

Abstract
Background
Metformin is the most common pharmacological treatment for type 2 diabetes. It is considered safe but has been associated with the development of lactic acidosis under circumstances where plasma concentrations exceed therapeutic levels. Metformin-induced lactic acidosis has been linked to the drug’s toxic effect on mitochondrial function. Current treatment strategies aim to remove the drug and correct for the acidosis. With a mortality of 20%, complementary treatment strategies are needed. In this study, it was investigated whether targeting mitochondria with pharmacological agents that bypass metformin-induced mitochondrial dysfunction can counteract the energetic deficit linked to toxic doses of metformin.
Methods
The redox agent methylene blue and the cell-permeable succinate prodrug NV118 were evaluated by measuring mitochondrial respiration and lactate production of human platelets exposed to metformin and co-treated with either of the two pharmacological bypass agents.
Results
The cell-permeable succinate prodrug NV118 increased mitochondrial respiration which was linked to phosphorylation by the ATP-synthase and alleviated the increase in lactate production induced by toxic doses of metformin. The redox agent methylene blue, in contrast, failed to mitigate the metformin-induced changes in mitochondrial respiration and lactate generation.
Conclusions
The cell-permeable succinate prodrug NV118 bypassed the mitochondrial dysfunction and counteracted the energy deficit associated with toxic doses of metformin. If similar effects of NV118 prove translatable to an in vivo effect, this pharmacological strategy presents as a promising complementary treatment for patients with metformin-induced lactic acidosis.
Electronic supplementary material
The online version of this article (10.1186/s40635-018-0186-1) contains supplementary material, which is available to authorized users.
Keywords: Cell-permeable succinate, Human platelets, Lactic acidosis, Metformin, Methylene blue, Mitochondrial respiration, Mitochondrial toxicity
Background
Metformin is the most common pharmacological treatment for type 2 diabetes [1]. Downregulation of hepatic gluconeogenesis and decreased glucose uptake through the gut, partially due to drug-induced decreased mitochondrial function, are key aspects of metformin’s antidiabetic effect. However, the exact mechanisms are not fully elucidated yet [2]. Metformin is considered safe but has been associated with cases of lactic acidosis. Lactic acidosis is defined as arterial lactate levels above 5 mM and a pH below 7.35 [1, 2]. Metformin-induced lactic acidosis (MILA) appears primarily in patients with renal failure, circumstances under which the drug’s blood concentration can exceed therapeutic levels. Pathological conditions affecting cardiovascular, respiratory, and liver function can further exacerbate the lactic acidosis as they can impair lactate metabolism and/or the acid-base balance [1, 3, 4]. Not surprisingly, the inhibitory action of metformin on mitochondrial function is also implicated in the development of MILA. Metformin inhibits the mitochondrial glycerophosphate dehydrogenase (mGPD) and complex I (CI) of the oxidative phosphorylation (OXPHOS) pathway [2, 5]. As a result, the cell increases glycolysis to compensate for the loss of mitochondrial ATP production, which is associated with increased lactate production and acidification. Current treatment strategies consist of supportive measures, forced clearance to remove the drug and correction of the acidosis [3]. With a mortality of around 20% [6, 7], there is a need for complementary treatment strategies for patients with MILA.
In this study, we hypothesized that restoration of the OXPHOS pathway by targeting mitochondria with pharmacological agents that bypass metformin-induced mitochondrial dysfunction can alleviate the changes in mitochondrial energy production associated with metformin intoxication. We investigated the redox agent methylene blue (MB), which has been described to facilitate electron transfer from NAD(P)H-dependent dehydrogenases to cytochrome C [8–12], and the cell-permeable succinate prodrug NV118, which readily passes through the cell membrane independent of active transporters, releases succinate, and, through oxidation at complex II (CII), donates electrons to the OXPHOS pathway [13]. Our hypothesis was tested by measuring mitochondrial respiration and lactate production of human platelets exposed to metformin and co-treated with either of the two pharmacological bypass agents.
Methods
Materials
All chemicals were obtained from Sigma-Aldrich Chemie GmbH (Schnelldorf, Germany). The cell-permeable succinate prodrugs were supplied by NeuroVive Pharmaceutical AB (Lund, Sweden) [13], and MB was purchased from Sigma-Aldrich Chemie GmbH (Schnelldorf, Germany).
Human platelet isolation
Venous blood from healthy volunteers was drawn in K2EDTA tubes (Vacutainer®, BD, Franklin Lakes, USA), and human platelets were isolated as previously described [14].
