

Abstract
Neuroinflammation is consistently found in many neurological disorders, but whether or not the inflammatory response independently affects neuronal network properties is poorly understood. Here, we report that intracerebroventricular injection of the prototypical inflammatory molecule lipopolysaccharide (LPS) in rats triggered a strong and long lasting inflammatory response in hippocampal microglia associated with a concomitant up-regulation of Toll-like receptor (TLR4) in pyramidal and hilar neurons. This, in turn, was associated with a significant reduction of the dendritic hyperpolarization-activated cyclic-AMP gated channel type 1 (HCN1) protein level while Kv4.2 channels were unaltered as assessed by western blot. Immunohistochemistry confirmed the HCN1 decrease in CA1 pyramidal neurons and showed that these changes were associated with a reduction of TRIP8b, an auxiliary subunit for HCN channels implicated in channel subcellular localization and trafficking.
At the physiological level, this effect translated into a 50% decrease in HCN1-mediated currents (Ih) measured in the distal dendrites of hippocampal CA1 pyramidal cells. At the functional level, the band-pass filtering properties of dendrites in the theta frequency range (4-12 Hz) and their temporal summation properties were compromised.
We conclude that neuroinflammation can independently trigger an acquired channelopathy in CA1 pyramidal cell dendrites that alters their integrative properties. By directly changing cellular function, this phenomenon may participate in the phenotypic expression of various brain diseases.
Keywords: cytokines, HMGB1, HCN1, channelopathy, lipopolysaccharide, rat, brain disease
Introduction
Chronic CNS pathologies, including epilepsy, Alzheimer’s and Parkinson’s diseases, multiple sclerosis, amyotrophic lateral sclerosis and neuropathic pain [1-6] as well as acute brain injuries with long term neurological sequelae such as infections, stroke and neurotrauma [7, 8] display similar neuronal network reorganizations, suggesting the presence of shared pathogenic mechanisms. A common hallmark of these conditions is the presence of neuroinflammation [9-12]. Neuroinflammation is evoked by infectious or non-infectious (sterile) brain injuries as well as increased neuronal activity (i.e. neurogenic inflammation [10]), and consists of the induction of molecules and pathways associated with innate immunity activation in glial cells, neurons, and endothelial cells of the blood brain barrier. To date, there has been a lack of studies examining the temporal resolution of the neuroinflammatory response responsible for tissue dysfunction that has been implicated in both acute and chronic CNS diseases [13]. Moreover, it is unclear whether or not neuroinflammation can trigger network reorganization by itself. It is however well established that pivotal components of the neuroinflammatory response such as interleukin (IL)-1β, tumor necrosis factor α and the danger signal protein High Mobility Group Box 1 (HMGB1), and their related downstream effector molecules, have strong neuromodulatory effects that are evoked by the activation of their cognate receptors expressed by neurons in diseased tissue [9, 10, 14]. Neuroinflammatory mediators can alter voltage- and ligand-gated channels [14], disrupt synaptic plasticity [15], lower seizure threshold [9] and even induce epileptiform discharges [16]. Neuroinflammation can also result in learning and memory deficits [15, 17, 18].
Similar to neuroinflammation, acquired channelopathies can change the way neurons process information and occur in numerous CNS disorders [1, 19-21]. We thus set out to test whether that a “pure” neuroinflammatory response induced by intracerebroventricular application of lipopolysaccaride (LPS), a prototypical activator of Toll-like receptor (TLR4) [22], could induce brain channelopathies. We focused on HCN channels as a model system because they are modified in CNS disorders associated with inflammatory conditions [3, 20, 23]. Moreover, the current conducted by HCN channels, Ih, regulates many important physiological functions in a cell-type dependent fashion including pacemaking activity, resting membrane potential, input resistance and synaptic integration [24]. HCN channels also play a key role in controlling dendritic integration in hippocampal CA1 pyramidal cells. The high density of Ih in distal dendrites [25] attenuates the temporal summation of excitatory inputs [26] and dampens neuronal excitability by decreasing the input resistance [27]. Importantly, Ih tunes the membrane to respond optimally to inputs in the theta frequency band, a brain rhythm central to numerous cognitive processes [28]. Therefore, alterations in HCN function may lead to network dysfunctions implicated in hyperexcitabilty and cognitive deficits.
We found that LPS-induced neuroinflammation triggers an HCN channelopathy associated with the induction of proinflammatory microglia and the up-regulation of neuronal TLR4, thereby resulting in drastic modifications of information processing in CA1 pyramidal cell dendrites. Our evidence suggests that by directly changing neuronal function, neuroinflammation may participate in the phenotypic expression of various brain diseases, underscoring a key target for therapeutic intervention.
Methods
Experimental animals
Adult male Sprague-Dawley rats (225–250 g; Charles-River, Calco, Italy; Charles-River, Lyon, France) were housed at constant temperature (23 ± 1°C) and humidity (60 ± 5%) with free access to food and water and a fixed 12 h light/dark cycle. In compliance with the ARRIVE guidelines, procedures involving animals and their care were conducted in conformity with the institutional guidelines that are in compliance with national (D.L. n.26, G.U. March 4, 2014) and international laws and policies (EEC Council Directive 86/609, OJ L 358, 1, December 12, 1987; Guide for the Care and Use of Laboratory Animals, U.S. National Research Council, 1996). All in vivo studies were carried out at the Mario Negri Institute in Milano except for the electrophysiological experiments that were carried out at INSERM, UMR 1106 in Marseille. Common procedures for animal handling and injections were followed at these two sites. All evaluations in animals were done by experienced investigators blinded to the treatment.
Intracerebroventricular injections
Rats (n=58) were deeply anesthetized with 1.5% isoflurane anesthesia, and stereotaxically injected with LPS (Lipopolysaccharide from Escherichia coli serotype 055:B5, Sigma, St. Louis, MO, USA) over 1 min bilaterally into the lateral ventricles (25 μg/2 μl each ventricle) at the following coordinates from bregma: mm, rat: bar nose – 2.5, AP – 1.0, L ± 1.5 and 3.7 below the dura mater. Sham-operated rats served as controls (n=39) and were similarly injected with the corresponding volume of vehicle (phosphate buffered saline, pH 7.4). Out of 58 rats LPS-injected, 14 rats were also injected with Cyanobacterial LPS (CyP; Bluegreen Biotech, Milano, Italy; 60 μg/3ml, icv bilaterally, 15 min before and 15 min after LPS), a potent and selective antagonist of TLR4 [29].
Immunohistochemistry
Rats were killed 6 h (n=4), 24 h (n=17) or 7 days (n=4) after LPS (Suppl Figure 1; Figure 1). Control animals were sacrificed at the same time points as the experimental rats (n=2/each time, except for n=13 at 24 h). Five rats were also injected with CyP and killed 24 h later. Rats were deeply anaesthetized using ketamine (75 mg/kg) and medetomidine (0.5 mg/kg) then perfused via the ascending aorta [30]. The brains were removed from skull and post-fixed for 48 h at 4°C. Serial coronal sections (30 μm) were cut on a vibratome throughout the temporal extension of the hippocampus (-2.0 to -4.0 mm from bregma; [31]). We prepared 3 series of 16 sections each and in each series we stained the slices as follows: the 1st slice for IL-1β (Suppl. Fig. 1 and 3a and Figure 1), the 2nd for HMGB1 (Figure 1), the 3rd for TLR4 (Figure 1), the 4th for HCN1 (Figure 3), the 5th for TRIP8b (Figure 3), and the 6th for Fluoro-Jade (not shown, refers to rats analyzed in Figure 3).
