

Abstract
Thiopeptides are members of the ribosomally synthesized and posttranslationally modified peptides (RiPP) family of natural products. Most characterized thiopeptides display nanomolar potency towards Gram-positive bacteria by blocking protein translation with several being produced at the industrial scale for veterinary and live-stock applications. Employing our custom bioinformatics program, RODEO, we expand the thiopeptide family of natural product by a factor of four. This effort revealed many new thiopeptide biosynthetic gene clusters with products predicted to be distinct from characterized thiopeptides and identified gene clusters for previously characterized molecules of unknown biosynthetic origin. To further validate our dataset of predicted thiopeptide biosynthetic gene clusters, we isolated and characterized a structurally unique thiopeptide featuring a central piperidine and rare thioamide moiety. Termed saalfelduracin, this thiopeptide displayed potent antibiotic activity towards several drug-resistant Gram-positive pathogens. A combination of whole-genome sequencing, comparative genomics, and heterologous expression experiments confirmed that the thioamide moiety of saalfelduracin is installed posttranslationally by the joint action of two proteins, TfuA and YcaO. These results reconcile the previously unknown origin of the thioamide in two long-known thiopeptides, thiopeptin and Sch 18640. Armed with these new insights into thiopeptide chemical-genomic space, we provide a roadmap for the discovery of additional members of this natural product family.
Graphical Abstract
INTRODUCTION
With the advent of modern genome sequencing technology and powerful bioinformatics tools to aid in their analysis, it is apparent that ribosomally synthesized and posttranslationally modified peptide (RiPP) natural products are underexplored and pervasive in all domains of life.1 Many bioactive RiPPs are antibiotics, making them potential leads for future drug development. One therapeutically promising class of RiPP are the thiopeptides, which routinely display nanomolar growth suppression activity towards clinically problematic Gram-positive pathogens such as methicillin-resistant Staphylococcus aureus, vancomycin-resistant enterococci, and Clostridium difficile.2,3 Thiostrepton has been employed in topical veterinary applications while LFF571, a semi-synthetic analog of GE2270A, displayed efficacy in treating C. difficile commensurate with vancomycin.4 Nosiheptide also has a long history of use in animal feed where it acts as a growth promoter at sub-therapeutic concentrations. Thiopeptides predominantly exert their antibiotic activity by inhibiting protein synthesis with two notable exceptions: the cyclothiazomycins are reported to inhibit RNA polymerase5 while lactazole is reported to lack antibiotic activity.6 The mechanism of action for thiopeptides acting as protein synthesis inhibitors correlates with the size of the primary macrocycle: 29-atom macrocycles (e.g. thiomuracin and GE2270A) bind elongation factor thermo unstable (EF-Tu)7 while 26- and 32-atom macrocycles (e.g. thiostrepton and berninamycin, respectively) bind to the interface of protein L11 and the 23S rRNA within the 50S ribosomal subunit8 (Figure 1).
Figure 1.

Representative structures of thiopeptides. The number of atoms in the primary macrocycle are given. All known thiopeptides are close structural analogs of these seven natural products.
The complex scaffolds that compose thiopeptide structures all adhere to a common biosynthetic logic. As RiPPs, thiopeptide biosynthesis begins with the ribosomal production of a precursor peptide. RiPP precursor peptides are bipartite and typically consist of an N-terminal leader region that contains a recognition sequence that is directly engaged by the maturation proteins. The C-terminal core region receives the posttranslational modifications to become the mature RiPP.9–11 Recent work has shown that thiopeptide precursor peptides are first acted upon by a trimeric heterocycle synthetase (Ocin-ThiF-dependent YcaO) which cyclodehydrates Cys and select Ser/Thr residues to azoline heterocycles, often with subsequent dehydrogenation to the corresponding azole.12,13 The azol(in)e-bearing intermediate is then dehydrated at other unmodified Ser and Thr residues in a tRNA-dependent manner by a split LanB-like dehydratase, giving rise to dehydroalanine (Dha) and dehydrobutyrine (Dhb) moieties, respectively. The last step that is common to all thiopeptide biosynthetic pathways involves a formal [4+2] cycloaddition between two Dha residues and the amide backbone, giving rise to a six-membered, central N-heterocycle. This cycloaddition forms the primary macrocycle found in all thiopeptides. Most frequently, this central heterocycle appears in mature thiopeptides as pyridine, but other oxidation states are known, such as hydroxypyridine, dehydropiperidine, and piperidine (Figure 1).10
Despite proven industrial and promising clinical applications, only a handful of truly distinct thiopeptide scaffolds have been characterized. Approximately 150 thiopeptides are known; however, the majority of these molecules are close analogs of each other (including naturally-occurring congeners) and fall within seven classes: nosiheptide/nocathiacin-like, thiocillin-like, thiostrepton-like, cyclothiazomycin-like, lactazole-like, thiomuracin/GE2270A-like, and berninamycin-like (Figure 1). All known thiopeptides are variations on these scaffolds by means of amino acid substitutions and/or ancillary modifications such as oxidation, methylation, glycosylation, amidation, and thioamidation.3
Based on our previous bioinformatic work on the pyridine-forming [4+2]-cycloaddition enzyme involved in thiomuracin biosynthesis14, we suspected that the thiopeptide genomic space was far greater than currently appreciated. Due to the recent magnitude of microbial genome sequencing that has taken place, multiple classes of natural products have been significantly expanded through genome-mining.15,16 Therefore, we endeavored to comprehensively catalog and classify all identifiable thiopeptide biosynthetic gene clusters in GenBank. This would allow us to identify biosynthetic gene clusters for thiopeptides of previous unknown genetic origin and the dataset could guide the discovery of new thiopeptides while also shedding light on currently enigmatic biosynthetic reactions.