High-resolution respirometry
High-resolution respirometry of human platelets was performed as previously described [5]. The corresponding doses of MB and NV118 used for further evaluation were determined based on dose-response experiments performed in rotenone-intoxicated human platelets. Both bypass strategies were then evaluated in a model of (a) CI inhibition caused by rotenone (2 μM) and (b) specific metformin-related mitochondrial dysfunction induced by exposure to either 10 mM metformin or 50 mM metformin for 60 min [5]. In the model of rotenone intoxication, MB was evaluated at 20 μM. In the model of metformin-induced mitochondrial dysfunction, the dose of MB was adjusted to 10 μM based on an additional dose-response performed with the lactate production assay. NV118 was evaluated in both models at 250 μM. Mitochondrial respiration coupled to phosphorylation, here referred to as coupled respiration, was evaluated by addition of the ATP-synthase inhibitor oligomycin (1 μg/ml) to block the phosphorylation pathway, and calculated as the difference in respiration before and after the inhibition of the ATP-synthase. Control experiments were performed without the addition of oligomycin to account for background drift of oxygen consumption. Complex III (CIII) was blocked with antimycin A (1 μg/ml), and complex IV (CIV) was inhibited with sodium azide (10 mM). Remaining respiration after addition of sodium azide was defined as non-mitochondrial respiration.
To exclude effects on mitochondrial respiration induced by the vehicles of the bypass strategies (NV118, DMSO; MB, double-deionized water), the effects of the corresponding vehicles were evaluated simultaneously (Fig. 1) or in a separately performed experiment (Fig. 2, vehicle controls of both bypass agents were pooled). In the model of specific metformin-related mitochondrial dysfunction, a vehicle control (double-deionized water) to the metformin treatment was included.
Fig. 1.
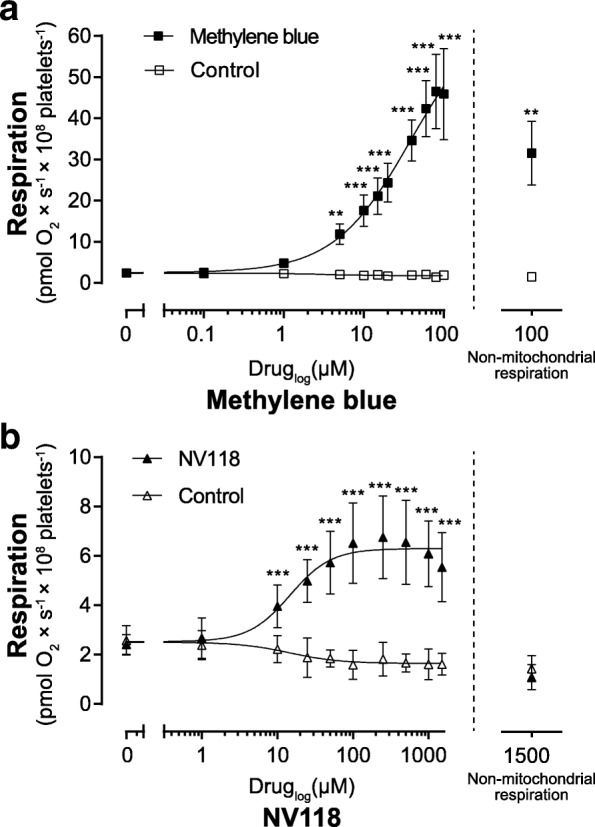
Dose-response of methylene blue and NV118 on oxygen consumption in rotenone-intoxicated human platelets. Respiration was measured in human platelets with complex I inhibition induced by rotenone (2 μM). The potential of the pharmacological bypass strategies methylene blue (a black square) and the cell-permeable succinate prodrug NV118 (b black triangle) to increase rotenone-inhibited respiration was evaluated by titrating increasing doses of drug or vehicle (a white square, double-deionized water; b white triangle, DMSO). After maximal respiration was reached, the contribution of non-mitochondrial respiration to total respiration was evaluated by addition of the complex III inhibitor antimycin A (1 μg/ml) followed by the complex IV inhibitor sodium azide (10 mM). The residual respiration shows the non-mitochondrial respiration at the highest dose of each drug. Data are expressed as mean ± SD. Non-linear curve fitting was applied for generation of the dose-response curves. n = 4. Two-way ANOVA with Bonferroni post hoc test was performed for analysis of differences. **p < 0.01, ***p < 0.001, compared to vehicle control
Fig. 2.