Figure 1. LPS-induced inflammatory microglia and overexpression of TLR4 in hippocampal neurons.
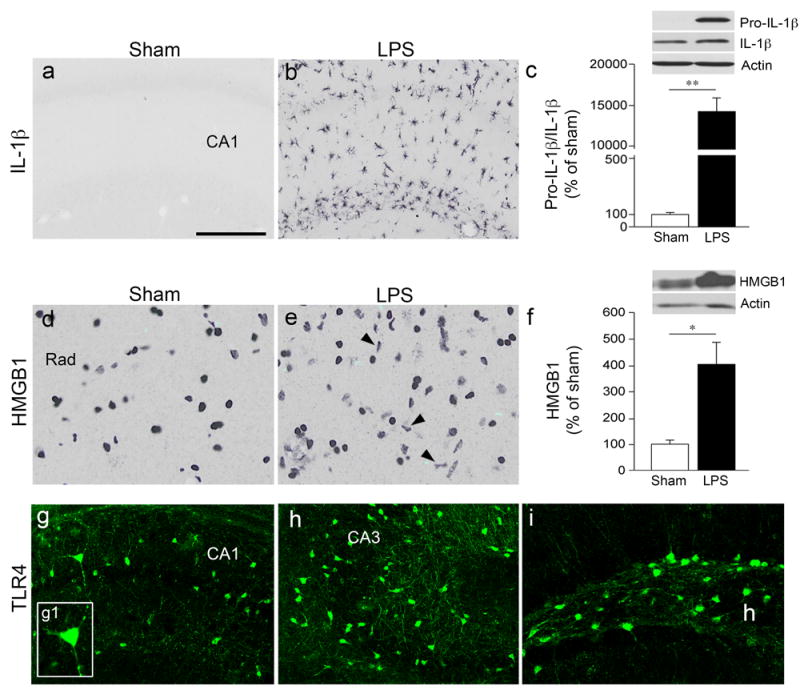
Representative pictures of increased cytoplasmatic IL-1β (a,b) and HMGB1 (d,e) immunoreactivity in the rat septal hippocampus 24 h after bilateral icv LPS (25 μg/2 μl) (b,e) or vehicle (a,d) injection (Sham, n=6 each group). Bargrams (c,f) depict protein quantification (mean±s.e.m., n=5 each group) obtained by the densitometric analysis of western blots probed with the respective antibodies and normalized to the respective actin band. *p<0.05, **p<0.01 by Mann–Whitney test.
(g-i) Representative images of TLR4 expression in neuronal cell bodies and dentrites of CA1 and CA3 pyramidal neurons and hilar (h) interneurons. TLR4 was not detectable in sham controls (not shown; see Refs 33,58). Rad, stratum radiatum (d,e). Scale bar: a,b,g-i: 50 μm; d,e: 33 μm.
Figure 3. LPS treatment leads to a reduction in HCN1 and TRIP8b expression in CA1 pyramidal neurons.
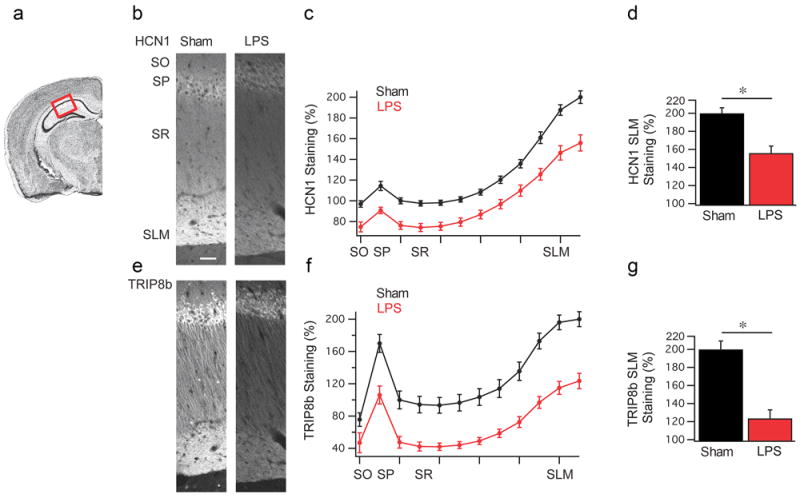
(a) Coronal view of the mouse hippocampus, highlighting the CA1 region used for quantification. (b) Representative HCN1 staining from rats 24 h after vehicle (n=6) (left) or LPS (n=6) (right) injection. SO: stratum oriens, SP: stratum pyramidale, SR: stratum radiatum, SLM: stratum lacunosum moleculare. (c) Quantification of the HCN1 staining shown in panel (b). (d) HCN1 staining in the SLM region was significantly reduced in LPS treated animals relative to vehicle controls (Sham). (e) Representative TRIP8b staining. (f) Quantification of TRIP8b staining. (g) TRIP8b staining was reduced in the SLM of LPS treated rats relative to controls (Sham). Data are mean±s.e.m. * p<0.05 by Mann–Whitney test. Scale bar: 50 μm.
Immunohistochemistry for IL-1β (1:200; Santa Cruz Biotechnology, Heidelberg, Germany), HMGB1 (1:1000; ABCAM, Cambridge, UK) and TLR4 (1:150; Santa Cruz Biotechnology, Heidelberg, Germany) was done as previously described in detail [30, 32, 33]. The IL-1β-immunostained area was measured in the whole temporal slice of the hippocampus (20X magnification). Three slices/rat (n=6 sham; n=6 LPS-24 h) were analyzed using ImageJ software. The area occupied by the specific IL-1β signal was averaged in the three slices/rat and expressed as % of the total assessed area (μm2) and this value was used for statistical analysis (Suppl Figure 1).
Quantification of HCN1 and TRIP8b staining
A cohort of rats was injected with LPS (n=6) or vehicle (n=6) and killed after 24 h for immunohistochemical analysis of HCN1 and TRIP8b using custom guinea pig anti-HCN1 (1:1000) [34]) and mouse anti-TRIP8b (1:1000, N212/17, Neuromab, CA, USA) antibodies using a previously described protocol [35, 36]. Imaging analysis was performed using a Nikon A1 confocal microscope at the Northwestern University Center for Advanced Microscopy (generously supported by NCI CCSG P30 CA060553 awarded to the Robert H, Lurie Comprehensive Cancer Center). Quantification was performed using custom written routines in MATLAB (Mathworks, Natick, MA, USA). Briefly, regions of interest (ROI) were drawn over each anatomical structure (stratum oriens, and stratum pyramidale) as well as a large ROI drawn over the stratum radiatum (SR) and stratum lacunosum moleculare (SLM). The large ROI encompassing the SR and SLM was then subdivided into ten equally spaced ROIs in order to quantify the gradient of HCN channel expression. In order to present the data from two distinct cohorts of animals analyzed independently (n=3/each group/each cohort), the data was then normalized by the expression in vehicle treated group with the equation Xnormalized = 100 + 100*(Xraw - SR1)/(SLM10 − SR1) where Xraw is the raw intensity of a given ROI, SR1 is the raw intensity of the first ROI in the stratum radiatum, and SLM10 is the intensity of the ROI in the last ROI of the SLM. This procedure ensured that vehicle injected animals ranged from 100% to 200% intensity and minimized between-cohort variability. In the same brains, adjacent slices were used for IL-1β and HMGB1 immunohistochemistry (Figure 1) as described above.
Fluoro-Jade staining
Three septal slices for each brain from vehicle (n=6) and LPS rats (24 h after LPS, n=6) (same rats as above) were matched for antero-posterior level and processed for Fluoro-Jade staining to detect degenerating neurons [37]. Images of the dorsal hippocampus were captured at 20X magnification using a BX61 microscope equipped with motorized platform (Olympus, Germany) and digitized. The presence or absence of Fluoro-Jade-stained neurons was analyzed in the entire hippocampal slice.
