

Abstract
Background
Somatostatin receptor targeting radiopeptides are successfully being used to image, stage, and monitor patients with neuroendocrine tumours. They are exclusively agonists that internalise upon binding to the relevant receptor. According to recent reports, antagonists may be preferable to agonists. To date, 99mTc-labelled somatostatin receptor antagonists have attracted little attention. Here, we report on a new somatostatin receptor subtype 2 (sst2) antagonist, SS-01 (p-Cl-Phe-cyclo(D-Cys-Tyr-D-Trp-Lys-Thr-Cys)D-Tyr-NH2), with the aim of developing 99mTc-labelled ligands for SPECT/CT imaging. SS-01 was prepared using Fmoc solid-phase synthesis and subsequently coupled to the chelators 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA), 6-carboxy-1,4,8,11-tetraazaundecane (N4), and 6-hydrazinonicotinic acid (HYNIC) to form the corresponding peptide-chelator conjugates SS-03, SS-04, and SS-05, respectively. SS-04 and SS-05 were radiolabelled with 99mTc and SS-03 with 177Lu. Binding affinity and antagonistic properties were determined using autoradiography and immunofluorescence microscopy. Biodistribution and small animal SPECT/CT studies were performed on mice bearing HEK293-rsst2 xenografts.
Results
The conjugates showed low nanomolar sst2 affinity and antagonistic properties. 177Lu-DOTA-SS-01 (177Lu-SS-03) and 99mTc-N4-SS-01 (99mTc-SS-04) demonstrated high cell binding and low internalisation, whereas 99mTc-HYNIC/edda-SS-01 (99mTc-SS-05) showed practically no cellular uptake in vitro. The 99mTc-SS-04 demonstrated impressive tumour uptake at early time points, with 47% injected activity per gram tumour (%IA/g) at 1 h post-injection. The tumour uptake persisted after 4 h and was 32.5 %IA/g at 24 h. The uptake in all other organs decreased much more rapidly leading to high tumour-to-normal organ ratios, which was reflected in high-contrast SPECT/CT images.
Conclusions
These data indicate a very promising 99mTc-labelled sst2-targeting antagonist. The results demonstrate high sensitivity of the 99mTc-labelling strategy, which was shown to strongly influence the receptor affinity, contrary to corresponding agonists. 99mTc-SS-04 exhibits excellent pharmacokinetics and imaging properties and appears to be a suitable candidate for SPECT/CT clinical translation.
Electronic supplementary material
The online version of this article (10.1186/s13550-018-0428-y) contains supplementary material, which is available to authorized users.
Keywords: NETs, Somatostatin receptor antagonists, 99mTc, SPECT/CT
Background
Somatostatin receptors are important biomarkers for imaging and targeted radionuclide therapy of human cancers. They belong to the large family of G-protein coupled receptors, which currently account for 30–40% of marketed drugs [1] and are not only overexpressed in neuroendocrine tumours in particular but also in non-neuroendocrine tumours [2]. The receptors make ideal targets for imaging, as they are easily accessible on the plasma membrane of the tumour cell. Their action is mediated through two mechanisms: G-protein activation and β-arrestin function. Among its signalling roles, the latter promotes receptor internalisation—an important mechanism for radiolabelled agonist ligand uptake, accumulation, and retention [3, 4].
In contrast, binding of neutral antagonists does not lead to internalisation but potentially involves many more binding sites than agonist-based approaches. This may lead to the binding of a higher number of radio-vectors and thus to a stronger signal originating from the tumour. Ginj et al. have shown that in both in vitro and in vivo animal models, somatostatin-based radioantagonists may indeed be superior to radioagonists [5]. These findings have recently been duplicated with antagonists conjugated with DOTA and NODAGA and labelled with the positron-emitting radiometals 68Ga and 64Cu and other 3+ (radio)metals [6–8] and further supported by first-in-human imaging and therapy studies [9–12]. Similarly, preclinical [13–16] and clinical [17] studies of bombesin-based radioantagonists showed that using antagonists may be advantageous over agonists for targeted imaging and therapy of GRP receptor-expressing tumours.