Effect of methylene blue and NV118 on coupled respiration in rotenone-intoxicated human platelets. a Representative traces of mitochondrial respiration in human platelets with complex I inhibition induced by rotenone (2 μM). Mitochondrial respiration due to coupled phosphorylation of rotenone-intoxicated platelets treated with vehicle (rotenone, dark gray trace), methylene blue (20 μM, dark blue trace) or the cell-permeable succinate prodrug NV118 (250 μM, dark green trace), here referred to as coupled respiration, was evaluated by subsequent addition of the ATP-synthase inhibitor oligomycin (1 μg/ml) to block the phosphorylation pathway, and calculated as the difference in respiration before and after the inhibition of the ATP-synthase. The protocol was continued by adding the complex III inhibitor antimycin A (1 μg/ml) followed by the complex IV inhibitor sodium azide (10 mM). Control experiments were performed without the addition of oligomycin to account for background drift of oxygen consumption in the presence of the bypass agents (light blue trace, light green trace). b Quantification of coupled respiration of rotenone-intoxicated platelets treated with vehicle (rotenone, black circle), methylene blue (20 μM, dark blue square), or the cell-permeable succinate prodrug NV118 (250 μM, dark green triangle) was calculated as the difference in respiration before and after inhibition of the ATP-synthase. c The coupled respiration, non-coupled respiration, and non-mitochondrial respiration was evaluated in rotenone-intoxicated platelets treated with vehicle, methylene blue, or NV118. Non-mitochondrial respiration was defined as respiration remaining after addition of sodium azide, which all other respiratory values were corrected for. Data are expressed as individual scatter plot and mean ± SD (b) or mean (c). Rotenone: n = 21, NV118 and methylene blue: n = 7. One-way ANOVA with Dunnet post hoc test was used for comparison of the drug’s effect on coupled respiration. ***p < 0.001. CII–V: complex II to V, CII–IV: complex II to IV, CIV: complex IV
Lactate production
Platelets were incubated with metformin (10 mM) or its vehicle (double-deionized water) in glucose-containing PBS (10 mM) for 60 min before co-treatment with either MB or the cell-permeable succinate prodrug was initiated. MB was administered either as a single dose (10 μM) or as one dose every 30 min (1 μM). The cell-permeable succinate prodrugs NV118, NV189, and NV241, or succinate, were given every 30 min (250 μM). Lactate levels in the medium were measured every 30 min over 4 h, and the lactate production was calculated from onset of intervention (60–240 min) as previously described [13]. Vehicle control and metformin alone were run on each occasion.
Data analysis
Dose-response evaluation of MB and NV118 on respiration in rotenone-intoxicated human platelets was performed with a group size of four replicates (n = 4). As the magnitude of the change in the evaluated parameter was not pre-specified, power calculation for sample size was not applied here. Unless indicated otherwise, all other evaluations were performed with a sample size of seven replicates (n = 7). This sample size was calculated from previously published data showing increased lactate production by human platelets exposed to metformin (10 mM) [5] and determined to be required to detect a targeted 25% reduction in lactate production in human platelets exposed to metformin (10 mM) for 4 h and co-treated with either pharmacological treatment, an alpha level of 5% and power of 80%. Data are expressed as scatter plot or mean ± SD. Lactate production was calculated using standard non-linear curve fitting. Statistical analysis was performed using GraphPad Prism version 7 (GraphPad Software, Inc., La Jolla, California, USA). Data from blood cell respirometry have been reported to be normally distributed [14], and parametric tests were used for analyses of differences. For multiple comparisons of two groups (Fig. 1), two-way ANOVA with Bonferroni post hoc test was performed. One-way ANOVA with Dunnet post hoc test was applied for one-factor comparison of three or more groups. A p value of 0.05 or less was considered to indicate significant difference. No blinding or randomization was performed.
Results
Dose-response of methylene blue and NV118 on respiration in rotenone-intoxicated human platelets
MB and NV118 dose dependently increased respiration in human platelets with rotenone-induced CI inhibition (Fig. 1). MB started to increase respiration at 5 μM (p < 0.01) and reached the maximum with a 33-fold increase at 80 μM (p < 0.001) as compared to control. MB also induced non-mitochondrial respiration which, at the highest concentration investigated here, was higher than control (p < 0.01), and responsible for 69% of total respiration (Fig. 1a). NV118 started to increase respiration at 10 μM (p < 0.001) with a maximum and fourfold increase at 250 μM (p < 0.001) and displayed no effect on non-mitochondrial respiration (Fig. 1b). Based on the dose-response and effect on non-mitochondrial respiration, 20 μM MB (15-fold increase compared to control) and 250 μM NV118 were selected for further evaluation in the model of rotenone intoxication. Neither of the vehicles of the pharmacological bypass strategies increased mitochondrial respiration (Fig. 1).
Effect of methylene blue and NV118 on coupled respiration in rotenone-intoxicated human platelets
Both MB and NV118 increased respiration in rotenone-intoxicated human platelets as compared to vehicle-treated controls (Fig. 2a). The increase in respiration with MB was not caused by increased coupled respiration (rotenone = 0.05 pmol O2 × s−1 × 108 platelets−1; MB = − 0.02 pmol O2 × s−1 × 108 platelets−1; p = 0.95) (Fig. 2b) but elevated non-coupled and non-mitochondrial respiration, accounting for 43.5 and 56.5% of total, drug-induced respiration respectively (Fig. 2a, c). NV118, in contrast, increased coupled respiration significantly as compare to vehicle-treated, rotenone-intoxicated human platelets (NV118 = 2.46 pmol O2 × s−1 × 108 platelets−1; p < 0.001) (Fig. 2b), accounting for 32.9% of total, drug-induced respiration and with no effect on non-mitochondrial respiration (Fig. 2a, c).
