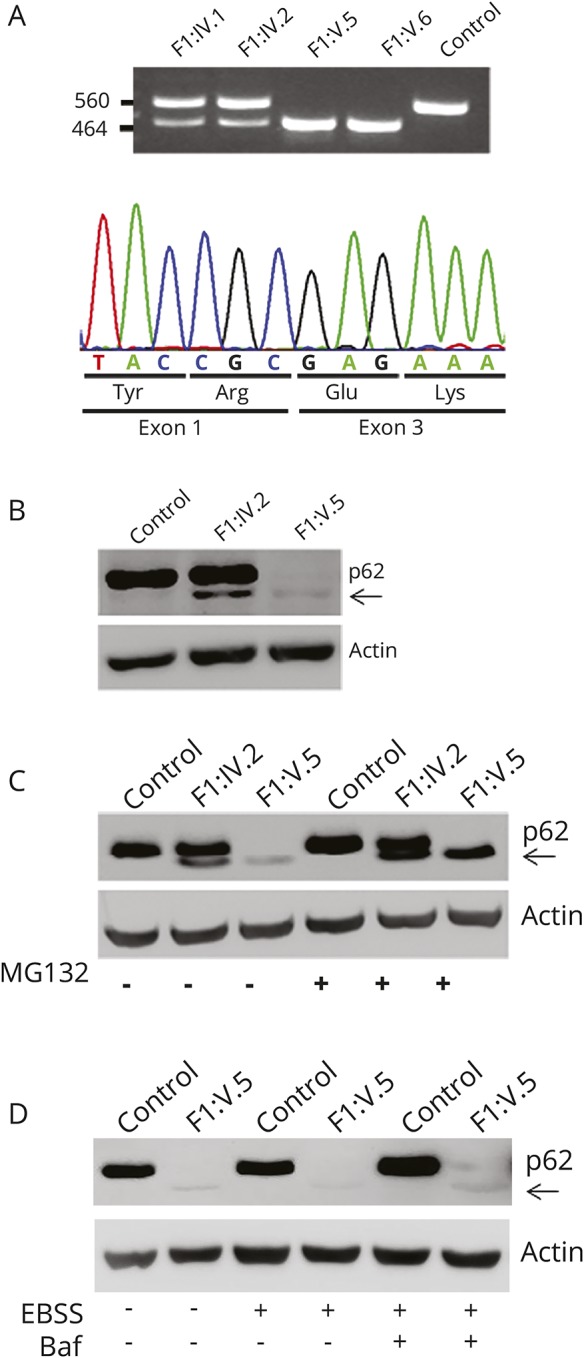
(A) The homozygous c.301+2T>A substitution causes skipping of coding exon 2. Amplification of the cDNA fragment encompassing coding exons 1 to 4 reveals an aberrant PCR product lacking exon 2 in the 2 affected siblings; wild-type and aberrant processed transcripts are observed in both parents (top). Sequencing of the mature SQSTM1 transcript from affected individual F1:V.5 documents skipping of coding exon 2 and the preservation of the phase of the reading frame (bottom). (B) Western blot analysis performed on skin fibroblasts from individual F1:V.5 (right), the unaffected mother (middle), and an unrelated individual (left), indicating that the mutated SQSTM1 protein lacking the amino acid portion encoded by exon 2 (arrow) has a dramatically reduced level compared to the wild-type protein. (C) Treatment of fibroblasts with MG132 (6 hours), an inhibitor of proteasome function, results in a marked increased level of the SQSTM1 mutant, indicating that this protein is rapidly degraded via proteasome. (D) Incubation with bafilomycin A1 (baf) and Earle balanced salt solution (EBSS) (6 hours) does not result in any significant change in the level of the mutated SQSTM1 protein, ruling out a major role of autophagy in the degradation of the SQSTM1 mutant.