Despite the fact that the first somatostatin-based radioantagonists have been studied in humans and proven to be superior to registered radioagonists, little is known about the influence of bioconjugation, labelling strategies, and other modifications on the in vitro and in vivo pharmacology of radiolabelled antagonists. Wadas et al. compared a potent agonist, TATE ([Tyr3,Thr8]octreotide) labelled with 64Cu using CB-TE2A, a cross-bridged cyclam-14 derivative, with the antagonist (sst2-ANT, p-NO2-Phe-cyclo(d-Cys-Tyr-d-Trp-Lys-Thr-Cys)d-Tyr-NH2), labelled using the same strategy [18]. They showed that the radioantagonist had lower tumour uptake despite the much higher receptor numbers found in the AR42J tumour model. AR42J cells endogenously express sst2 receptors. The authors also reported a much lower receptor affinity for the antagonist, which might be the reason for the difference rather than the origin of the receptor (natural or transfected), as proposed by the authors.
Even more intriguing is a study by Dude et al. [19], who compared the antagonist 68Ga-NODAGA-JR11 with the two agonists 68Ga-DOTATOC and 68Ga-DOTATATE. Surprisingly, the authors found that 68Ga-NODAGA-JR11 has the lowest tumour uptake in their human ZR-75-1 breast tumour model. In addition, the number of receptors using 68Ga-DOTATOC was more than twofold higher than 68Ga-DOTATATE and significantly higher than the antagonist.
More clinically relevant, however, is the fact that the phenomenon of higher tumour uptake of antagonists was also observed in human tumours. Reubi et al. performed quantitative autoradiography in neuroendocrine and non-neuroendocrine tumour specimens using the 125I-DOTA-JR11 antagonist and 125I-Tyr3octreotide agonist and found up to tenfold higher uptake of the antagonist radioligand compared to the agonist. The authors concluded that all renal cell cancers, most breast cancers, non-Hodgkin lymphomas, and medullary thyroid cancers appear to be novel targets for in vivo targeting with sst2 radioantagonists [20].
These different observations emphasise the need to study the influence of overall structure on pharmacology in vitro and in vivo in different cell and tumour models. In particular, modifications allowing 99mTc-labelling have only been studied in one case [21], although 99mTc-labelled radiopharmaceuticals are still the mainstay of nuclear medicine. We report here the synthesis of a new somatostatin-based antagonist, 4-Cl-Phe-cyclo(D-Cys-Tyr-D-Trp-Lys-Thr-Cys)D-Tyr-NH2 (SS-01), which was designed for labelling with 99mTc using two different strategies, namely N4: 6-carboxy-1,4,8,11-tetraazaundecane (SS-04) and HYNIC: 6-hydrazinopyridine-3-carboxylic acid (SS-05), as metal-binding domains. The choice of the chemical structure of the antagonist was based on our previous experiences. We and others observed that the chirality change of amino acids 1 and 2 (aa1, aa2) and C-terminal amidation compared to octreotide type octapeptides transforms an agonist into an antagonist [22, 23]. In addition, we chose the most easily accessible amino acids leading to antagonistic peptides. To serve as a control, we also modified the peptide with DOTA for labelling with 177Lu (177Lu-SS-03).
Methods
The supplier information for reagents, radiolabelling protocols, and log D determination, as well as details about instrumentation, are provided in the Additional file 1.
Synthesis of chelator-peptide conjugates, radiochemistry
The peptides were assembled on the Rink-Amide methylbenzylhydryl (MBHA) resin employing standard Fmoc strategy. The coupling reactions were performed on a semiautomatic peptide synthesiser (RinkCombichem, Bubendorf, Switzerland) with a threefold excess of Fmoc-amino acids, using DIC/HOBt as activating agents in DMF/NMP for 2 h (see Additional file 1 for details, including labelling protocols and corresponding high-performance liquid chromatography (HPLC) data).