RESULTS AND DISCUSSION
Thiopeptide genome-mining
The bioinformatics algorithm RODEO (Rapid ORF Description and Evaluation Online) has previously been leveraged to define the genomic landscape of the lasso peptide class of RiPPs.17,18 Given that all thiopeptides share a core set of biosynthetic genes (i.e. Ocin-ThiF-like protein, azoline-forming YcaO cyclodehydratase, azoline dehydrogenase, split LanB dehydratase, and [4+2]-cycloaddition enzyme), homologous gene clusters can be readily identifiable using position-specific iterative BLAST (PSI-BLAST).19 As the presence of a [4+2]-cycloaddition enzyme discriminates thiopeptides from closely related goadsporin-type linear RiPPs,14,20 we chose the encoding gene from the thiomuracin, thiostrepton, nosiheptide, cyclothiazomycin, thiocillin, and berninamycin biosynthetic gene clusters as input for PSI-BLASTP searches to generate a comprehensive, non-redundant list of all [4+2]-cycloaddition enzymes in GenBank.
One difficulty in analyzing thiopeptide [4+2]-cycloaddition enzymes is they bear the initially misleading annotation as members of the “Lant_Dehydr_C” protein superfamily (PF14028).21 Indeed, members of PF14028 carry out the elimination reaction required for Dha/Dhb formation as exemplified by the C-terminal domain of NisB from nisin biosynthesis.22 During the evolution of thiopeptide biosynthesis, it is likely that this domain evolved to catalyze the non-spontaneous [4+2]-cycloaddition reaction characteristic of thiopeptides, as best-evidenced by substantial primary sequence and tertiary structural similarity between the two enzyme types.14
To aid in the differentiation of bona fide [4+2]-cycloaddition enzymes from other members of PF14028, we generated custom profile Hidden Markov Models (pHMMs) for each of the four major subclasses of enzyme found in mature thiopeptides (Figure 2A & Supplementary Dataset 1). Using these models as classification standards, we identified 508 thiopeptide [4+2] enzymes in GenBank with 372 being unique at an amino acid identity cutoff of 95% (Supplementary Dataset 2). This represents a nearly 4-fold expansion of the thiopeptide class of natural products compared to previous estimates.14 This aggregated list of [4+2]-cycloaddition enzymes was used as input for RODEO, in conjunction with the custom pHMMs for the remaining proteins minimally required for thiopeptide biosynthesis: Ocin-ThiF-like protein, azoline-forming YcaO cyclodehydratase,23 and split LanB enzymes involved in Dha/Dhb formation (Supplementary Dataset 1).12 Clusters bearing the majority of core modifying enzymes were included for further analyses.
Figure 2.
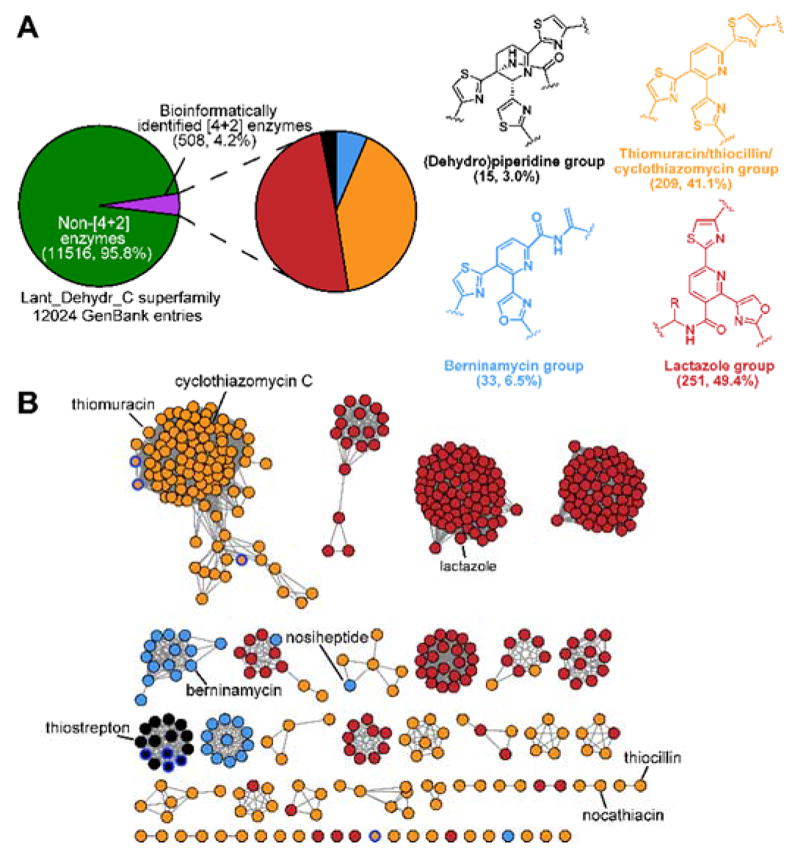
Analysis of thiopeptide [4+2] enzymes found in GenBank. (A) Thiopeptide [4+2] enzymes constitute ~4% of the Lant_Dehydr_C enzyme superfamily. The majority of [4+2] enzymes closely match the lactazole- or thiomuracin-like pHMMs. (B) Sequence similarity network of thiopeptide [4+2] enzymes (100% identical sequences are conflated, alignment score 43, colored as in panel A). While thiomuracin-, thiostrepton-, and berninamycin-like [4+2] enzymes tend to cluster together, the lactazole-like [4+2] enzymes form numerous unexplored groups at this alignment value. Blue outline nodes co-occur with additional TfuA/YcaO-encoding genes.
Most characterized thiopeptides bear a central pyridine heterocycle and the [4+2]-cycloaddition enzyme responsible for its synthesis falls into the thiomuracin-like class of pyridine synthases. Unexpectedly, nearly half of all [4+2]-cycloaddition enzymes matched most closely with the pHMM for lactazole-like pyridine synthases, despite the only known member being lactazole itself (Figure 2).6 Thiopeptide biosynthetic gene clusters containing a lactazole-like [4+2]-cycloaddition enzyme span several large clades that do not contain any characterized members (Figure 2B). These data suggest that there exists a great number of undiscovered thiopeptides that will contain a lactazole-like central pyridine but are otherwise structurally divergent. The remaining three classes of [4+2]-cycloaddition enzymes, thiomuracin-, thiostrepton-, and berninamycin-like, tend to form groupings that correlate with central heterocycle oxidation/substitution status. Analysis of our dataset suggests there is less structural diversity yet to be discovered within these clades compared to thiopeptides biosynthetic gene clusters that harbor a lactazole-like [4+2]-cycloaddition enzyme.