Family 2
We performed WES on the DNA from the proband (IV.1). On the assumption of autosomal recessive inheritance and the presence of consanguinity, we prioritized all the homozygous private/rare variants with predicted functional effect residing within blocks of homozygosity. With this filtering approach, a truncating dinucleotide change, c.934_936delinsTGA in SQSTM1, predicting premature termination (p.Arg312*) of the protein emerged as the best disease-causing variant (figure 1B). The variant was located within an ≈8-Mb region of homozygosity, had not previously been reported, and was validated by Sanger sequencing, which also confirmed cosegregation with the disease in the family.
Family 3
We analyzed the extended family by homozygosity mapping using high-resolution SNP genotyping on 6 affected individuals. The analysis identified a single shared region of ≈2 Mb on chromosome 5q35.3 delimited by rs340121 and rs2545083. Copy number variation analysis ruled out the occurrence of any potentially pathogenic structural change. Filtering by position the functionally relevant rare/private variants annotated by WES in an affected member (IV.11) identified a homozygous frameshift variant in SQSTM1 (c.875_876insT) predicted to cause premature termination (p.Asp292Aspfs*9). The variant cosegregated with the phenotype, with all affected family members being homozygous (figure 1B), and was absent from public databases.
Biochemical analyses
While the c.875_876insT and c.934_936delinsTGA changes predicted to result in a prematurely truncated protein, the splice site change, c.301+2T>A, could either result in a functionally defective protein or promote accelerated degradation. To characterize the functional effect of the splice site change, we analyzed the SQSTM1 levels in primary skin fibroblasts obtained from the 2 sibs and their parents of family 1, documenting an almost completely abolished expression of the mutant protein (figure 3B). Treatment with MG132, a proteosomal inhibitor, resulted in restored levels of mutant protein, while bafilomycin A1, an inhibitor of autophagy, was not associated to any significant change in expression, providing evidence of accelerated degradation of the mutant via proteosome (figure 3, C and D, data available from Dryad, figure 1, doi.org/10.5061/dryad.r1vp879). Besides the accelerated degradation, we tested possible proper residual function of the SQSTM1 mutant. Because SQSTM1 is known to play a role in selective autophagy by directing autophagic uptake of proteins via its interaction with LC3,16 we evaluated whether mutant SQSTM1 could bind to LC3 by confocal microscopy analysis. In contrast to cells expressing wild-type SQSTM1, we did not appreciate the colocalization of SQSTM1 bodies and LC3 in patient fibroblasts treated with MG132 to block the accelerated degradation of the disease-associated SQSTM1 mutant, documenting its impaired binding to LC3 (data available from Dryad, figure 2, doi.org/10.5061/dryad.r1vp879).
To explore whether the accelerated degradation and impaired LC3 binding of the SQSTM1 mutant might affect the autophagic pathway, we first analyzed levels and processing of LC3. Primary control and patient fibroblasts were incubated in EBSS for 2, 4, and 6 hours, with and without the addition of the lysosomal inhibitor bafilomycin A1. We evaluated LC3I and LC3II levels by Western blot analysis and observed LC3II accumulation in SQSTM1-deficient cells compared with controls in the absence of the inhibitor but no significant differences between control and patient cells after treatment with bafilomycin A1 (figure 4A). These results were confirmed by confocal laser scanner microscopy analysis (figure 4B) and suggested a delay in or defective activation of the autophagic process. To further explore the extent of dysregulation of the autophagic flux, we monitored the emission of fluorescence of a BSA derivative (DQ-BSA) conjugated to a self-quenched fluorophore in patient fibroblasts and control cells left in starvation for 5 and 16 hours to induce autophagy. We did not observe dequenched DQ-BSA in patient cells over the time period (figure 5A). Overall, the collected data indicated a slowdown of the autophagic flux in cells expressing the mutant protein, possibly because of a less efficient priming of the process promoting autophagosome formation.
Figure 4. Investigation of the autophagic flux.

(A) Microtubule-associated protein 1A/1B-light chain (3LC3) levels were analyzed in control and patient fibroblasts treated with Earle balanced salt solution (EBSS) for different times. Western blot analysis shows that LC3II accumulates in patients' fibroblasts. After incubation of the cells in EBSS and treatment with 200 nmol/L bafilomycin (2 hours), no significant differences were seen between control and patient cells. (B) Confocal laser scanner microscopy analysis was performed in the same experimental conditions reported in panel A and confirms the same findings. Bars correspond to 40 μm.