Effect of methylene blue and NV118 on coupled respiration and lactate metabolism in metformin-induced mitochondrial dysfunction in human platelets
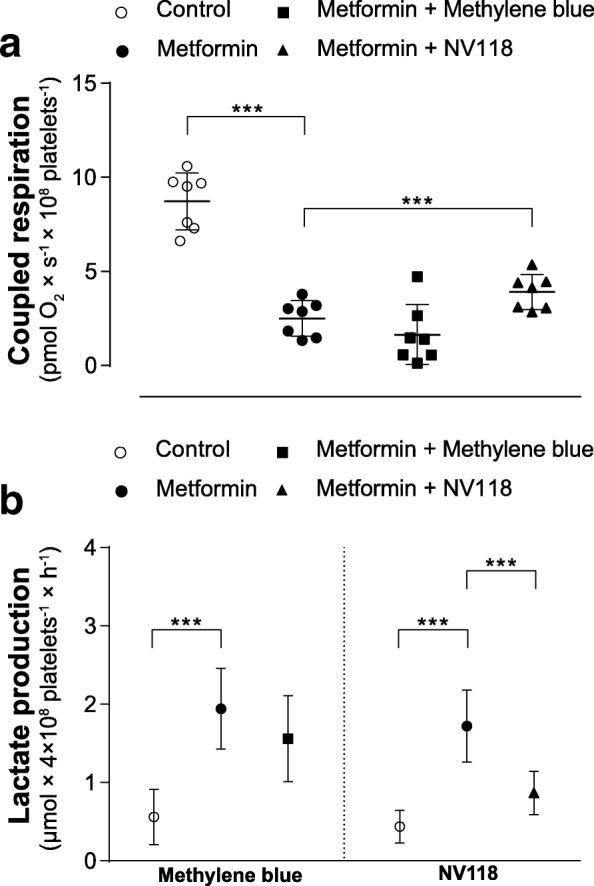
Both 10 mM metformin (− 28%, p < 0.05) (Additional file 1) and 50 mM metformin (− 69%, p < 0.001) (Fig. 3a) reduced coupled respiration in human platelets significantly compared to control. Samples treated with MB showed a tendency towards decreased coupled respiration compared to metformin alone (10 mM metformin: − 13%, p = 0.49; 50 mM metformin: − 32%, p = 0.41) (Additional file 1, Fig. 3a). NV118, to the contrary, increased coupled respiration by 20% after exposure to 10 mM metformin (p = 0.37) (Additional file 1) and by 46% after exposure to 50 mM metformin (p < 0.001) (Fig. 3a) as compared to metformin alone.
Fig. 3.
Effect of methylene blue and NV118 on coupled respiration and lactate production in metformin-intoxicated human platelets. a Mitochondrial respiration was measured in human platelets with mitochondrial dysfunction induced by 60 min exposure to metformin (black circle; 50 mM). After subsequent addition of methylene blue (10 μM, black square) or the cell-permeable succinate prodrug NV118 (250 μM, black triangle), mitochondrial respiration due to coupled phosphorylation, here referred to as coupled respiration, was evaluated by addition of the ATP-synthase inhibitor oligomycin (1 μg/ml) to block the phosphorylation pathway, and calculated as the difference in respiration before and after the inhibition of the ATP-synthase. Subsequently, the complex III inhibitor antimycin A (1 μg/ml) followed by the complex IV inhibitor sodium azide (10 mM) were added. Control experiments were performed without the addition of oligomycin to account for background drift of oxygen consumption. A vehicle control to metformin was run with each experiment (white circle). b Lactate production of human platelets incubated with metformin (10 mM, black circle) was measured every 30 min over 4 h with or without co-treatment of methylene blue (10 μM, single dose, black square) or NV118 (250 μM, every 30 min, black triangle) starting at 60 min. A vehicle control was run with each experiment (white circle). Data are expressed as individual scatter plot and mean ± SD (a) or mean ± SD (b). All experiments were performed with n = 7, with exception of the control and metformin group of the lactate production assay run with methylene blue (n = 9) (b). One-way ANOVA with Dunnet post hoc test was used. ***p < 0.001, compared to metformin
Metformin significantly increased lactate production compared to vehicle control (p < 0.001) (Fig. 3b). Co-treatment with a single dose of MB (10 μM) did not reduce lactate production significantly (− 20%, p = 0.30) (Fig. 3b) and neither did supplementation with a low dose of MB (1 μM) every 30 min (− 11%, p = 0.69) (Additional file 2). In contrast, co-treatment with NV118 (250 μM) every 30 min alleviated the metformin-induced increase in lactate production significantly (− 50%, p < 0.001) (Fig. 3b). Qualitatively, similar results were obtained for NV189 and NV241, two compounds of the same drug class as NV118, whereas succinate did not mitigate the metformin-induced increase in lactate production (Additional file 2).
Bypass mechanism of methylene blue and NV118 in human platelets with rotenone intoxication and metformin-induced mitochondrial dysfunction
In MB-treated cells, respiration decreased primarily when CIV was inhibited but only marginally when CIII was blocked (Fig. 2a). In cells treated with NV118, mitochondrial respiration decreased when CIII was inhibited but no further decline was seen when CIV was blocked (Fig. 2a). This was the case for cells with rotenone intoxication and also for cells with metformin-induced mitochondrial dysfunction.