The vast majority (95.1%) of organisms that encode thiopeptide biosynthetic gene clusters belong to either the Actinobacteria or Bacilli bacterial classes (Figure S1). The remaining 4.9% of thiopeptides in our dataset are encoded by a limited number of bacterial phyla not yet reported to naturally produce thiopeptides, including Bacteroidetes, Chloroflexi, Deinococcus-Thermus, and Proteobacteria. Examination of a maximum likelihood tree based on the [4+2]-cycloaddition protein sequence suggests that lactazole-like thiopeptides diverged early from other members of this family (Figure S2–S3). Parametric G+C content comparison of the gene encoding the [4+2]-cycloaddition enzyme versus the genome suggests that most identified clusters were vertically acquired with a few exceptions within both Firmicutes and Actinomyces. This analysis highlights the dominance of Actinobacteria and Firmicutes with their characteristic high and low G+C contents, respectively (Figure S4).
Analysis of precursor peptide diversity and co-occurrence of biosynthetic genes in identified clusters
Thiopeptide precursor peptides were identified and scored using our previously described RODEO algorithm.18 Upon scoring potential thiopeptide precursors, we noticed there was more variability in the leader peptide cleavage site compared to lasso peptides. Therefore, to develop a robust model for predicting the site of thiopeptide leader peptide cleavage, we scanned all potential thiopeptide precursors and identified three distinct motifs that were located at the junction of known leader/core sequences. These three motifs were implemented into the RODEO thiopeptide scoring algorithm (Figure S5).24 In total, 614 sequences were identified as probable thiopeptide precursor peptides, which is greater than the number of [4+2] enzymes owing to some gene clusters encoding more than one precursor peptide. Upon removal of precursor peptides with identical core sequences, 246 unique peptides remained (Supplementary Dataset 2). When the full-length precursor peptides were visualized in the form of a sequence similarity network (SSN), where sequences are displayed as nodes and similarity to another sequence by an edge, two trends emerged: (i) certain thiopeptide precursor subclasses are driven together by sequence similarity in the leader region, despite having highly variable core sequences and, (ii) the majority of predicted thiopeptide precursors are sequence divergent from currently known thiopeptides (Figure 3A). Aligned sequence logos of the core region of the major thiopeptide subclasses highlight the dominant trends in these groups (Figure S6). These trends support the hypothesis that like other RiPPs, thiopeptide structure is diversified mainly through variation of the core sequence while preserving the leader region which is critical for recognition by the biosynthetic proteins.
Figure 3.

Bioinformatic analysis of thiopeptide precursor peptides. (A) Sequence similarity network of precursor peptides (conflated at 100% identity, alignment score of 8). Sequence logos of the top 14 most populous clusters are in Figure S6. Nodes are colored based on Cys content as in panel B. (B) Histogram of Cys content in thiopeptide precursors. (C) Histogram of Ser+Thr content in thiopeptide precursors. (D) Histogram of maximum possible (cyclo)dehydrations excluding critical dehydrations incorporated into pyridine/(dehydro)piperidine. This value is calculated by Cys+Ser+Thr-2 (if the C-terminus is C/S/T, the residue is not counted given these are not known to undergo dehydration).
The sequences of the thiopeptide core region show a wide distribution in Cys content, ranging from 1–10 with a mean of 4.6 ± 2.1 Cys. Ser/Thr content also varies widely, appearing bimodal with most core sequences containing 7 or 13 Ser+Thr residues (Figure 3). Correlating Cys and Ser/Thr counts to determine the maximum number of (cyclo)dehydrations yields a nearly normal distribution around 10–11 residues, although there is an enrichment of sequences that contain 15 dehydratable (i.e. Cys+Ser+Thr) residues. Lastly, the lengths of the thiopeptide core regions is highly variable (range: 10–45 residues); however, the majority contain 17–25 residues (Figure S7). Based on the fact that the majority of groups on both the [4+2]-cycloaddition enzyme and precursor peptide SSNs lack a characterized member, we predict that many unusual thiopeptides remain to be discovered. The data presented here provide a starting point for pursuing such compounds (Figure S6, Supplementary Dataset 2).
To further validate our thiopeptide dataset, we assigned biosynthetic gene clusters for several known thiopeptides previously of unknown genetic origin based on their deduced precursor peptide sequence and tailoring genes. Examples include sulfomycin,25 geninthiocin,26 A10255,27 and Sch 40832 (Figure S8 & Supplementary Dataset 2).28 The latter thiopeptide is distinctive in that it bears the only 6-membered N-heterocycle fused to an imidazoyl-like moiety. Contrary to the previously reported precursor peptide for Sch 40832, our genomic data show that the fused imidazoyl ring derives from Cys instead of Ser.29
Thiopeptide structural diversity is not solely determined by core sequence, but also from additional modification by ancillary tailoring enzymes. Many thiopeptide gene clusters feature ancillary tailoring enzymes, but, with the exception of methyltransferases, no particular class appears in >25% of analyzed gene clusters. This observation is in accord with the known breadth of modification to thiopeptide scaffolds (Supplementary Table S1). Besides methylation, modification by cytochrome P450 enzymes appears to be the next most common ancillary tailoring step. Another prevalent protein family of interest to thiopeptide biosynthesis are members of the radical S-adenosylmethionine (SAM) superfamily. Genes encoding P450 or radical SAM enzymes are present in ~20% of thiopeptide biosynthetic clusters.30,31
When superimposed on the [4+2]-cycloaddition enzyme SSN (Figure 2, S3), it becomes apparent that thiopeptide biosynthetic gene clusters encoding P450 or radical SAM enzymes are not constrained to any particular group. Presumably, acquisition of these and other additional modification enzymes provided a gain-of-function, as it is pervasive amongst thiopeptide biosynthetic gene clusters (Figure S3). The modular nature of RiPP biosynthesis allows for a few dominating modifications, but also a magnitude of diverse modifications that are less frequently encountered.