Real-time reverse transcription PCR and western blot analysis
Twenty-four hours after LPS injection, a new cohort of experimental rats and their vehicle-injected controls were decapitated, and the dorsal hippocampi from both hemispheres were rapidly dissected out at 4°C, then frozen on dry ice, and stored at −80°C until assayed. RT-qPCR of HCN1 transcript and western blotting for the various proteins were performed as described before [19, 38-40]. mRNA from microdissected septal hippocampal CA1 region was isolated using the Dynabeads mRNA Direct™ Micro Kit (Invitrogen) according to the manufacturer’s protocol. cDNA was synthesized by reverse transcription using the RevertAid™ Premium First strand cDNA Synthesis Kit (Fermentas) following the manufacturer’s protocol. HCN1 subunit transcript quantification was performed by real-time RT-PCR as described before [39]. Synaptophysin as a neuron specific gene was used as a reference gene. Briefly, relative quantification of the starting mRNA copy numbers was carried out according to the ΔΔCt-method using 2x Maxima SYBR Green qPCR Master Mix (Invitrogen), 5pM each oligonucleotide primer (see below) and 1.25 μl synthesized cDNA in a 6.25 μl volume.
|
| |||
| Gene | Gene accession number | Sequence | |
|
| |||
| Synaptophysin | NM_012664.3 | (fw) | AACACGAACCATAAGTTGCCAA |
|
| |||
| (rv) | TCAGGACTCAACACCTCAGTGG | ||
|
| |||
| HCN1 | NM_053375.1 | (fw) | CAGACGGCTCTTACTTTGGAGAA |
|
| |||
| (rv) | GCTCGAACACTGGCAGTGC | ||
|
| |||
Quantitative PCR was performed as follows (n=5 LPS; n=5 Sham): 2 min at 50°C, 10 min at 95°C, then 40 cycles of 15 s at 95°C and 1 min at 59°C (ABI Prism 7900HT, Applied Biosystems, Foster City, CA, USA).
For western blot analysis (n=18 LPS; n=9 LPS+CyP; n=19 Sham), we used antibodies against HCN1 (1:1000; Antibodies Inc, Davis, CA, USA [19, 38-40]), TRIP8b (1:1000, N212/17, Neuromab, CA, USA [35, 36]), total Kv4.2 and triply P-Kv4.2 (1:200 and 1:300, respectively; Chemicon, Labon [40]), ERK1/2 and P-ERK (1:10000 and 1:5000, respectively; Cell Signaling Technology, Beverly, MA, USA [40]), IL-1β (1:500; ABCAM, Cambridge, UK [41]), HMGB1 (1:1000; ABCAM, Cambridge, UK [33]), and actin (1:5000, Sigma-Aldrich, St. Louis, MO, USA) which was used to normalize the relevant protein bands.
Electrophysiology
Twenty-four hours after LPS injection, a new cohort of LPS-injected rats (n=6) and their vehicle-injected controls (n=8) were decapitated, and the dorsal hippocampi were rapidly dissected out at 4°C. Hippocampal slices were prepared from the dorsal hippocampus. ACSF contained (in mM) NaCl 126, KCl 3.5, CaCl2 2, MgCL2 1.3, NaH2PO4 1.2, NaHCO3 26, D-Glucose 10, and NBQX (1 μM), D-APV (50 μM) and bicuculline (10 μM) to block AMPA, NMDA and GABAA receptors, respectively. CA1 pyramidal neurons were recorded at 34 ± 1°C with a solution containing (in mM) KMeSO4 120, KCl 20, EGTA 0.2, MgCl2 2, HEPES 10, Na2ATP 4, Tris GTP 0.3, Phosphocreatine 14, biocytin 0.4% and KOH to adjust to pH 7.3. All cells and recordings sites were morphologically identified post hoc [19]. Wavelets at varying frequencies were injected in current-clamp mode while the membrane was maintained at -70 mV [38]. Impedance (Z) was calculated as the ratio of the voltage/current Fourier transforms for each frequency. The resonance frequency, Fres, is the frequency for which |Z| is maximum; the amplification ratio, Q, is defined as the |Z|Fres/|Z|1 Hz ratio. The impedance phase was calculated as ϕ = tan-1(Im(Z)/Re(Z)), where Im(Z) and Re(Z). Fϕ is defined as the frequency f where ϕ(f) = 0. The total inductive phase, ΦL, was defined as the area of ϕ(f) where ϕ(f) > 0. Trains of 5 currents in the form of an alpha function At.e-0.1t were injected to mimic the occurrence of series of EPSPs at 20 Hz and 50 Hz. The summation ratio was defined as the amplitude of the fifth EPSP divided by the amplitude of the first. Currents mediated by Ih were recorded in voltage-clamp mode by applying hyperpolarizing voltage steps (starting from a holding potential of -50 mV up to -140 mV). The amplitude of Ih was determined by subtracting the instantaneous current at the beginning of the voltage step from the steady-state current at the end. The activation time constant was obtained using exponential fit. Boltzmann fits were made to obtain V1/2, the midpoint activation voltage using the tail current.
Statistical analysis of data
SPSS or Prism software was used for statistical analysis of data. Data are expressed as mean ± s.e.m. We used Mann–Whitney test for 2 independent groups and Kruskal-Wallis followed by Dunn’s post-hoc test for more than 2 independent groups. p<0.05 was considered significant.
Results
LPS-induced neuroinflammation
On the basis of preliminary dose-response experiments (6.25-25 μg LPS each ventricle), we choose for all the experiments the 50 μg total dose icv (25 μg each side) since this was the minimal dose inducing widespread neuroinflammation in the hippocampus in 100% of rats. LPS injection (50 μg total dose) induced a time-dependent neuroinflammatory response in the hippocampus involving the induction of IL-1β in microglia (Suppl Figure 1). This response was observed 6 h after LPS in the hippocampal region more adjacent to the ventricular area (Suppl Figure 1, panel b vs a) while at 24 h (c) the IL-1β signal was strongly expressed throughout the whole hippocampus. As far as the similarity of our model with an infection scenario, the pattern of microglia activation, associated with increased cytokines, in the hippocampus is very similar to that observed in a murine model of viral infection[42]. IL-1β immunoreactivity declined to levels that were identical to the sham control at 1 week post-LPS (d vs a). IL-1β induction was strongly reduced by icv injection of CyP, as assessed at 24 h (Suppl Figure 3a). Co-localization experiments showed that IL-1β was expressed in OX42-positive microglia (e) but not in GFAP-positive astrocytes (f). We did not find evidence of neuronal cell damage in the hippocampus and overlying cortex of LPS-injected rats as assessed by Fluoro-Jade staining in adjacent slices (not shown).
We chose the time of maximal neuroinflammatory response (24 h post-LPS) to quantify the hippocampal changes in IL-1β and HMGB1 expression, two generators of the neuroinflammatory response endowed with neuromodulatory effects [14, 43, 44]. In agreement with the immunohistochemical analysis (Figure 1, panels b,e vs a,d), we found a significant increase in both protein levels over their basal values, as assessed by western blot (Figure 1c,f). The expression of TLR4, which mediates the LPS effects [22], was induced in CA1 and CA3 pyramidal cell bodies and dendrites, and in hilar interneurons (Figure 1g-i),while it was undetectable in sham controls (not shown).