Discussion
In the present study, we demonstrated that in human platelets, the cell-permeable succinate prodrug NV118 increased mitochondrial respiration in a model of rotenone intoxication which was linked to increased coupled respiration. NV118 further improved coupled respiration significantly in a model of specific metformin-induced mitochondrial dysfunction and counteracted the phenotype that metformin had caused on lactate metabolism, as it alleviated the metformin-induced increase in lactate production in human platelets. With NV118, mitochondrial respiration increased downstream of CI and upstream of CIII, indicating CII as entry point to the OXPHOS pathway. Although the redox agent MB enhanced mitochondrial respiration downstream of CIII, it did not improve coupled respiration in human platelets in either model of drug-induced mitochondrial toxicity, nor did it mitigate the metformin-induced increase in lactate generation. In this study, human platelets were used as a substitute for more metabolically active tissues, both in regard to metformin’s mitochondrial toxicity and the treatment effect of the bypass strategies. If the counteracting effect of NV118 proves translatable to an in vivo effect, this strategy could potentially contribute to resolving the systemic energy failure associated with toxic doses of metformin and thus, correct the lactic acidosis.
Succinate is oxidized by CII of the OXPHOS pathway (Fig. 4) [13, 15]. Succinate oxidation and subsequent electron transfer along the respiratory chain enables proton translocation across the inner mitochondrial membrane at CIII and CIV, build-up of the proton motive force, and an increase in mitochondrial ATP production. CII serves as an alternative entry point to CI for electrons to the OXPHOS pathway. Thus, the cell-permeable succinate prodrugs can bypass the CI inhibition induced by metformin (Fig. 4b). This has been demonstrated in the present study and in a study by Hinke et al. [16] in which succinate improved mitochondrial activity and rescued pancreatic β-cells from metformin-induced toxicity [16]. The improvement of OXPHOS relieved pressure from the metformin-induced increase in glycolytic ATP production with the results that less lactate was produced (Fig. 4a, b). Because NV118 lacks sufficient plasma stability, we were not able to investigate its effect in vivo. Large animal models of rotenone-induced CI-dysfunction and metformin-induced mitochondrial dysfunction like those by Karlsson et al. [17] and Protti et al. [18] would be suitable models for in vivo proof of concept of this drug class. This pharmacological bypass strategy could potentially find additional applications in intensive care. Conditions such as sepsis, traumatic brain injury, primary mitochondrial disease or drug-induced lactic acidosis are indications with a high need for further treatment development, where impaired, CI-related mitochondrial dysfunction has been described and succinate has shown success as potential treatment in vitro and in vivo [15, 19–26].
Fig. 4.

Schematic illustration of the bypass mechanism of NV118 and methylene blue in metformin-induced mitochondrial dysfunction. a At high concentrations, metformin induces inhibition of complex I (CI) and the mitochondrial glycerophosphate dehydrogenase (mGPD). As a result, ATP generation at complex V (CV/ATP-synthase) is decreased causing increased glycolysis to compensate for the reduced mitochondrial ATP production. Pyruvate metabolism is shifted towards lactate generation to meet the increased NAD+ demand, which is accompanied by intracellular and extracellular acidification. b Cell-permeable succinate prodrugs, such as NV118, can permeate the cell membrane independent of active transporters. Through intracellular metabolism, the succinate core is released and made available for oxidation by complex II (CII). Oxidation of succinate at CII restores downstream electron flow and mitochondrial respiration which is linked to phosphorylation (ATP generation). c The redox agent methylene blue (MB) is reduced by NAD(P)H-dependent dehydrogenases. The reduced form of methylene blue (MBH2, leucomethylene blue) then donates the electrons to cytochrome C (labeled C), recycling MB. Electron donation by MBH2 to cytochrome C restores downstream electron flow and mitochondrial respiration which is not coupled to phosphorylation. ADP: adenosine diphosphate, ATP: adenosine triphosphate, CIII: complex III, CIV: complex IV, cGPD: cytosolic glycerophosphate dehydrogenase, DHAP: dihydroxyacetone phosphate, G3P: glycerol-3-phosphate, NAD+/NADH: nicotinamide adenine dinucleotide, NADP+/NADPH: nicotinamide adenine dinucleotide phosphate, Q: Quinone, TCA: tricarboxylic acid