Figure 5. Analysis of autophagic flux and formation of inclusion bodies containing ubiquitinated proteins.

(A) Primary fibroblasts were incubated with bovine serum albumin derivative (dye-conjugated [DQ] BSA) conjugated to a self-quenched fluorophore (10 µg/mL) for 1 hour at 37°C in complete culture medium and then left in a starvation medium for 5 and 16 hours to induce autophagy. Cells were mounted on coverslips and immediately analyzed by confocal microscopy. Dequenched DQ-BSA was not observed over the time period of 16 hours in patient cells. (B) Primary fibroblasts obtained from one of the 2 affected sibs (right) and an unaffected individual (left) were treated with puromycin (5 μg/mL) for 2 hours to induce formation of polyubiquitin-positive bodies. Cells were then fixed and stained with SQSTM1 (green) and FK1 (red) antibodies. Images are representative of 300 analyzed cells. In all panels, bars correspond to 40 μm.
Several reports have described SQSTM1 as a component of protein aggregates containing polyubiquitinated proteins.16,17 To test the effect of loss of SQSTM1 function in the formation and degradation of inclusion bodies containing polyubiquitinated protein aggregates, control and patient fibroblasts were stained with an anti-ubiquitin monoclonal antibody recognizing polyubiquitinated proteins after treatment with puromycin (2 hours) to induce formation of ubiquitin-containing bodies. As expected, we observed efficient induction of SQSTM1-positive ubiquitin-containing bodies in control cells, while a significantly reduced number was detected in patient cells (figure 5B), indicating impaired formation of inclusion bodies containing polyubiquitinated proteins in primary fibroblasts lacking SQSTM1 function.
Because of the important role of SQSTM1 in selective autophagy of damaged mitochondria,18 we also assessed the consequences of the impaired SQSTM1 function on mitochondrial clearance in patient fibroblasts. We evaluated the mitochondrial network by TOMM20 staining. In regular medium, control and patient cells displayed a widespread and regular tubular organization of mitochondria. However, after CCCP-promoted induction of mitochondrial depolarization, we observed clustering of mitochondria around the nuclei in control fibroblasts but no appreciable modifications in patient fibroblasts (data available from Dryad, figure 3A, doi.org/10.5061/dryad.r1vp879). We also examined mitochondrial ATP production and respiratory chain enzyme activities, observing significantly reduced total cellular ATP content in patient fibroblasts compared to controls in regular medium (−28%) and an even more reduced content (−48%) in galactose medium, reflecting a lower efficiency of ATP production by oxidative phosphorylation (OXPHOS) in stress condition. We confirmed these data by indirect evaluation of the OXPHOS status in patient fibroblasts by measuring the ATP synthesis in purified mitochondria, which displayed decreased activity (≈40% compared to controls, data available from Dryad, figure 3B, doi.org/10.5061/dryad.r1vp879), confirming the generalized partial reduction of mitochondrial respiratory chain enzyme activities previously documented by muscle biopsy.
In vivo analysis
Given the evidence that suppression of genes causative for Gordon-Holmes syndrome can be modeled reliably in the developing zebrafish embryo, especially with regard to the core phenotype of cerebellar hypoplasia,19 we used this model system to examine the consequences of suppression of SQSTM1 function in vivo. The zebrafish genome contains a single sqstm1 ortholog (NP_001299842; 44% identity and 58% similarity with the human gene). We designed a splice-blocking MO targeting the donor splice site of exon 2 to mimic the effect of the disease-causing variant affecting transcript processing, driving suppression of sqstm1 with high efficiency due to skipping of exon 2 of the endogenous mRNA. Then, we evaluated the effect of the MO-induced sqstm1 knockdown on the structural integrity of the cerebellum at 3 days postfertilization. Across duplicate experiments (50–100 embryos per injection), we found structural cerebellar defects in ≈60% of embryos, ranging from depletion of the axonal connections across the midline of the cerebellum to complete atrophy (figure 6). The observed phenotype was specific: the larvae displayed no other overt pathology that might indicate toxicity (body length, timing of swim bladder, edema, or any other apparent gross morphologic defects). Furthermore, coinjection of human wild-type SQSTM1 mRNA was able to rescue the phenotype (figure 6). Different from what was observed for the wild-type allele, coinjection of the disease-associated SQSTM1Δ69–100 cDNA with sqstm1 MO was unable to rescue the phenotype (figure 6), further supporting the loss-of-function effect of the disease-causing mutation. Testing of a second, nonoverlapping MO against the donor site of exon 4 confirmed these findings (data not shown).