LPS effects on HCN1 channel expression
HCN1 mRNA levels were not modified by LPS as assessed by quantitative real-time reverse transcription PCR in the dorsal hippocampus 24 h after LPS injection (Figure 2a). However, we found a significant 37% reduction in HCN1 protein level measured by western blot (Figure 2b; Suppl Figure 2a,b) which was blocked by icv injection of CyP (Suppl Figure 3b). HCN1 downregulation has consistently been found in human and experimental epilepsy [38, 45-47] where it is driven by the up-regulation of the transcriptional Neuron Restrictive Silencer Factor (NRSF) [47]. Moreover, HCN1 channel changes in epilepsy have been associated with the downregulation of Kv4.2, another important dendritic channel [19]. Notably, LPS did not increase NRSF (Figure 2c) nor did it change Kv4.2 or its phosphorylated (pKv4.2) form (Figure 2d,e), or the related kinase ERK/pERK (Figure 2f,g). LPS therefore affects specific sets of dendritic channel proteins, and mediates downregulation of HCN1 by post-transcriptional modifications.
Figure 2. LPS-induced HCN1 protein downregulation.
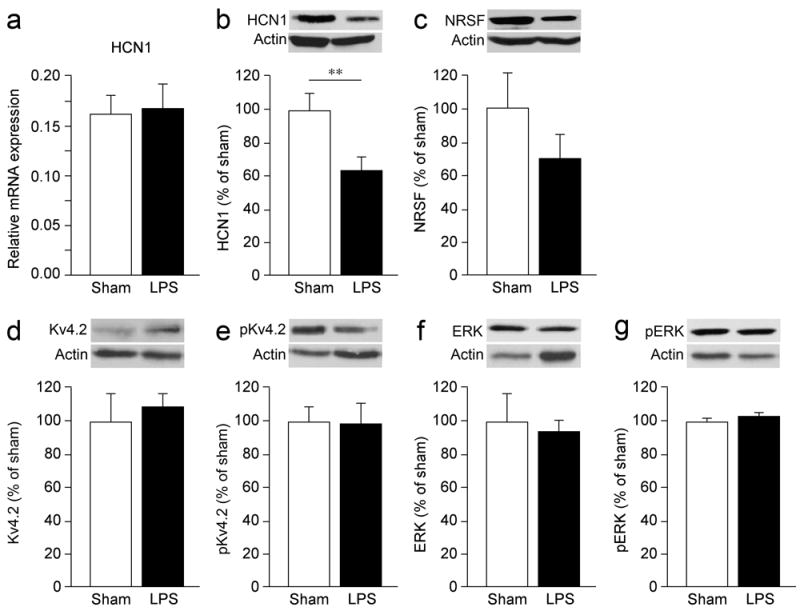
(a) LPS did not affect HCN1 mRNA levels as assessed by RT-qPCR while it induced a significant reduction in the levels of HCN1 protein measured by western blot (b) (see also Suppl. Figure 2a,b) in hippocampal homogenates, 24 h following injection. LPS did not significantly change in the same hippocampal homogenates the protein level of NRSF (c) or Kv4.2 (d) and ERK (f) and their phosphorylated forms (e,g). Bargraphs (mean±s.e.m., n=5 each group) show the protein value obtained by the densitometric analysis of the blots probed with the respective antibodies and normalized to the respective actin band. ** p<0.01 by Mann–Whitney test.
Analysis of HCN1 channel dendritic trafficking
Given the reduction in HCN1 hippocampal protein, we next investigated the distribution of the channel in CA1 pyramidal neurons by immunohistochemistry [46] in rats killed 24 h after LPS or vehicle injection. Consistent with our western blot results, we measured a significant reduction in HCN1 expression in CA1 pyramidal neurons (Figure 3b-d). We next examined if TRIP8b, the HCN channel auxiliary subunit, was similarly changed since TRIP8b plays an important role in subcellular distribution and trafficking of HCN channels [48, 49], We measured a decrease in TRIP8b expression in CA1 pyramidal neurons by immunohistochemistry (Figure 3e-g) but the total protein level did not change as assessed by western blot (Suppl Figure 2c,d). Our data therefore indicate that there is a specific loss of both HCN1 and TRIP8b proteins in CA1 pyramidal neurons.
Functional changes in HCN1 properties induced by LPS
We next investigated the impact of HCN1 alterations on Ih currents since HCN1 is the main subunit of the channel carrying this current in the hippocampus [25, 50]. Using whole cell recordings of distal dendrites of CA1 pyramidal cells at the stratum radiatum-stratum lacunosum moleculare border in vehicle (333 ± 8 μm from the soma, n=8) and LPS-treated (320 ± 8 μm from the soma, n=6) rats, we found a 50% decrease in the amplitude of Ih current (Table 1; Figure 4a,b) 24 h following LPS treatment. These results are consistent with the reduction of HCN1 protein levels. In keeping with the reduction in Ih, the input resistance was increased by 20%, and the resting membrane potential was hyperpolarized by 3 mV after LPS (Table 1). Downregulation of HCN1 is also often associated with alterations in channel kinetics [38, 45, 47]. We found that the time constants were increased by 50% and the membrane potential for half-maximal activation was shifted by -7mV (Table 1; Figure 4a,b). These changes in Ih kinetics further decrease the availability of the channel in the dendrites. We conclude that LPS triggers a dendritic channelopathy by slowing and dampening the Ih current.
Table 1.
| Sham (n=8) | LPS (n=6) | |
|---|---|---|
|
|
|
|
| Distance (μm) | 333 ± 8 | 320 ± 8 |
|
|
|
|
| RMP (mV) | -61.9 ± 0.9 | -64.9 ± 0.8 * |
|
|
|
|
| Rin (MΩ) | 57 ± 5 | 76 ± 8 * |
|
|
|
|
| C (pF) | 76 ± 9 | 78 ± 11 |
|
|
|
|
| RS (MΩ) | 28 ± 2 | 29 ± 1 |
|
|
|
|
| Ih amp (pA) | 642 ± 44 | 320 ± 50 * |
|
|
|
|
| Ih (pA/pF) | 14.3 ± 1.3 | 5.8 ± 0.9 * |
|
|
|
|
| τ fast (ms) | 23 ± 1 | 35 ± 3 * |
|
|
|
|
| τ slow (ms) | 151 ± 12 | 215 ± 34 * |
|
|
|
|
| V1/2 (mV) | -99.6 ± 0.9 | -106.9 ± 2.2 * |
|
|
|
|
| Fres (Hz) | 6.0 ± 0.4 | 5.0 ± 0.4 * |
|
|
|
|
| Q | 1.40 ± 0.09 | 1.22 ± 0.03 * |
|
|
|
|
| Fϕ (Hz) | 4.9 ± 0.3 | 3.5 ± 0.3 * |
|
|
|
|
| ΦL (rad.Hz) | 0.46 ± 0.05 | 0.19 ± 0.04 * |
Recording distance from the soma and basic properties of dendrites. Properties of Ih current (amplitude and kinetics). Ih amplitude was reduced (also when normalized to capacitance), and kinetics slower in LPS as compared to sham. Resonance and phase responses were decreased in LPS as compared to sham.
p<0.05 vs Sham (vehicle-injected rats).
Figure 4. Decreased Ih, theta resonance and phase lead and increased temporal summation after LPS.