Repurposing drugs is attractive because pharmacodynamics and pharmacokinetics, safety profiles and contraindications are known. MB has been used clinically as treatment for methemoglobinemia and as an anti-malaria agent [27, 28]. Because of its potential beneficial effects on mitochondrial function, MB has recently attracted attention as treatment for neurodegenerative disorders and drug-induced side-effects [8, 11, 29, 30]. MB has been described to shuttle electrons from NAD(P)H-dependent dehydrogenase to cytochrome C (Fig. 4c) [8, 10–12, 31]. In the present study, we evaluated both bypass strategies based on immediate effects on coupled respiration and related changes in lactate metabolism. Due to the severity of MILA, there is a need for treatments acting instantly. Under these conditions, MB was unable to improve coupled respiration and alleviate the increased lactate generation associated with metformin (Fig. 4a, c). Others have previously described beneficial effects of MB on electron shuttling to the OXPHOS pathway [8, 10, 12, 32]. However, in these studies, either the oxidation of NADH at CI was evaluated in isolated, sonicated mitochondria or sub-mitochondrial particles where coupling of the electron transfer to phosphorylation pathways cannot be assessed [8, 12, 32] or it was not evaluated whether the increase in mitochondrial respiration of the fully integrated respiratory chain was linked to increased coupled respiration [10, 12]. As demonstrated in these studies, we also detected an increase in mitochondrial respiration. However, we have shown that the increased electron transport and mitochondrial respiration was not used to or is not sufficient to increase the phosphorylation pathway (Fig. 4c). By increasing CIV-linked mitochondrial respiration, MB would support proton translocation across the inner mitochondrial membrane and contribute to the proton motive force at CIV. CIV pumps less protons than complex I and III alone and thus contributes to a lesser degree to the build-up of the proton motive force which drives the phosphorylation pathway [33]. This could potentially explain the lack of beneficial effect of MB observed in the present study. On the basis of our experiments, we cannot exclude that MB potentially supports mitochondrial function through reduction of oxidative stress or stimulation of mitochondrial biogenesis and degradation of impaired mitochondria, effects that would not be immediate and might be more relevant at later stage of intervention [11, 29, 30]. MB has also been described to stabilize hemodynamic parameters through inhibition of the guanylate cyclase and has been used successfully in a small number of case reports of drug-induced shock with this objective [34].
Conclusions
In the present study, using human platelets as model, we demonstrated that treatment with the cell-permeable succinate prodrug NV118 can ameliorate the metformin-induced impairment of the OXPHOS pathway and alleviate the associated increase in production of lactate. The redox agent MB did neither correct the inhibition of coupled mitochondrial respiration nor attenuate the increased lactate generation induced by metformin. Thus, the pharmacological bypass of metformin-induced mitochondrial dysfunction with cell-permeable succinate prodrugs presents as a promising complementary treatment strategy for patients with MILA.
Additional files
Effect of methylene blue (10 μM) and the cell-permeable succinate prodrug NV118 (250 μM) on coupled respiration after exposure to 10 mM metformin for 60 min. (PDF 42 kb)
Effect of methylene blue (1 μM), the cell-permeable succinate prodrugs NV189 and NV241 (250 μM), and succinate (250 μM) on lactate production in metformin-induced mitochondrial dysfunction in human platelets. (PDF 238 kb)
Acknowledgements
The cell-permeable succinate prodrugs NV118, NV241, and NV189 were kindly provided by NeuroVive Pharmaceutical AB (Lund, Sweden).
Funding
This study was funded by the Swedish government project and salary funding for clinically oriented medical research (ALF-grants; F 2014/354), Regional research and development grants (Southern healthcare region, Sweden; 170083), the Crafoord Foundation (2017-0776), and the Royal Physiographic Society in Lund (20141112).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- C
Cytochrome C
- cGPD
Cytosolic glycerophosphate dehydrogenase
- CI
Complex I
- CII
Complex II
- CIII
Complex III
- CIV
Complex IV
- CV
Complex V
- DHAP
Dihydroxyacetone phosphate
- G3P
Glycerol-3-phosphate
- MB
Methylene blue
- MBH2
Leucomethylene blue
- mGPD
Mitochondrial glycerophosphate dehydrogenase
- MILA
Metformin-induced lactic acidosis
- NAD+/NADH
Nicotinamide adenine dinucleotide
- NADP+/NADPH
Nicotinamide adenine dinucleotide phosphate
- OXPHOS
Oxidative phosphorylation
- Q
Quinone
Authors’ contributions
SP, JKE, FS, EE, and MJH conceived and directed the study. SP, IC, and EÅF evaluated the properties of the compounds. SP and JKE performed the statistical analysis. SP, JKE, FS, EE, and MJH prepared the figures. SP drafted the manuscript. SP had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors critically reviewed the manuscript and approved the final version.
Ethics approval and consent to participate
All protocols were approved by the regional ethics board of Lund University, Sweden (permit no. 2013/181). Venous blood from healthy volunteers was drawn according to clinical standard after written informed consent was acquired.
Consent for publication
Not applicable.
Competing interests
S.P., J.K.E., I.C., E.Å.F., F.S., E.E., and M.J.H have, or have had, salary from and/or equity interest in NeuroVive Pharmaceutical AB, a company active in the field of mitochondrial medicine. S.P., J.K.E., E.E., and M.J.H have filed patent applications for the use of succinate prodrugs for treatment of lactic acidosis or drug-induced side-effects due to complex I-related impairment of mitochondrial oxidative phosphorylation (WO/2015/155238) and protected carboxylic acid-based metabolites for treatment of mitochondrial disorders (WO/2017/060400, WO/2017/060418, WO/2017/060422).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sarah Piel, Phone: +46 46 222 0605, Email: sarah.piel@med.lu.se.