Among these infrequently co-occurring genes, we found that 2% of thiopeptide biosynthetic clusters feature a gene encoding a TfuA-like protein (PF07812, Supplementary Table S1). Previous work has shown that ~10% of all YcaO proteins are encoded next to TfuA.32 Recently, the TfuA-YcaO pair from Methanosarcina acetivorans, a methanogenic archaeon, was found to be responsible for a posttranslational thioamidation modification. Conversion of a key glycine residue to thioglycine within methyl-coenzyme M reductase has been shown to be ubiquitous amongst methanogens.33,34 Nature employs several distinct strategies to form carbon-sulfur bonds.35 In contrast to methanobactins, whose thioamides are formed in a YcaO-independent fashion36,37, thioviridamide biosynthetic gene clusters encode un-characterized TfuA-YcaO pairs,38–41 and hence we reasoned that thiopeptides encoding these two proteins may also feature a thioamide modification. Two known thiopeptides have been reported that feature a single thioamide modification at equivalent positions in Sch 18640 and thiopeptin.42,43 Although the genome sequences for the producing organisms were not available, our bioinformatics survey detected a thiostrepton-like biosynthetic gene cluster in Amycolatopsis saalfeldensis NRRL B-24474 that contained a tfuA-ycaO gene pair. Subsequently, this strain was screened for the production of a new thiopeptide.
Isolation and characterization of a new thiopeptide
Reactivity-based screening augments typical media/growth condition testing for the production of compound of interest by exploiting selective chemical reactivity of functional groups.44 With few exceptions, all known thiopeptides contain Dha or Dhb residues that are susceptible to 1,4-nucleophilic addition by reaction with a soft nucleophile. The predicted A. saalfeldensis precursor peptide contains 10 Ser+Thr residues. Based on the structure of thiostrepton, we predicted the following: two Ser residues would be employed for the [4+2]-cycloaddition, one Thr would be used for linking to the quinaldic acid moiety, and one Thr would be left unmodified as in thiostrepton. This analysis thus predicted the presence of 6 Michael acceptors in the final structure (5 Dha, 1 Dhb). Upon reacting the methanolic extract of A. saalfeldensis cultures with dithiothreitol, we identified by mass spectrometry (MS) a species that contained upwards of six DTT adducts (ions with 4–5 labels were the most intense), consistent with the prediction (Figure S9).
Further inspection of the A. saalfeldensis biosynthetic gene cluster revealed significant similarity to other (dehydro)piperidine-containing thiopeptides. In particular, genes for the biosynthesis of the hallmark quinaldic acid moiety are present and conservation of Thr12 in the precursor peptide suggested a secondary, ester-linked macrocycle similar to that seen in thiostrepton. Missing from the A. saalfeldensis gene cluster was the asparagine synthetase responsible for C-terminal amidation45, implying the compound would display a carboxylate terminus (Figure 4). High-resolution electrospray ionization tandem MS (HR-ESI-MS/MS) confirmed the predicted chemical formula (C69H77N18O18S6+). This formula is consistent with: (i) the 6 Dha/Dhb groups identified by reactivity towards dithiothreitol, (ii) the bioinformatically predicted C-terminal carboxylate, and (iii) the bioinformatically predicted thioamidation (replacement of an oxygen atom with sulfur) akin to Sch 18640 and thiopeptin. Tandem MS fragmentation of the quinaldic acid linkage involving residue Ala1 and Thr12 resembled that of known thiopeptides (Supplementary Table S2, Figure S10). Notably, the best alternative chemical formula, which involves oxidation (addition of oxygen) was inconsistent with our observed mass.
Figure 4.
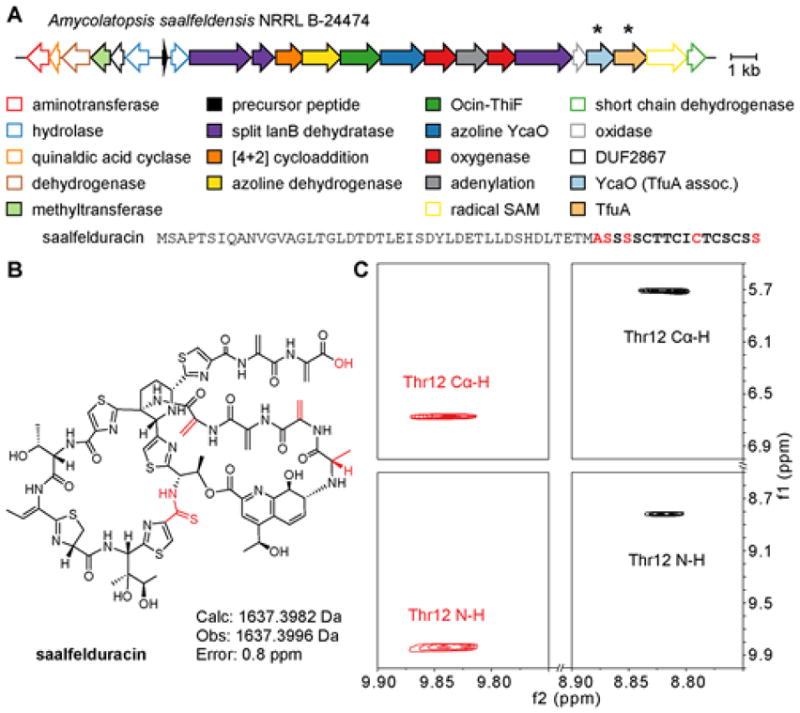
Discovery of saalfelduracin (A) Saalfelduracin biosynthetic gene cluster and predicted precursor peptide (B) Proposed structure of saalfelduracin with motifs distinct from thiostrepton in red (C) 1H shift of the Thr12 amide and Cα proton in saalfelduracin (red), compared to thiostrepton (black).