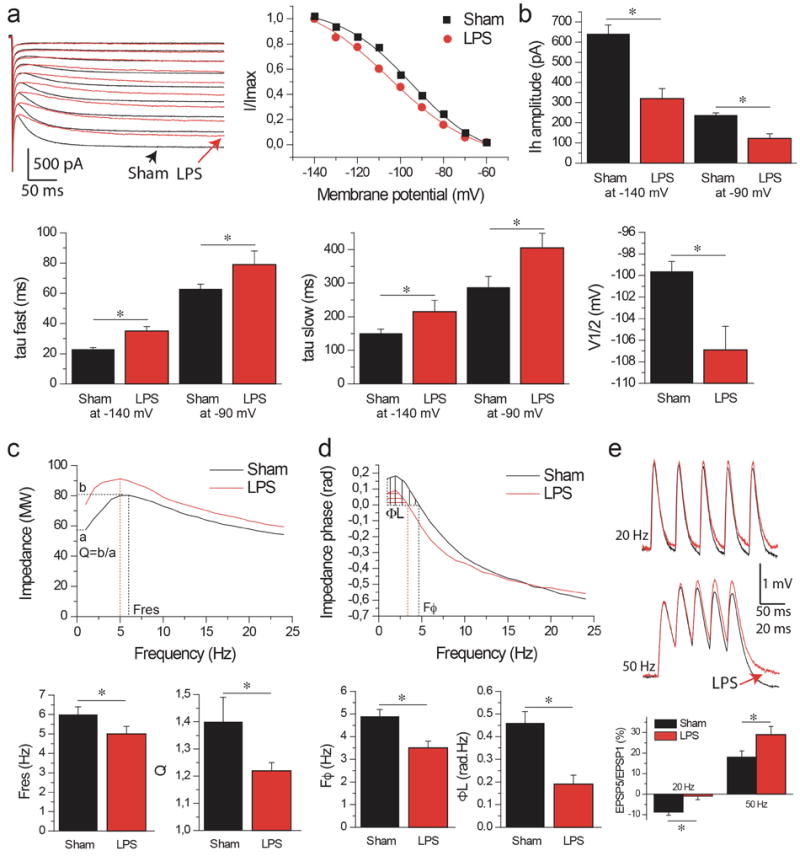
Rats were used for electrophysiological analysis 24 h after LPS (n=6) or vehicle injection (Sham, n=8).
(a) Left panel: Ih activation in CA1 pyramidal cell distal dendrites in sham (black) and LPS-treated (red) animals. Right panel: Ih activation curves fitted with a Boltzman function. Note the decrease in amplitude and the shift of the activation curve to more hyperpolarized values in LPS as compared to Sham. (b) Histogram of Ih amplitude, fast and slow time constant (tau) and V1/2 in Sham and LPS animals. All parameter changes will decrease Ih availability in dendrites of LPS animals. (c) Top panel: Impedance magnitude as a function of the input frequency in Sham (black trace) and LPS (red trace) animals. Bottom panels: Summary histograms of resonance frequency, Fres, and amplification ratio, Q. Resonance is decreased in LPS animals. (d) Same as (c) for phase responses. Note that the phase advance due to Ih activation is decreased in LPS animals. (e) Temporal summation of EPSPs at 20 and 50 Hz. Note that the summation is increased in LPS animals. Data are mean±s.e.m. *p<0.05 by Mann–Whitney test.
We then investigated the functional consequences of this channelopathy, focusing on resonance and summation of synaptic inputs as two key functional readouts of Ih. Ih acts as an inductance L in a RCL electrical circuit [51, 52] and provides resonance (band passfilter) properties in the theta frequency range (4-12 Hz), i.e. filtering out frequencies outside the theta frequency range. LPS treatment altered resonance properties by decreasing Fres, the resonance frequency, and Q, the amplification factor (Table 1; Figure 4c). HCN channels also modify the temporal response of the membrane [38, 51, 52]. They create an apparent negative time delay up to a critical frequency Fφ, i.e., the voltage response of the membrane appears to precede the inputs generating this response (phase lead). Beyond Fφ, the voltage response follows the input (phase lag). LPS treatment also changed the temporal response of the membrane, shifting Fφ to lower frequencies and decreasing the magnitude of phase advance (Table 1, Fig 4d). LPS therefore altered the filtering properties and time responses of dendrites.
Another important function of Ih in the dendrites is to shorten temporal summation of synaptic inputs, in order to normalize excitatory inputs when reaching the soma regardless of their dendritic origin [26]. Temporal summation was largely increased after LPS (Figure 4e, at 20 Hz: Sham, -8.8 ± 1.5%, n=8; LPS, -1.0 ± 1.7%, n=6; at 50 Hz: Sham, 18 ± 3%, LPS, 29 ± 4%, p<0.05 between LPS and Sham at 20 and 50 Hz). We conclude that inflammation triggers an acquired HCN1 channelopathy, reducing Ih availability, thus changing the integrative properties of CA1 pyramidal cell dendrites.
Discussion
This study provides novel evidence that neuroinflammation, by itself, induces a channelopathy affecting HCN1 and the related Ih current, with a direct impact upon membrane integrative properties. The lack of changes in Kv4.2 channel, which is modified together with HCN1 in pathologic conditions such as epilepsy [19], suggests that neuroinflammation targets specific sets of channel proteins.
Following LPS treatment, we found reduced HCN1 and TRIP8b immunostaining in the hippocampal CA1 region. However, western blotting of whole hippocampal lysate established a reduction in levels of HCN1 but not TRIP8b proteins. The most parsimonious explanation for our data is that there is a reduction in TRIP8b expression specifically in CA1 pyramidal neurons (our immunohistochemical data), but not in distinct cell types in which TRIP8b interacts with other HCN isoforms [53], and western blot data is not sensitive enough to resolve this difference. LPS induced the HCN1 protein reduction in the absence of corresponding mRNA changes thus suggesting that the channels were degraded. This is consistent with the evidence that loss of TRIP8b leads to increased HCN channel trafficking to lysosomes [48]. We showed previously that a dissociation between TRIP8b and HCN channels after status epilepticus ultimately leads to a reduction in the distal dendritic enrichment pattern of HCN channel trafficking [46]. Here we have observed a different phenomenon, whereby the distribution of HCN1 channels is more uniformly altered. Moreover, while increased NRSF levels reduce HCN1 transcription in epilepsy [47], neither NRSF nor HCN1 mRNA were significantly changed by LPS. These results therefore indicate that different mechanisms mediate the HCN1 reduction induced by the two insults- LPS and status epilepticus.
Although the specific molecular cascade responsible for HCN1/Ih downregulation is unresolved, the upstream inducer is a TLR4-dependent signaling. The TLR4 involvement is supported by the blockade of LPS-induced HCN1 changes using the specific receptor antagonist CyP. Several epigenetic and post-translational mechanisms have been implicated in HCN1 downregulation [45, 47, 54]. LPS might affect dendritic HCN1 channels via mitogen activated protein kinases, such as p38 or c-Jun terminal kinases [55], upon its direct activation of neuronal TLR4. Due to the very short half-life of unbound LPS in tissue [56], its lasting effects on dendritic Ih currents suggests that a persistent LPS-mediated TLR4 activation is not required. It is likely that the LPS effect on HCN1 also involves soluble inflammatory mediators released by microglia following NF-kB-dependent transcriptional gene induction, a phenomenon which may persist after the acute activation of the LPS-TLR4 axis. Two potential candidates are IL-1β and HMGB1, which are both increased in the hippocampus following LPS injection. Their convergent IL-1R1 and TLR4 signaling activation in neurons has been previously reported to modulate neuronal functions, for example by inducing post-translational changes in glutamate or GABAA receptor-gated channels [43, 44, 57]. Notably, TLR4 expression was undetectable by immunohistochemistry in sham rats [33, 58]. However, basal level of TLR4 expression were measured using RTqPCR [59, 60]. It is likely therefore that immunohistochemistry is not sensitive enough to detect the basal cellular expression of TLR4. Moreover, in vitro experiments demonstrated that LPS can rapidly induce TLR4 in both CNS and non CNS cell types [61-63].