Figure 6. In vivo analysis in zebrafish embryos.

(A) Representative dorsal images of anti–α-tubulin (acetylated state)–stained larvae from 4 experimental conditions: control, sqstm1 morpholino (MO) injected, sqstm1_MO+SQSTM1_WT, and sqstm1_MO+SQSTM1_p.69-100del. Blowups from the highlighted area in panel A showing in detail the cerebellar structures in each of the 4 respective conditions presented. (B) Quantification of the qualitative scoring assaying the cerebellar integrity across 3 biological replicas.
Discussion
Here, we report the clinical characterization of an early-onset, variably progressive, neurodegenerative disorder affecting 11 individuals from 3 unrelated families caused by 3 distinct inactivating homozygous mutations in SQSTM1. We describe the natural history of this condition for the 2 siblings from family 1 who have been followed up for >20 years. Functional studies performed in patient fibroblasts demonstrated that defective SQSTM1 function is associated with both a slowdown of the autophagic flux and impaired production of ubiquitin-positive protein aggregates in response to misfolded protein stress. Finally, morpholino-induced sqstm1 down-modulation in zebrafish was shown to cause a reproducible phenotype characterized by structural anomalies of the cerebellum.
Continuous turnover of proteins and organelles is crucial for the maintenance of cellular homeostasis.20 In neurons, which cannot dilute protein aggregates or dysfunctional organelles by cell division, efficient degradation pathways are essential for maintaining cellular health and long-term survival.21–25 SQSTM1 contributes to these clearance systems26 and has been reported to localize to a variety of ubiquitin-positive neuropathologic inclusions27–29 and to protect against misfolded protein stress by promoting the formation of ubiquitin-positive inclusions.17 Consistently, the presently collected data documented defective stress-promoted ubiquitin-positive protein aggregate formation in SQSTM1-deficient cells. Accumulated evidence also indicates that SQSTM1 actively contributes to the autophagic degradation of ubiquitinated proteins.16,17,30 While defective cargo assembly as a result of impaired SQSTM1 function might not exert a cytotoxic effect in dividing cells, it is likely to have adverse consequences on postmitotic cells.
Besides its function as an autophagy adaptor, SQSTM1 contributes to the assembly of the molecular platforms responsible for autophagosomes formation via self-oligomerization and plays a priming role in autophagosome biogenesis.31 The present data suggest that lack of SQSTM1 may weaken the phagophore assembly and cause a slowdown of the autophagosome turnover, as shown by the accumulation of LC3 after 2 hours of starvation and reduced responsiveness of cells after treatment with bafilomycin.