To confirm the presence of the thioamide, we purified a larger quantity the A. saalfeldensis thiopeptide, now termed saalfelduracin, and subjected the compound to 1H, 1H-1H TOCSY, and 1H-1H NOESY NMR experiments. Analysis of the spectra confirmed the predicted structure. Spin systems consistent with all modifications, including the quinaldic acid, dehydrated amino acids, and side chains were identified (Supplementary Table S3, Figures S11–14). In contrast to other members of the (dehydro)piperidine thiopeptides, we only observed the formation of a central piperidine central heterocycle. Assignment of the thioamide at position Cys11-Thr12 was determined through analysis of amide protons. The amide NH of Thr12 was downshifted significantly (9.83 ppm), as well as the CαH (6.67 ppm), due to the electronic effects of the thiocarbonyl (Figure 4C). These chemical shift values are consistent with reported thioamide chemical shifts from Sch 18640 and thiopeptin.43,46,47 A commercial standard of thiostrepton was obtained and the NMR spectrum showed that the Thr12 NH and CαH protons were significantly shielded (~1 ppm) relative to the corresponding nuclei of saalfelduracin (Figure 4C).
To evaluate the antibacterial activity of saalfelduracin, we measured the minimum inhibitory concentration (MIC) against a panel of human pathogenic bacteria. Saalfelduracin was not active against any Gram-negative bacterium tested, but the compound was strongly growth inhibitory towards against Gram-positive bacteria, including vancomycin-resistant Enterococcus and methicillin-resistant Staphylococcus aureus (Table 1). The potency and spectrum of activity of saalfelduracin is approximately equivalent to thiostrepton.
Table 1.
Antibiotic activity of saalfelduracin and thiostrepton as determined by microbroth dilution assay. MIC, minimum inhibitory concentration.
| Strain | MIC (μg/mL) | |
|---|---|---|
| saalfelduracin | thiostrepton | |
| Staphylococcus aureus USA300 | 0.25 | 0.12 |
| Enterococcus faecium U503 | 0.12 | 0.12 |
| Bacillus anthracis str. Sterne | 0.25 | 0.25 |
| Escherichia coli MC4100 | >64 | >64 |
| Pseudomonas aeruginosa PAO1 | >64 | >64 |
| Acinetobacter baumannii ATCC 19606 | >64 | >64 |
Characterization of thiopeptide thioamidation
Intrigued by the rarity of thioamides as a posttranslational modification, we sought to determine if the TfuA-YcaO pair was responsible for installing thioamides in thiopeptide natural products. The TfuA and YcaO proteins encoded by A. saalfeldensis belong to protein families PF07812 and PF02624, respectively. Unlike the YcaOs responsible for azoline formation via the ATP-dependent cyclodehydration of Cys/Ser/Thr residues, TfuA-associated YcaO proteins are shorter and do not contain the highly conserved Pro-rich C-terminal sequence.48,49 We reasoned that the other thiopeptides reported to contain thioamides (i.e. Sch 18640 and thiopeptin) would have biosynthetic gene clusters that also contain tfuA-ycaO genes; however, the genomes of the producing organisms were not available. Therefore, we obtained the natural producers of Sch 18640 and thiopeptin (Micromonospora arborensis NRRL 8041 and Streptomyces tateyamensis ATCC 21389, respectively), sequenced the genomes, and assembled the reads into ~200 contigs for each organism. Homologs of the TfuA-like protein from A. saalfeldensis and the thioviridamide producer Streptomyces olivoviridis were used to query the sequenced genomes using translated BLAST (TBLASTN). A single matching sequence was identified adjacent to a YcaO homolog in both organisms (Figure 5). Further analysis of the local region confirmed these appear to be valid thiopeptide biosynthetic gene clusters given that the requisite genes for an azoline-forming cyclodehydratase, split LanB enzymes, and [4+2]-cycloaddition enzyme were identified. Short genes encoding the precursor peptides were found as well that perfectly matched the inferred core sequences of Sch 18640 and thiopeptin.
Figure 5.
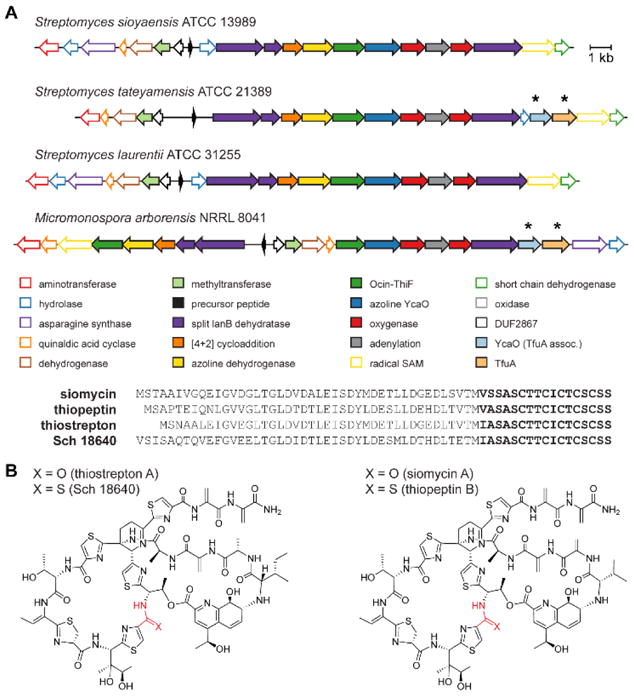
(A) Organization of thiopeptide biosynthetic gene clusters and precursor peptides with core sequences in bold. Whole-genome sequencing of S. tateyamensis and M. arborensis revealed analogous thiopeptide gene clusters with additional ycaO-tfuA genes, denoted by asterisks. (B) Structures of thiopeptides with thioamide analogs (red).