Johannes K. Ehinger, Email: johannes.ehinger@med.lu.se
Imen Chamkha, Email: imen.chamkha@med.lu.se.
Eleonor Åsander Frostner, Email: eleonor.asander_frostner@med.lu.se.
Fredrik Sjövall, Email: fredrik.sjovall@med.lu.se.
Eskil Elmér, Email: eskil.elmer@med.lu.se.
Magnus J. Hansson, Email: magnus.hansson@med.lu.se
References
- 1.Lalau JD, Arnouts P, Sharif A, De Broe ME. Metformin and other antidiabetic agents in renal failure patients. Kidney Int. 2015;87:308–322. doi: 10.1038/ki.2014.19. [DOI] [PubMed] [Google Scholar]
- 2.Rena G, Hardie DG, Pearson ER (2017) The mechanisms of action of metformin. Diabetologia. 10.1007/s00125-017-4342-z [DOI] [PMC free article] [PubMed]
- 3.Lalau J-D. Lactic acidosis induced by metformin. Drug Saf. 2010;33:727–740. doi: 10.2165/11536790-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Lalau JD, Kajbaf F, Protti A, Christensen MM, De Broe ME, Wiernsperger N (2017) Metformin-associated lactic acidosis (MALA): moving towards a new paradigm. Diabetes Obes Metab. 10.1111/dom.12974 [DOI] [PubMed]
- 5.Piel S, Ehinger JK, Elmer E, Hansson MJ. Metformin induces lactate production in peripheral blood mononuclear cells and platelets through specific mitochondrial complex I inhibition. Acta Physiol (Oxford, England) 2015;213:171–180. doi: 10.1111/apha.12311. [DOI] [PubMed] [Google Scholar]
- 6.Lalau J-D, Race J-M. Lactic acidosis in metformin-treated patients. Drug Saf. 1999;20:377–384. doi: 10.2165/00002018-199920040-00006. [DOI] [PubMed] [Google Scholar]
- 7.Vecchio S, Protti A. Metformin-induced lactic acidosis: no one left behind. Crit Care. 2011;15:107–107. doi: 10.1186/cc9404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee KK, Boelsterli UA. Bypassing the compromised mitochondrial electron transport with methylene blue alleviates efavirenz/isoniazid-induced oxidant stress and mitochondria-mediated cell death in mouse hepatocytes. Redox Biol. 2014;2:599–609. doi: 10.1016/j.redox.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee KK, Imaizumi N, Chamberland SR, Alder NN, Boelsterli UA. Targeting mitochondria with methylene blue protects mice against acetaminophen-induced liver injury. Hepatology (Baltimore, Md) 2015;61:326–336. doi: 10.1002/hep.27385. [DOI] [PubMed] [Google Scholar]
- 10.Poteet E, et al. Neuroprotective actions of methylene blue and its derivatives. PLoS One. 2012;7:e48279. doi: 10.1371/journal.pone.0048279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tucker D, Lu Y, Zhang Q (2017) From mitochondrial function to neuroprotection-an emerging role for methylene blue. Mol Neurobiol. 10.1007/s12035-017-0712-2 [DOI] [PMC free article] [PubMed]
- 12.Wen Y, et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J Biol Chem. 2011;286:16504–16515. doi: 10.1074/jbc.M110.208447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ehinger JK, et al. Cell-permeable succinate prodrugs bypass mitochondrial complex I deficiency. Nat Commun. 2016;7:12317. doi: 10.1038/ncomms12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sjovall F, et al. Mitochondrial respiration in human viable platelets-methodology and influence of gender, age and storage. Mitochondrion. 2013;13:7–14. doi: 10.1016/j.mito.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 15.Jalloh I, et al. Focally perfused succinate potentiates brain metabolism in head injury patients. J Cereb Blood Flow Metab. 2017;37:2626–2638. doi: 10.1177/0271678X16672665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinke SA, Martens GA, Cai Y, Finsi J, Heimberg H, Pipeleers D, Van de Casteele M. Methyl succinate antagonises biguanide-induced AMPK-activation and death of pancreatic beta-cells through restoration of mitochondrial electron transfer. Br J Pharmacol. 2007;150:1031–1043. doi: 10.1038/sj.bjp.0707189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karlsson M, et al. Changes in energy metabolism due to acute rotenone-induced mitochondrial complex I dysfunction—an in vivo large animal model. Mitochondrion. 2016;31:56–62. doi: 10.1016/j.mito.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Protti A, et al. Skeletal muscle lactate overproduction during metformin intoxication: an animal study with reverse microdialysis. Toxicol Lett. 2016;255:43–46. doi: 10.1016/j.toxlet.2016.05.010. [DOI] [PubMed] [Google Scholar]