Conflicting data have been obtained on the consequences of defective SQSTM1 function on mitochondrial function and clearance.11,18,32,33 In line with previous findings,11 we observed an aberrant mitochondria perinuclear clustering after inducing mitochondrial depolarization and defective OXPHOS function, which was consistent with a mild reduction of complexes I-II-IV activity in muscle biopsy of F1:V.5 and a small lactate peak in the ventricular CSF in the same patient. These data suggest that secondary mitochondrial dysfunction might contribute to pathogenesis, as documented in other neurodegenerative diseases.34
Major features of the neurodegenerative disorder shared by the 11 affected individuals from the 3 families include ataxia, dysmetria, dysarthria, gaze palsy, cognitive decline, dyskinesia, dystonia, and, in 2 patients, hypergonadotropic hypogonadism. These are in line with the recent report by Haack et al.,11 who described 3 different homozygous inactivating mutations in SQSTM1 in 9 patients from 4 unrelated families. Overall, the striking homogeneity in the 20 affected patients indicates that the core features of this newly recognized, recessive neurodegenerative disorder include childhood-onset cerebellar ataxia (6–15 years), oculomotor involvement, cognitive decline, and dystonia and/or dyskinetic movements (table). The cerebellar ataxia occurred invariantly. Ophthalmoparesis characteristically involves the vertical gaze first and all ocular movements subsequently. Dystonia or dyskinesia has been recorded in 9 and 8 patients, respectively (table), but severe and generalized dystonia was documented in 1 family only,11 in association with a pyramidal syndrome and iron accumulation in basal ganglia. In the 2 siblings of the present family 1, dyskinesia responded well to low doses of l-DOPA, 5-hydroxy-tryptophan, and trihexyphenidyl treatment, with no adverse effects or any requirement of increased dosage during the follow-up, although any attempt to withdraw was associated with reappearance of the symptoms. Of note, dopamine transporter scan revealed a small bilateral reduction of dopamine transporter uptake in the basal ganglia in both siblings. Cognitive impairment is another consistent clinical feature of the disorder; it was recorded in all but 1 individual. In the 2 affected siblings of family 1, it was documented since the onset of disease and appeared stable over time. Neuropsychological assessment demonstrated impairment of executive functions in both sibs and compromised mnesic and attentive functions in the oldest. Accordingly, SPECT showed hypoperfusion of right and left frontal lobes in both patients, extended to the right parietal cortex in the first brother, in the absence of any evidence of atrophy in the corresponding areas at the brain MRI.
A major distinctive finding of the disorder is the relatively mild pattern of cerebellar atrophy despite the particularly severe form of cerebellar syndrome that appears to represent an informative feature in differential diagnosis in the rapidly growing, heterogeneous group of spinocerebellar ataxias. In terms of the course of the disease, it was progressive in most patients, with some losing the ability to walk before the age of 20 years, even though loss of motor or cognitive performances does not represent an invariant feature. In the absence of any treatment details, it is difficult to ascertain whether the relatively stable course in the 2 siblings of family 1 is the result of their early treatment with antioxidants and l-DOPA or simply due to a particularly slow progression of disease. Hypergonadotropic hypogonadism was documented in both the affected siblings of family 1 but could not be investigated in affected members from families 2 and 3 and was not mentioned among the previously reported SQSTM1 mutation–positive patients.11 Hypogonadism has remarkable association with variable neurologic disorders, including cerebellar ataxia,35–37 and has been recently reported as an associated feature of a neurodegenerative syndrome characterized by ataxia, dementia, and hypogonadotropic hypogonadism caused by mutations in RNF216 and OTUD4,19 both encoding proteins implicated in ubiquitin-mediated protein degradation. A dedicated assessment is required to establish definitely any association between hypogonadism and this neurodegenerative disorder.
Heterozygous SQSTM1 mutations have previously been reported in a subset of patients with amyotrophic lateral sclerosis and frontotemporal lobar degeneration, PDB, and distal myopathy with rimmed vacuoles,5,8,10,38,39 which suggests that SQSTM1 is critical for several pathophysiologic pathways and that the individual molecular lesion, environmental factors, and other modifier loci are important in determining expressivity. The present findings provide further data on the relevance of compromised SQSTM1 function in neurodegeneration, linking a distinctive early-onset and variably progressive neurodegenerative trait to the defective clearance of polyubiquitinated aggregates.
Acknowledgment
The authors thank the participating families, Serenella Venanzi (Istituto Superiore di Sanità, Rome) for technical support, and Francesco Cecconi (Università Tor Vergata, Rome) for his helpful comments on the manuscript.