In general, a high level of conservation was seen for the gene clusters of thioamide-containing (dehydro)piperidine thiopeptides. Most protein homologs encoded by these gene clusters were >30% sequence identical across species, with the greatest sequence conservation values found for the enzymes involved in the modifications common to all thiopeptides (Supplementary Table S4 and Figure S15). Concurrently, synteny analysis of the biosynthetic gene clusters also indicated conservation of the relative gene order, with exceptions occurring near the precursor peptide gene and the presumed gene cluster boundaries (Figure S16). Taken together, these data suggest that variation in the precursor sequence, as well as the genetic content of the gene cluster periphery, are the primary mechanisms by which thiopeptide structural diversity is introduced.
With the finding that all thioamide-containing thiopeptides contain tfuA-ycaO genes in their respective biosynthetic gene clusters, there was a clear genetic link to the modification, but no direct biochemical evidence that either gene was responsible for thiopeptide thioamidation. To evaluate the role of these genes in thioamide installation, we sought to convert the thiopeptide thiostrepton (naturally produced by Streptomyces laurentii, as well as a limited number of other strains) to its known thioamidated analog, Sch 18640 (naturally produced by M. arborensis) through heterologous expression. We reasoned that the M. arborensis TfuA-YcaO pair might be able to process a non-cognate thiopeptide precursor if the sequence was sufficiently similar to its native substrate. Notably, mature thiostrepton and Sch 18640 only differ by thioamidation, although there are a number of amino acid substitutions in the leader regions of the precursor peptides (the same is true for siomycin and thiopeptin, Figure 5). As indicated, the saalfelduracin precursor sequence is more divergent, however, the location of the thioamide (Cys11-Thr12) is unchanged.
To determine whether genetic introduction of a TfuA-YcaO pair into a thiostrepton-only producer would result in a thioamidated analog, a bacteriophage integrase system was employed to mediate chromosomal insertion of tfuA and ycaO. Via a phage integrase protein, plasmids bearing the attP locus will insert the vector as long as the host chromosome contains an attB locus.50–52 The bacteriophage ΦC31 integrase targets attB sequences found in many Streptomyces species, including S. laurentii NRRL B-24298, which strictly produces thiostrepton.53 Our integrative plasmid also included an engineered, strong, constitutive promoter (ermE*p) for driving the expression of the non-cognate tfuA-ycaO genes (Figure S17 and Supplementary Table S5–S6).
Conjugation and subsequent integration of S. laurentii with the integrative plasmid containing tfuA and ycaO were subsequently screened by MS to monitor thioamide congener formation. Upon introduction of the M. arborensis tfuA/ycaO, a metabolite 16 Da heavier than thiostrepton was observed (Figure 6). This +16 Da metabolite was also observed after introduction of either the S. tateyamensis or A. saalfeldensis tfuA/ycaO gene pairs. Both tfuA and ycaO were necessary for the +16 Da metabolite to be observed, given that omission of either tfuA or ycaO only yielded a mass consistent with thiostrepton (Figure S18). HR-ESI-MS confirmed the +16 Da species to be chemically identical to Sch 18640 (m/z calculated: 1680.4768 Da; observed: 1680.4781 Da; error: 0.8 ppm). These data suggest that all three tested TfuA-YcaO pairs were flexible enough to act upon a non-cognate substrate. Future work will be necessary to delineate at which point in the biosynthetic pathway thioamidation takes place. The precise role of this modification, if any, also awaits further exploration.
Figure 6.
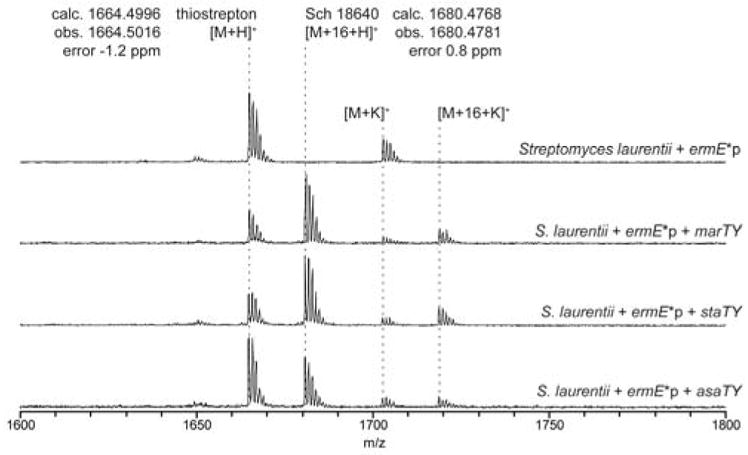
MALDI-TOF MS of S. laurentii integration mutants. Colonies of S. laurentii with chromosomal copies of ycaO-tfuA genes under strong constitutive promotion from M. arborensis (marTY), S. tateyamensis (staTY), and A. saalfeldensis (asaTY) were analyzed by MALDI-TOF MS. Conjugants produced a thioamidated species (m/z 1680), 16 Da greater than unmodified thiostrepton (m/z 1664) and consistent with Sch 18640. High-resolution masses were obtained on an Orbitrap instrument.
CONCLUSION
Thiopeptides are among the most posttranslationally modified RiPP natural products identified, originally being misidentified as non-ribosomal peptides. The remarkable structural complexity of thiopeptides is due to a modular “plug and play” logic to the modifications displayed by thiopeptides. Indeed, the core modifications of thiopeptides appear to be a natural hybrid of two progenitor classes of RiPPs: linear azole-containing peptides and lanthipeptides, with an additional [4+2]-cycloaddition reaction giving rise to a central N-heterocycle. Mutations to the precursor peptide genes further amplify the diversity of thiopeptides.