The decrease in Ih current is consistent with the downregulation of HCN1. In addition, the negative shift of the activation curve also contributes to the decrease in Ih. The negative shift may be due to decreased cAMP binding to the channel [24] as well as altered p38 mitogen-activated protein kinase or protein phosphatase 2B activity [45, 64]. The loss of Ih results in important alterations in dendritic information processing. The summation of excitatory inputs is increased after LPS, which, together with the increase in input resistance, would enhance cell excitability. This is consistent with the increased cortical excitability evoked in rats after LPS exposure, which was mediated by the release of IL-1β [16]. In addition, Ih is assumed to linearize dendritic synaptic inputs when they reach the soma, thus providing a precise temporal window for synaptic integration [26]. Decreased Ih would lead therefore to a loss of temporal precision and inadequate integration of synaptic inputs. These changes, combined with the reduction in the band pass filtering properties of the dendrites- which will become less tuned to inputs in the theta frequency range- and the change in the phase response, may alter cognitive processes [38, 65]. The HCN1 reduction could therefore contribute to the cognitive deficits induced by LPS in animals and provoked by sepsis in humans- which are chiefly mediated by IL-1β and HMGB1 [17, 66-68], and more in general in CNS disorders associated with neuroinflammation [38, 69, 70]. Of note, neuroinflammation and HCN1 downregulation are co-existing phenomena in epilepsy [20] and PD [3] and LPS also reduces Ih currents in myocytes [71], suggesting that these channels are particularly sensitive to an inflammatory challenge.
HCN1 currents also play a key role for pacing the activity of some neurons [24]. It is therefore possible that HCN1 downregulation in the CA1 pyramidal cells will alter the genesis of rhythmic activities [3]. Although causality remains to be demonstrated, it is interesting to note that HCN1 downregulation, decrease in theta rhythm and cognitive deficits co-occur in experimental epilepsy [38, 65].
We conclude that neuroinflammation may directly account for some of the key neuronal functional reorganizations associated with neurological disorders, thus potentially contributing and reinforcing the development of phenotypic traits. This evidence supports the use of anti-inflammatory drugs as potential disease-modifying therapies to complement the symptomatic drugs specifically used for a given neurological disorder.
Supplementary Material
Suppl Figure 1.IL -1β expressing microglia in the rat hippocampus after LPS (a-d) Representative photomicrographs showing the progressive changes in IL-1β immunoreactivity in the rat septal hippocampus at various times (n=4-12 rats) after bilateral icv LPS injection (25 μg/2 μl) vs vehicle injection (n=2-8 rats). Six hours after LPS injection (b), IL-1β immunostaining was increased in the hippocampal regions adjacent to the ventricular area. IL-1β signal was strongly induced throughout the whole hippocampus at 24 h (c) post-LPS, then declining by 1 week (d) to undetectable levels as in sham controls (a). The merge images (e,f) show co-localization of IL-1β signal with the microglia marker OX-42 (yellow signal in e) but not with the astrocytic marker GFAP (f). Bargram in (g) depicts the quantification of IL-1β expression 24 h post-LPS (area occupied by the specific signal/total area analyzed). CA1, CA3: pyramidal cell layers; h: hilus. Scale bar: a-d, 220 μm; e,f: 25 μm.
Suppl Figure 2.Treatment with LPS causes a reduction in total hippocampal HCN1 protein but not TRIP8b
Western blots were performed from whole hippocampi of rats treated with either vehicle or LPS (n=5 each experimental group). A significant reduction in HCN1 (a,b) was observed without a difference in TRIP8b (c,d). Data are mean±s.e.m. * p<0.05 by Mann–Whitney test.
Suppl Figure 3. Blockade of LPS effects by the selective TLR4 antagonist Cyanobacterial LPS
Rats were injected icv with Cyanobacterial LPS (CyP; 60 μg/3 μl in PBS, bilaterally) 15 min before and 15 min after icv LPS injection (25 μg/2 μl in PBS bilaterally). Rats were sacrificed 24 h after LPS injection. (a) A significant reduction of IL-1β staining was observed by immunohistochemistry in rats treated with LPS+CyP vs LPS alone (n=5 rats each group). CA1, CA1 pyramidal neurons; CA3, CA3 pyramidal neurons; h, hilus. Scale bar: 250 μm. (b) LPS-induced reduction of HCN1 was prevented by CyP as assessed by western blot. Data are mean±s.e.m. (n= 8-9 rats). *p<0.05 by Kruskal-Wallis followed by Dunn’s post-hoc test.
Acknowledgments
This work was supported by INSERM, ANR MINOS and ANTARES (C.B.), and the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement n°602102 (EPITARGET to C.B. and A.V.), C.F. was supported by an Alberta Heritage Foundation for Medical Research (AHFMR) Fellowship. Additionally, this work was supported by Fondazione Italo Monzino (A.V.), National Institutes of Health Grant 2R01NS059934, R01MH106511 and R21MH104471 (D.M.C). We thank Amy L. Brewster and Francesco Noé for their contribution to the initial experiments.
Footnotes
Disclosures
The authors declare no conflict of interest.
References
- 1.Beck H, Yaari Y. Plasticity of intrinsic neuronal properties in CNS disorders. Nat Rev Neurosci. 2008;9:357–369. doi: 10.1038/nrn2371. [DOI] [PubMed] [Google Scholar]
- 2.Santos SF, Pierrot N, Octave JN. Network excitability dysfunction in Alzheimer’s disease: insights from in vitro and in vivo models. Rev Neurosci. 2010;21:153–171. doi: 10.1515/revneuro.2010.21.3.153. [DOI] [PubMed] [Google Scholar]
- 3.Chan CS, Glajch KE, Gertler TS, et al. HCN channelopathy in external globus pallidus neurons in models of Parkinson’s disease. Nat Neurosci. 2011;14:85–92. doi: 10.1038/nn.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng K, Howells J, Pollard JD, Burke D. Up-regulation of slow K(+)channels in peripheral motor axons: a transcriptional channelopathy in multiple sclerosis. Brain. 2008;131:3062–3071. doi: 10.1093/brain/awn180. [DOI] [PubMed] [Google Scholar]
- 5.Israelson A, Arbel N, Da Cruz S, et al. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron. 2010;67:575–587. doi: 10.1016/j.neuron.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boadas-Vaello P, Castany S, Homs J, et al. Neuroplasticity of ascending and descending pathways after somatosensory system injury: reviewing knowledge to identify neuropathic pain therapeutic targets. Spinal Cord. 2016;54:330–340. doi: 10.1038/sc.2015.225. [DOI] [PubMed] [Google Scholar]
- 7.Alia C, Spalletti C, Lai S, et al. Neuroplastic Changes Following Brain Ischemia and their Contribution to Stroke Recovery: Novel Approaches in Neurorehabilitation. Front Cell Neurosci. 2017;11:76. doi: 10.3389/fncel.2017.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giza CC, Prins ML. Is being plastic fantastic? Mechanisms of altered plasticity after developmental traumatic brain injury. Dev Neurosci. 2006;28:364–379. doi: 10.1159/000094163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol. 2011;7:31–40. doi: 10.1038/nrneurol.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xanthos DN, Sandkuhler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. 2014;15:43–53. doi: 10.1038/nrn3617. [DOI] [PubMed] [Google Scholar]