Glossary
- BSA
bovine serum albumin
- CCCP
protonophore carbonyl cyanide m-chlorophenyl hydrazone
- DQ
dye-conjugated
- EBSS
Earle balanced salt solution
- Ig
immunoglobulin
- LC3
microtubule-associated protein 1A/1B-light chain 3
- MO
morpholino oligonucleotide
- OXPHOS
oxidative phosphorylation
- PDB
Paget disease of bone
- SNP
single nucleotide polymorphism
- WES
whole-exome sequencing
Author contributions
Valentina Muto, Elisabetta Flex, Serena Cecchetti, Giovanna Carpentieri, Teresa Rizza, Michela Di Nottia, Simone Martinelli, and Rosalba Carrozzo performed the in vitro studies. Zachary Kupchinsky, Alice Traversa, Maria M. Kousi, and Nicholas Katsanis performed the in vivo studies. Valentina Muto, Hamid Galehdari, Neda Mazaheri, Mohammad Yahya Vahidi Mehrjardi, Yalda Jamshidi, Aaron Jeffries, Viviana Caputo, Andrea Ciolfi, Francesca Pantaleoni, Reza Azizi Malamiri, Jawaher Zeighami, and Marco Tartaglia performed WES/genotyping and genomic data analysis and validation. Mohammadreza Dehghani, Alireza Sedaghat, Guido Primiano, Amir Sherafat, Daniela Di Giuda, Gholam Reza Shariati, and Serenella Servidei performed the clinical assessment of patients and gathered the clinical data. Reza Maroofian, Serenella Servidei, and Marco Tartaglia conceived and coordinated the project. Valentina Muto, Serenella Servidei, and Marco Tartaglia wrote the manuscript. All the authors critically reviewed the manuscript.
Study funding
This work was supported in part by Fondazione Bambino Gesù (Vite Coraggiose) and the Italian Ministry of Health (Ricerca Corrente 2016 and 2017) to M.T.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011;7:279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Götzl JK, Lang CM, Haass C, Capell A. Impaired protein degradation in FTLD and related disorders. Ageing Res Rev 2016;32:122–139. [DOI] [PubMed] [Google Scholar]
- 3.Frake RA, Ricketts T, Menzies FM, Rubinsztein DC. Autophagy and neurodegeneration. J Clin Invest 2015;125:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nixon RA, Yang DS. Autophagy failure in Alzheimer's disease: locating the primary defect. Neurobiol Dis 2011;43:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Majcher V, Goode A, James V, Layfield R. Autophagy receptor defects and ALS-FTLD. Mol Cel Neurosci 2015;66:43–52. [DOI] [PubMed] [Google Scholar]
- 6.Seibenhener ML, Geetha T, Wooten MW. Sequestosome 1/p62: more than just a scaffold. FEBS Lett 2007;581:175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J 2015;282:4672–4678. [DOI] [PubMed] [Google Scholar]
- 8.Rea SL, Majcher V, Searle MS, Layfield R. SQSTM1 mutations: bridging Paget disease of bone and ALS/FTLD. Exp Cel Res 2014;325:27–37. [DOI] [PubMed] [Google Scholar]
- 9.Usategui-Martin R, Garcia-Aparicio J, Corral-Gudino L, Calero-Paniagua I, Del Pino-Montes J, González Sarmiento R. Polymorphisms in autophagy genes are associated with Paget disease of bone. PLoS One 2015;10:e0128984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goode A, Butler K, Long J, Shaw B, Searle MS, Layfield R. Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy 2016;12:1094–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haack TB, Ignatius E, Calvo-Garrido J, et al. Absence of the autophagy adaptor SQSTM1/p62 causes childhood-onset neurodegeneration with ataxia, dystonia, and gaze palsy. Am J Hum Genet 2016;99:735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vázquez CL, Colombo MI. Assays to assess autophagy induction and fusion of autophagic vacuoles with a degradative compartment, using monodansylcadaverine (MDC) and DQ-BSA. Methods Enzymol 2009;452:85–95. [DOI] [PubMed] [Google Scholar]
- 13.Carrozzo R, Torraco A, Fiermonte G, et al. Riboflavin responsive mitochondrial myopathy is a new phenotype of dihydrolipoamide dehydrogenase deficiency: the chaperon-like effect of vitamin B2. Mitochondrion 2014;18:49–57. [DOI] [PubMed] [Google Scholar]