Despite their intrinsic potential for structural divergence, most known thiopeptides are close structural homologs of one another. To enable a more rational exploration of the thiopeptide chemical-genomic space, we cataloged and classified all observable biosynthetic gene clusters in GenBank. Leveraging this dataset, we connected a protein modification universally present in methanogenic archaea to a comparatively rare and understudied modification in peptidic natural products. From this, we used a genomics-driven approach to isolate a new thiopeptide and reconcile the enigmatic biosynthetic pathways of several previously known thiopeptides. Lastly, we demonstrated the activity of the genes responsible for thiopeptide thioamidation, which adds yet another characterized modification to the growing list of reactions seen in these complex natural products.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by the National Institutes of Health (GM097142 to D.A.M.), the Chemistry–Biology Interface Training Program (GM070421 to C.J.S.), and the Seemon Pines Fellowship from the Department of Chemistry at the University of Illinois at Urbana-Champaign (to G.A.H.).
This work was supported in part by the National Institutes of Health (GM097142 and GM123998 to D.A.M.), the Chemistry–Biology Interface Training Program (GM070421 to C.J.S.), and the Seemon Pines Fellowship from the Department of Chemistry at the University of Illinois at Urbana-Champaign (to G.A.H.). We thank M.N. Goettge and W.W. Metcalf for the gift of microbiological materials and assistance and A.J DiCaprio for NMR data acquisition.
Footnotes
Notes
The authors declare no competing financial interests.
The Supporting Information, including experimental methods and supporting figures, is available free of charge on the ACS Publications website.
References
- 1.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian K-D, Fischbach MA, Garavelli JS, Goransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Muller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJT, Rebuffat S, Ross RP, Sahl H-G, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Sussmuth RD, Tagg JR, Tang G-L, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA. Nat Prod Rep. 2013;30:108. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bagley MC, Dale JW, Merritt EA, Xiong X. Chem Rev. 2005;105:685. doi: 10.1021/cr0300441. [DOI] [PubMed] [Google Scholar]
- 3.Just-Baringo X, Albericio F, Álvarez M. Mar Drugs. 2014;12:317. doi: 10.3390/md12010317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mullane K, Lee C, Bressler A, Buitrago M, Weiss K, Dabovic K, Praestgaard J, Leeds JA, Blais J, Pertel P. Antimicrob Agents Chemother. 2015;59:1435. doi: 10.1128/AAC.04251-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hashimoto M, Murakami T, Funahashi K, Tokunaga T, Nihei K-i, Okuno T, Kimura T, Naoki H, Himeno H. Bioorganic Med Chem. 2006;14:8259. doi: 10.1016/j.bmc.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi S, Ozaki T, Asamizu S, Ikeda H, Ōmura S, Oku N, Igarashi Y, Tomoda H, Onaka H. Cell Chem Biol. 2014;21:679. doi: 10.1016/j.chembiol.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 7.Heffron SE, Jurnak F. Biochemistry. 2000;39:37. doi: 10.1021/bi9913597. [DOI] [PubMed] [Google Scholar]
- 8.Reusser F. Biochemistry. 1969;8:3303. doi: 10.1021/bi00836a026. [DOI] [PubMed] [Google Scholar]
- 9.Burkhart BJ, Hudson GA, Dunbar KL, Mitchell DA. Nat Chem Biol. 2015;11:564. doi: 10.1038/nchembio.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burkhart BJ, Schwalen CJ, Mann G, Naismith JH, Mitchell DA. Chem Rev. 2017;117:5389. doi: 10.1021/acs.chemrev.6b00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hudson GA, Mitchell DA. Curr Opin Microbiol. 2018;45:61. doi: 10.1016/j.mib.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hudson GA, Zhang Z, Tietz JI, Mitchell DA, van der Donk WA. J Am Chem Soc. 2015;137:16012. doi: 10.1021/jacs.5b10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Z, Hudson GA, Mahanta N, Tietz JI, van der Donk WA, Mitchell DA. J Am Chem Soc. 2016;138:15511. doi: 10.1021/jacs.6b08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cogan DP, Hudson GA, Zhang Z, Pogorelov TV, van der Donk WA, Mitchell DA, Nair SK. Proc Natl Acad Sci USA. 2017;114:12928. doi: 10.1073/pnas.1716035114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bachmann BO, Van Lanen SG, Baltz RH. J Ind Microbiol Biotechnol. 2014;41:175. doi: 10.1007/s10295-013-1389-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ziemert N, Alanjary M, Weber T. Nat Prod Rep. 2016;33:988. doi: 10.1039/c6np00025h. [DOI] [PubMed] [Google Scholar]
- 17.Blin K, Wolf T, Chevrette MG, Lu X, Schwalen CJ, Kautsar SA, Suarez Duran HG, de los Santos Emmanuel LC, Kim HU, Nave M, Dickschat JS, Mitchell DA, Shelest E, Breitling R, Takano E, Lee SY, Weber T, Medema MH. Nucleic Acids Res. 2017;45:W36. doi: 10.1093/nar/gkx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tietz JI, Schwalen CJ, Patel PS, Maxson T, Blair PM, Tai HC, Zakai UI, Mitchell DA. Nat Chem Biol. 2017;13:470. doi: 10.1038/nchembio.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Onaka H, Nakaho M, Hayashi K, Igarashi Y, Furumai T. Microbiology (Reading, England) 2005;151:3923. doi: 10.1099/mic.0.28420-0. [DOI] [PubMed] [Google Scholar]