- 11.Banjara M, Ghosh C. Sterile Neuroinflammation and Strategies for Therapeutic Intervention. Int J Inflamm. 2017;2017:8385961. doi: 10.1155/2017/8385961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Algattas H, Huang JH. Traumatic Brain Injury pathophysiology and treatments: early, intermediate, and late phases post-injury. Int J Mol Sci. 2013;15:309–341. doi: 10.3390/ijms15010309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ransohoff RM, Schafer D, Vincent A, et al. Neuroinflammation: Ways in Which the Immune System Affects the Brain. Neurotherapeutics. 2015;12:896–909. doi: 10.1007/s13311-015-0385-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vezzani A, Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology. 2015;96:70–82. doi: 10.1016/j.neuropharm.2014.10.027. [DOI] [PubMed] [Google Scholar]
- 15.Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84:87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- 16.Rodgers KM, Hutchinson MR, Northcutt A, et al. The cortical innate immune response increases local neuronal excitability leading to seizures. Brain. 2009;132:2478–86. doi: 10.1093/brain/awp177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazarati A, Maroso M, Iori V, et al. High-mobility group box-1 impairs memory in mice through both toll-like receptor 4 and Receptor for Advanced Glycation End Products. Exp Neurol. 2011;232:143–8. doi: 10.1016/j.expneurol.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riazi K, Galic MA, Pittman QJ. Contributions of peripheral inflammation to seizure susceptibility: cytokines and brain excitability. Epilepsy Res. 2010;89:34–42. doi: 10.1016/j.eplepsyres.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 19.Bernard C, Anderson A, Becker A, et al. Acquired dendritic channelopathy in temporal lobe epilepsy. Science. 2004;305:532–535. doi: 10.1126/science.1097065. [DOI] [PubMed] [Google Scholar]
- 20.Noam Y, Bernard C, Baram TZ. Towards an integrated view of HCN channel role in epilepsy. Curr Opin Neurobiol. 2011;21:873–9. doi: 10.1016/j.conb.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar P, Kumar D, Jha SK, et al. Ion Channels in Neurological Disorders. Adv Protein Chem Struct Biol. 2016;103:97–136. doi: 10.1016/bs.apcsb.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Bryant CE, Spring DR, Gangloff M, Gay NJ. The molecular basis of the host response to lipopolysaccharide. Nat Rev Microbiol. 2010;8:8–14. doi: 10.1038/nrmicro2266. [DOI] [PubMed] [Google Scholar]
- 23.Baruscotti M, Bottelli G, Milanesi R, et al. HCN-related channelopathies. Pflugers Arch. 2010;460:405–415. doi: 10.1007/s00424-010-0810-8. [DOI] [PubMed] [Google Scholar]
- 24.Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- 25.Brewster AL, Chen Y, Bender RA, et al. Quantitative analysis and subcellular distribution of mRNA and protein expression of the hyperpolarization-activated cyclic nucleotide-gated channels throughout development in rat hippocampus. Cereb Cortex 1991. 2007;17:702–712. doi: 10.1093/cercor/bhk021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magee JC. Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci. 1999;2:848. doi: 10.1038/12229. [DOI] [PubMed] [Google Scholar]
- 27.George MS, Abbott LF, Siegelbaum SA. HCN hyperpolarization-activated cation channels inhibit EPSPs by interactions with M-type K(+)channels. Nat Neurosci. 2009;12:577–584. doi: 10.1038/nn.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buzsaki G. Rhythms of the Brain. New York: Oxford University Press; 2006. [Google Scholar]
- 29.Macagno A, Molteni M, Rinaldi A, et al. A cyanobacterial LPS antagonist prevents endotoxin shock and blocks sustained TLR4 stimulation required for cytokine expression. J Exp Med. 2006;203:1481–92. doi: 10.1084/jem.20060136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ravizza T, Gagliardi B, Noé F, et al. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2008;29:142–60. doi: 10.1016/j.nbd.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 31.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic Press; New York: 2007. [DOI] [PubMed] [Google Scholar]
- 32.Walker LE, Frigerio F, Ravizza T, et al. Molecular isoforms of high-mobility group box 1 are mechanistic biomarkers for epilepsy. J Clin Invest. 2017;127:2118–2132. doi: 10.1172/JCI92001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Maroso M, Balosso S, Ravizza T, et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16:413–9. doi: 10.1038/nm.2127. [DOI] [PubMed] [Google Scholar]
- 34.Shin M, Chetkovich DM. Activity-dependent regulation of h channel distribution in hippocampal CA1 pyramidal neurons. J Biol Chem. 2007;282:33168–33180. doi: 10.1074/jbc.M703736200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heuermann RJ, Jaramillo TC, Ying S-W, et al. Reduction of thalamic and cortical Ih by deletion of TRIP8b produces a mouse model of human absence epilepsy. Neurobiol Dis. 2016;85:81–92. doi: 10.1016/j.nbd.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han Y, Heuermann RJ, Lyman KA, et al. HCN-channel dendritic targeting requires bipartite interaction with TRIP8b and regulates antidepressant-like behavioral effects. Mol Psychiatry. 2017;22:458–465. doi: 10.1038/mp.2016.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmued LC, Albertson C, Slikker W. Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- 38.Marcelin B, Chauvière L, Becker A, et al. h channel-dependent deficit of theta oscillation resonance and phase shift in temporal lobe epilepsy. Neurobiol Dis. 2009;33:436–447. doi: 10.1016/j.nbd.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 39.Marcelin B, Lugo JN, Brewster AL, et al. Differential dorso-ventral distributions of Kv4.2 and HCN proteins confer distinct integrative properties to hippocampal CA1 pyramidal cell distal dendrites. J Biol Chem. 2012;287:17656–61. doi: 10.1074/jbc.C112.367110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lugo JN, Barnwell LF, Ren Y, et al. Altered phosphorylation and localization of the A-type channel, Kv4.2 in status epilepticus. J Neurochem. 2008;106:1929–1940. doi: 10.1111/j.1471-4159.2008.05508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dubé CM, Ravizza T, Hamamura M, et al. Epileptogenesis provoked by prolonged experimental febrile seizures: mechanisms and biomarkers. J Neurosci. 2010;30:7484–94. doi: 10.1523/JNEUROSCI.0551-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kirkman NJ, Libbey JE, Wilcox KS, et al. Innate but not adaptive immune responses contribute to behavioral seizures following viral infection. Epilepsia. 2010;51:454–64. doi: 10.1111/j.1528-1167.2009.02390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Viviani B, Bartesaghi S, Gardoni F, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balosso S, Liu J, Bianchi ME, Vezzani A. Disulfide-containing High Mobility Group Box-1 promotes N-methyl-d-aspartate receptor function and excitotoxicity by activating Toll-like receptor 4-dependent signaling in hippocampal neurons. Antioxid Redox Signal. 2014;21:1726–1740. doi: 10.1089/ars.2013.5349. [DOI] [PubMed] [Google Scholar]