- 14.Flex E, Niceta M, Cecchetti S, et al. Biallelic mutations in TBCD, encoding the tubulin folding cofactor D, perturb microtubule dynamics and cause early-onset encephalopathy. Am J Hum Genet 2016;99:962–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis EE, Frangakis S, Katsanis N. Interpreting human genetic variation with in vivo zebrafish assays. Biochim Biophys Acta 2014;1842:1960–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007;282:24131–24145. [DOI] [PubMed] [Google Scholar]
- 17.Bjørkøy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on Huntingtin-induced cell death. J Cel Biol 2005;171:603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geisler S, Holmstrom KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cel Biol 2010;12:119–131. [DOI] [PubMed] [Google Scholar]
- 19.Margolin DH, Kousi M, Chan YM, et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med 2013;368: 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mizushima N. Autophagy: process and function. Genes Dev 2007;21:2861–2873. [DOI] [PubMed] [Google Scholar]
- 21.Carroll B, Hewitt G, Korolchuk VI. Autophagy and ageing: implications for age-related neurodegenerative diseases. Essays Biochem 2015;55:119–131. [DOI] [PubMed] [Google Scholar]
- 22.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006;441:880–884. [DOI] [PubMed] [Google Scholar]
- 23.Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006;441:885–889. [DOI] [PubMed] [Google Scholar]
- 24.Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy. J Neurosci 2011;31:7817–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nedelsky NB, Todd PK, Taylor JP. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim Biophys Acta 2008;1782:691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cel 2009;33:517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuusisto E, Salminen A, Alafuzoff I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport 2001;12:2085–2090. [DOI] [PubMed] [Google Scholar]
- 28.Kuusisto E, Salminen A, Alafuzoff I. Early accumulation of p62 in neurofibrillary tangles in Alzheimer's disease: possible role in tangle formation. Neuropathol Appl Neurobiol 2002;28:228–237. [DOI] [PubMed] [Google Scholar]
- 29.Zatloukal K, Stumptner C, Fuchsbichler A, et al. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 2002;160:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007;131:1149–1163. [DOI] [PubMed] [Google Scholar]
- 31.Rogov V, Dötsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cel 2014;53:167–178. [DOI] [PubMed] [Google Scholar]
- 32.Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010;6:1090–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okatsu K, Saisho K, Shimanuki M, et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 2010;15:887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu H, Dai C, Fan Y, et al. From autophagy to mitophagy: the roles of P62 in neurodegenerative diseases. J Bioenerg Biomembr 2017;49:413–422. [DOI] [PubMed] [Google Scholar]
- 35.Amor DJ, Delatycki MB, Gardner RJ, Storey E. New variant of familial cerebellar ataxia with hypergonadotropic hypogonadism and sensorineural deafness. Am J Med Genet 2001;99:29–33. [DOI] [PubMed] [Google Scholar]
- 36.Holmes G. A form of familial degeneration of the cerebellum. Brain 1908;30:466–489. [Google Scholar]
- 37.Georgopoulos NA, Papapetropoulos S, Chroni E, et al. Spinocerebellar ataxia and hypergonadotropic hypogonadism associated with familial sensorineural hearing loss. Gynecol Endocrinol 2004;19:105–110. [DOI] [PubMed] [Google Scholar]
- 38.Ralston SH, Layfield R. Pathogenesis of Paget disease of bone. Calcif Tissue Int 2012;91:97–113. [DOI] [PubMed] [Google Scholar]
- 39.Bucelli RC, Arhzaouy K, Pestronk A, et al. SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology 2015;85:665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video of the 2 brothers of family 1 at the ages of 41 and 35 years documenting the cerebellar syndrome. Both siblings exhibit ataxia, hypotonia, incoordination, and gaze palsy.Download Supplementary Video 1 (9.5MB, mp4) via http://dx.doi.org/10.1212/879940_Video_1
Data Availability Statement
Any data not published within the article will be shared on request.