- 21.Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman A. Nucleic Acids Res. 2016;44:D279. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garg N, Salazar-Ocampo LMA, van der Donk WA. Proc Natl Acad Sci USA. 2013;110:7258. doi: 10.1073/pnas.1222488110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunbar KL, Tietz JI, Cox CL, Burkhart BJ, Mitchell DA. J Am Chem Soc. 2015;137:7672. doi: 10.1021/jacs.5b04682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. Nucleic Acids Res. 2009;37:W202. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abe H, Kushida K, Shiobara Y, Kodama M. Tetrahedron Lett. 1988;29:1401. [Google Scholar]
- 26.Yun BS, Hidaka T, Furihata K, Seto H. J Antibiot (Tokyo) 1994;47:969. doi: 10.7164/antibiotics.47.969. [DOI] [PubMed] [Google Scholar]
- 27.Boeck LD, Berry DM, Mertz FP, Wetzel RW. J Antibiot (Tokyo) 1992;45:1222. doi: 10.7164/antibiotics.45.1222. [DOI] [PubMed] [Google Scholar]
- 28.Puar MS, Chan TM, Hegde V, Patel M, Bartner P, Ng KJ, Pramanik BN, MacFarlane RD. J Antibiot (Tokyo) 1998;51:221. doi: 10.7164/antibiotics.51.221. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Qu X, He X, Duan L, Wu G, Bi D, Deng Z, Liu W, Ou HY. PLoS One. 2012;7:e45878. doi: 10.1371/journal.pone.0045878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahanta N, Hudson GA, Mitchell DA. Biochemistry. 2017;56:5229. doi: 10.1021/acs.biochem.7b00771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng Q, Fang H, Liu W. Org Biomol Chem. 2017;15:3376. doi: 10.1039/c7ob00466d. [DOI] [PubMed] [Google Scholar]
- 32.Schwalen CJ, Hudson GA, Kosol S, Mahanta N, Challis GL, Mitchell DA. J Am Chem Soc. 2017;139:18154. doi: 10.1021/jacs.7b09899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nayak DD, Mahanta N, Mitchell DA, Metcalf WW. eLife. 2017;6 doi: 10.7554/eLife.29218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahanta N, Liu A, Dong S, Nair SK, Mitchell DA. Proc Natl Acad Sci USA. 2018;115:3030. doi: 10.1073/pnas.1722324115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunbar KL, Scharf DH, Litomska A, Hertweck C. Chem Rev. 2017;117:5521. doi: 10.1021/acs.chemrev.6b00697. [DOI] [PubMed] [Google Scholar]
- 36.Kenney GE, Dassama LMK, Pandelia ME, Gizzi AS, Martinie RJ, Gao P, DeHart CJ, Schachner LF, Skinner OS, Ro SY, Zhu X, Sadek M, Thomas PM, Almo SC, Bollinger JM, Krebs C, Kelleher NL, Rosenzweig AC. Science. 2018;359:1411. doi: 10.1126/science.aap9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sébastien C, Cornelia CA, KMN, Agnieszka L, Klaus R, Ludger B, Christian H. Angew Chemie Int Ed. 2013;52:10564. [Google Scholar]
- 38.Hayakawa Y, Sasaki K, Nagai K, Shinya K, Furihata K. J Antibiot (Tokyo) 2006;59:6. doi: 10.1038/ja.2006.2. [DOI] [PubMed] [Google Scholar]
- 39.Frattaruolo L, Lacret R, Cappello AR, Truman AW. ACS Chem Biol. 2017;12:2815. doi: 10.1021/acschembio.7b00677. [DOI] [PubMed] [Google Scholar]
- 40.Kjaerulff L, Sikandar A, Zaburannyi N, Adam S, Herrmann J, Koehnke J, Muller R. ACS Chem Biol. 2017;12:2837. doi: 10.1021/acschembio.7b00676. [DOI] [PubMed] [Google Scholar]
- 41.Kawahara T, Izumikawa M, Kozone I, Hashimoto J, Kagaya N, Koiwai H, Komatsu M, Fujie M, Sato N, Ikeda H, Shin-Ya K. J Nat Prod. 2018;81:264. doi: 10.1021/acs.jnatprod.7b00607. [DOI] [PubMed] [Google Scholar]
- 42.Miyairi N, Miyoshi T, Aoki H, Kosaka M, Ikushima H. Antimicrob Agents Chemother. 1972;1:192. doi: 10.1128/aac.1.3.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Puar MS, Ganguly AK, Afonso A, Brambilla R, Mangiaracina P, Sarre O, MacFarlane RD. J Am Chem Soc. 1981;103:5231. [Google Scholar]
- 44.Cox CL, Tietz JI, Sokolowski K, Melby JO, Doroghazi JR, Mitchell DA. ACS Chem Biol. 2014;9:2014. doi: 10.1021/cb500324n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liao R, Liu W. J Am Chem Soc. 2011;133:2852. doi: 10.1021/ja1111173. [DOI] [PubMed] [Google Scholar]
- 46.Hensens OD, Albers-Schönberg G. Tetrahedron Lett. 1978;19:3649. [Google Scholar]
- 47.Hensens OD, Albers-Schonberg G. J Antibiot (Tokyo) 1983;36:814. doi: 10.7164/antibiotics.36.814. [DOI] [PubMed] [Google Scholar]
- 48.Dunbar KL, Melby JO, Mitchell DA. Nat Chem Biol. 2012;8:569. doi: 10.1038/nchembio.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dunbar KL, Chekan JR, Cox CL, Burkhart BJ, Nair SK, Mitchell DA. Nat Chem Biol. 2014;10:823. doi: 10.1038/nchembio.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bierman M, Logan R, O’Brien K, Seno ET, Rao RN, Schoner BE. Gene. 1992;116:43. doi: 10.1016/0378-1119(92)90627-2. [DOI] [PubMed] [Google Scholar]
- 51.Groth AC, Olivares EC, Thyagarajan B, Calos MP. Proc Natl Acad Sci USA. 2000;97:5995. doi: 10.1073/pnas.090527097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herrmann S, Siegl T, Luzhetska M, Petzke L, Jilg C, Welle E, Erb A, Leadlay PF, Bechthold A, Luzhetskyy A. Appl Environ Microbiol. 2012;78:1804. doi: 10.1128/AEM.06054-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trejo WH, Dean LD, Pluscec J, Meyers E, Brown WE. J Antibiot (Tokyo) 1977;30:639. doi: 10.7164/antibiotics.30.639. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.