- 19.Ferreira FL, Ladriere L, Vincent JL, Malaisse WJ. Prolongation of survival time by infusion of succinic acid dimethyl ester in a caecal ligation and perforation model of sepsis. Horm Metab Res. 2000;32:335–336. doi: 10.1055/s-2007-978647. [DOI] [PubMed] [Google Scholar]
- 20.Giorgi-Coll S, Amaral AI, Hutchinson PJA, Kotter MR, Carpenter KLH. Succinate supplementation improves metabolic performance of mixed glial cell cultures with mitochondrial dysfunction. Sci Rep. 2017;7:1003. doi: 10.1038/s41598-017-01149-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirby DM, Thorburn DR, Turnbull DM, Taylor RW. Biochemical assays of respiratory chain complex activity. Methods Cell Biol. 2007;80:93–119. doi: 10.1016/S0091-679X(06)80004-X. [DOI] [PubMed] [Google Scholar]
- 22.Loeffen JL, Smeitink JA, Trijbels JM, Janssen AJ, Triepels RH, Sengers RC, van den Heuvel LP. Isolated complex I deficiency in children: clinical, biochemical and genetic aspects. Hum Mutat. 2000;15:123–134. doi: 10.1002/(SICI)1098-1004(200002)15:2<123::AID-HUMU1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 23.Malaisse WJ, Nadi AB, Ladriere L, Zhang T-M. Protective effects of succinic aciddimethyl ester infusion in experimental endotoxemia. Nutrition (Burbank, Los Angeles County, Calif) 1997;13:330–341. [PubMed] [Google Scholar]
- 24.Pham A, Xu L, Moe O. Drug-induced metabolic acidosis [version 1; referees: 3 approved] vol 4. vol 1460. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Protti A, Carre J, Frost MT, Taylor V, Stidwill R, Rudiger A, Singer M. Succinate recovers mitochondrial oxygen consumption in septic rat skeletal muscle. Crit Care Med. 2007;35:2150–2155. doi: 10.1097/01.ccm.0000281448.00095.4d. [DOI] [PubMed] [Google Scholar]
- 26.Skladal D, et al. The clinical spectrum of mitochondrial disease in 75 pediatric patients. Clin Pediatr. 2003;42:703–710. doi: 10.1177/000992280304200806. [DOI] [PubMed] [Google Scholar]
- 27.Ehrhardt K, Davioud-Charvet E, Ke H, Vaidya AB, Lanzer M, Deponte M. The antimalarial activities of methylene blue and the 1,4-naphthoquinone 3-[4-(trifluoromethyl)benzyl]-menadione are not due to inhibition of the mitochondrial electron transport chain. Antimicrob Agents Chemother. 2013;57:2114–2120. doi: 10.1128/AAC.02248-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong A, Koutsogiannis Z, Greene S, McIntyre S. A case of hemolysis and methemoglobinemia following amyl nitrite use in an individual with G6PD deficiency. J Acute Med. 2013;3:23–25. doi: 10.1016/j.jacme.2012.12.005. [DOI] [Google Scholar]
- 29.Atamna H, Atamna W, Al-Eyd G, Shanower G, Dhahbi JM. Combined activation of the energy and cellular-defense pathways may explain the potent anti-senescence activity of methylene blue. Redox Biol. 2015;6:426–435. doi: 10.1016/j.redox.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shin SY, Kim TH, Wu H, Choi YH, Kim SG. SIRT1 activation by methylene blue, a repurposed drug, leads to AMPK-mediated inhibition of steatosis and steatohepatitis. Eur J Pharmacol. 2014;727:115–124. doi: 10.1016/j.ejphar.2014.01.035. [DOI] [PubMed] [Google Scholar]
- 31.Atamna H, Mackey J, Dhahbi JM. Mitochondrial pharmacology: electron transport chain bypass as strategies to treat mitochondrial dysfunction. BioFactors (Oxford, England) 2012;38:158–166. doi: 10.1002/biof.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Atamna H, Nguyen A, Schultz C, Boyle K, Newberry J, Kato H, Ames BN. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. 2008;22:703–712. doi: 10.1096/fj.07-9610com. [DOI] [PubMed] [Google Scholar]
- 33.Sazanov LA. A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat Rev Mol Cell Biol. 2015;16:375. doi: 10.1038/nrm3997. [DOI] [PubMed] [Google Scholar]
- 34.Warrick BJ, Tataru AP, Smolinske S. A systematic analysis of methylene blue for drug-induced shock. Clin Toxicol. 2016;54:547–555. doi: 10.1080/15563650.2016.1180390. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of methylene blue (10 μM) and the cell-permeable succinate prodrug NV118 (250 μM) on coupled respiration after exposure to 10 mM metformin for 60 min. (PDF 42 kb)
Effect of methylene blue (1 μM), the cell-permeable succinate prodrugs NV189 and NV241 (250 μM), and succinate (250 μM) on lactate production in metformin-induced mitochondrial dysfunction in human platelets. (PDF 238 kb)
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.