- 45.Jung S, Jones TD, Lugo JN, et al. Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci. 2007;27:13012–13021. doi: 10.1523/JNEUROSCI.3605-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shin M, Brager D, Jaramillo TC, et al. Mislocalization of h channel subunits underlies h channelopathy in temporal lobe epilepsy. Neurobiol Dis. 2008;32:26–36. doi: 10.1016/j.nbd.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McClelland S, Flynn C, Dubé C, et al. Neuron-restrictive silencer factor-mediated hyperpolarization-activated cyclic nucleotide gated channelopathy in experimental temporal lobe epilepsy. Ann Neurol. 2011;70:454–64. doi: 10.1002/ana.22479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lewis AS, Vaidya SP, Blaiss CA, et al. Deletion of the hyperpolarization-activated cyclic nucleotide-gated channel auxiliary subunit TRIP8b impairs hippocampal Ih localization and function and promotes antidepressant behavior in mice. J Neurosci. 2011;31:7424–7440. doi: 10.1523/JNEUROSCI.0936-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lyman KA, Han Y, Chetkovich DM. Animal models suggest the TRIP8b-HCN interaction is a therapeutic target for major depressive disorder. Expert Opin Ther Targets. 2017;21:235–237. doi: 10.1080/14728222.2017.1287899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Surges R, Brewster AL, Bender RA, et al. Regulated expression of HCN channels and cAMP levels shape the properties of the h current in developing rat hippocampus. Eur J Neurosci. 2006;24:94–104. doi: 10.1111/j.1460-9568.2006.04880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Narayanan R, Johnston D. Long-term potentiation in rat hippocampal neurons is accompanied by spatially widespread changes in intrinsic oscillatory dynamics and excitability. Neuron. 2007;56:1061–1075. doi: 10.1016/j.neuron.2007.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Narayanan R, Johnston D. The h channel mediates location dependence and plasticity of intrinsic phase response in rat hippocampal neurons. J Neurosci. 2008;28:5846–5860. doi: 10.1523/JNEUROSCI.0835-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Notomi T, Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1-4, in the rat brain. J Comp Neurol. 2004;471:241–276. doi: 10.1002/cne.11039. [DOI] [PubMed] [Google Scholar]
- 54.Jung S, Warner LN, Pitsch J, et al. Rapid loss of dendritic HCN channel expression in hippocampal pyramidal neurons following status epilepticus. J Neurosci. 2011;31:14291–14295. doi: 10.1523/JNEUROSCI.1148-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–64. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 56.Flegel WA, Baumstark MW, Weinstock C, et al. Prevention of endotoxin-induced monokine release by human low- and high-density lipoproteins and by apolipoprotein A-I. Infect Immun. 1993;61:5140–5146. doi: 10.1128/iai.61.12.5140-5146.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roseti C, van Vliet EA, Cifelli P, et al. GABA currents are decreased by IL-1beta in epileptogenic tissue of patients with temporal lobe epilepsy: implications for ictogenesis. Neurobiol Dis. 2015;82:311–320. doi: 10.1016/j.nbd.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 58.Mishto M, Raza ML, de biase D, et al. The immunoproteasome Beta5i subunit is key contributor to ictogenesis in a rat model of chronic epilepsy. Brain Behav Immun. 2015;49:188–96. doi: 10.1016/j.bbi.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 59.Iori V, Maroso M, Rizzi M, et al. Receptor for Advanced Glycation Endproducts is upregulated in temporal lobe epilepsy and contributes to experimental seizures. Neurobiol Dis. 2013;58:102–14. doi: 10.1016/j.nbd.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 60.Chen K, Sun Y, Diao Y, et al. α7 nicotinic acetylcholine receptor agonist inhibits the damage of rat hippocampal neurons by TLR4/Myd88/NF κB signaling pathway during cardiopulmonary bypass. Mol Med Rep. 2017;16:4770–4776. doi: 10.3892/mmr.2017.7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garay-Malpartida HM, Mourão RF, Mantovani M, et al. Toll-like receptor 4 (TLR4) expression in human and murine pancreatic beta-cells affects cell viability and insulin homeostasis. BMC Immunol. 2011;12:18. doi: 10.1186/1471-2172-12-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang J, Feng X, Zeng Y, et al. Lipopolysaccharide (LPS)-induced autophagy is involved in the restriction of Escherichia coli in peritoneal mesothelial cells. BMC Microbiol. 2013;13:255. doi: 10.1186/1471-2180-13-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Badshah H, Ali T, Kim MO. Osmotin attenuates LPS-induced neuroinflammation and memory impairments via the TLR4/NFκB signaling pathway. Sci Rep. 2016;6:24493. doi: 10.1038/srep24493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen K, Aradi I, Thon N, et al. Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability. Nat Med. 2001;7:331–337. doi: 10.1038/85480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chauvière L, Rafrafi N, Thinus-Blanc C, et al. Early deficits in spatial memory and theta rhythm in experimental temporal lobe epilepsy. J Neurosci. 2009;29:5402–10. doi: 10.1523/JNEUROSCI.4699-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cunningham C, Sanderson DJ. Malaise in the water maze: untangling the effects of LPS and IL-1beta on learning and memory. Brain Behav Immun. 2008;22:1117–27. doi: 10.1016/j.bbi.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Terrando N, Rei Fidalgo A, Vizcaychipi M, et al. The impact of IL-1 modulation on the development of lipopolysaccharide-induced cognitive dysfunction. Crit Care. 2010;14:R88. doi: 10.1186/cc9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Valdés-Ferrer SI, Rosas-Ballina M, Olofsson PS, et al. High-mobility group box 1 mediates persistent splenocyte priming in sepsis survivors: evidence from a murine model. Shock. 2013;40:492–495. doi: 10.1097/SHK.0000000000000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Uhlhaas PJ, Singer W. Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron. 2006;52:155–168. doi: 10.1016/j.neuron.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 70.Vlooswijk MC, Jansen JF, de Krom MC, et al. Functional MRI in chronic epilepsy: associations with cognitive impairment. Lancet Neurol. 2010;9:1018–27. doi: 10.1016/S1474-4422(10)70180-0. [DOI] [PubMed] [Google Scholar]
- 71.Zorn-Pauly K, Pelzmann B, Lang P, et al. Endotoxin impairs the human pacemaker current If. Shock. 2007;28:655–661. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl Figure 1.IL -1β expressing microglia in the rat hippocampus after LPS (a-d) Representative photomicrographs showing the progressive changes in IL-1β immunoreactivity in the rat septal hippocampus at various times (n=4-12 rats) after bilateral icv LPS injection (25 μg/2 μl) vs vehicle injection (n=2-8 rats). Six hours after LPS injection (b), IL-1β immunostaining was increased in the hippocampal regions adjacent to the ventricular area. IL-1β signal was strongly induced throughout the whole hippocampus at 24 h (c) post-LPS, then declining by 1 week (d) to undetectable levels as in sham controls (a). The merge images (e,f) show co-localization of IL-1β signal with the microglia marker OX-42 (yellow signal in e) but not with the astrocytic marker GFAP (f). Bargram in (g) depicts the quantification of IL-1β expression 24 h post-LPS (area occupied by the specific signal/total area analyzed). CA1, CA3: pyramidal cell layers; h: hilus. Scale bar: a-d, 220 μm; e,f: 25 μm.
Suppl Figure 2.Treatment with LPS causes a reduction in total hippocampal HCN1 protein but not TRIP8b
Western blots were performed from whole hippocampi of rats treated with either vehicle or LPS (n=5 each experimental group). A significant reduction in HCN1 (a,b) was observed without a difference in TRIP8b (c,d). Data are mean±s.e.m. * p<0.05 by Mann–Whitney test.
Suppl Figure 3. Blockade of LPS effects by the selective TLR4 antagonist Cyanobacterial LPS
Rats were injected icv with Cyanobacterial LPS (CyP; 60 μg/3 μl in PBS, bilaterally) 15 min before and 15 min after icv LPS injection (25 μg/2 μl in PBS bilaterally). Rats were sacrificed 24 h after LPS injection. (a) A significant reduction of IL-1β staining was observed by immunohistochemistry in rats treated with LPS+CyP vs LPS alone (n=5 rats each group). CA1, CA1 pyramidal neurons; CA3, CA3 pyramidal neurons; h, hilus. Scale bar: 250 μm. (b) LPS-induced reduction of HCN1 was prevented by CyP as assessed by western blot. Data are mean±s.e.m. (n= 8-9 rats). *p<0.05 by Kruskal-Wallis followed by Dunn’s post-hoc test.