Binding affinity measurements
Cell membrane pellets were prepared from human sst1-expressing Chinese hamster ovary cells (kindly provided by Drs. T. Reisine and G. Singh, University of Pennsylvania, Philadelphia, Pa.); sst2-, sst3-, and sst4-expressing CCL39 cells (kindly provided by Dr. D. Hoyer, Novartis Pharma, Basel, Switzerland); and sst5-expressing human embryonic kidney 293 (HEK293) cells (kindly provided by Prof. Stefan Schulz, University of Jena, Germany) and stored at − 80 °C. Quantitative receptor autoradiography was performed on 20-μm-thick membrane pellet sections and quantitated as previously described [22, 24].
Immunofluorescence microscopy
An immunofluorescence microscopy-based internalisation assay was performed on HEK293-rsst2 cells, as previously described [22]. Briefly, the cells were treated with different antagonist chelator-peptide conjugates and/or [Tyr3]octreotide (TOC) (the sst2 agonist) at 37 °C for 30 min. After fixation and permeabilisation, cells were stained with the sst2-specific primary antibody R2-88 (provided by Dr. Agnes Schonbrunn, McGovern Medical School, University of Texas Health Science Center, Houston, Texas, USA) as described previously [25, 26]. The cells were imaged using a Leica DM RB immunofluorescence microscope, and the images were acquired using an Olympus DP10 camera.
Receptor-binding, internalisation, and dissociation kinetics
The receptor binding, internalisation, and dissociation rates of 177Lu-SS-03, 99mTc-SS-04, and 99mTc-SS-05 were studied in HEK293-rsst2 cells seeded in six-well plates, as described previously [6]. Briefly, the radiopeptide (0.25 pmol/well) was added and the cells were incubated at 37 °C. At different time points (0.5, 1, 2, and 4 h), the cellular uptake was stopped by washing twice with ice-cold PBS. The membrane-bound and internalised fractions were collected with ice-cold glycine buffer, pH 2.8 and 1 M NaOH, respectively.
For the dissociation experiments, the plates were placed on ice for 30 min. The radiopeptide (0.25 pmol/well) was added to the cells and allowed to bind for 2 h at 4 °C. The cells were then quickly washed with ice-cold PBS, and fresh pre-warmed (37 °C) medium was added. The cells were incubated at 37 °C for 10, 20, 30, 60, 120, and 240 min. The medium was collected for quantification, and the cells were treated as described previously [6].
The activity in each fraction was measured in a γ-counter (Cobra II). Nonspecific uptake was determined in the presence of 250 pmol/well [Tyr3]octreotide. The results were expressed as a percentage of the applied radioactivity.
Biodistribution studies in HEK293-rsst2-bearing mice
All animal experiments were conducted in compliance with Swiss animal protection laws and associated regulations. The protocol was approved by the Cantonal Veterinary Ethics Committee of the University of Basel (approval #789). Female athymic nude mice (4–6 weeks old) were injected subcutaneously (s.c.) in the right shoulder with 107 HEK293-rsst2 cells in 100 μL sterile PBS. The tumours were allowed to grow for 14–18 days (tumour weight 100–200 mg).
The mice were injected into the tail vein with 100 μL/10 pmol/0.37 MBq of 177Lu-SS-03 or 99mTc-SS-04 and were euthanised at 1, 4, and 24 h post-injection (p.i.). Organs of interest and blood were collected, rinsed of excess blood, blotted dry, weighed, and counted in a γ-counter. To determine nonspecific uptake, three animals were pre-injected with 20 nmol of the relevant unlabelled peptide in 0.9% NaCl solution (0.1 mL); after 5 min, the radiopeptide was injected and the percentage of injected activity per gram (%IA/g) was calculated for each tissue. The total counts injected per animal were determined by extrapolation from counts of an aliquot taken from the injected solution as a standard.
SPECT/CT imaging study
Mice bearing HEK-rsst2 tumours were euthanised 4 h after intravenous injection of 15 MBq (150 pmoles) of 99mTc-SS-04 and imaged supine, head first, using a SPECT/CT system dedicated to imaging small animals (NanoSPECT/CT™ Bioscan Inc.). Topogram and helical CT scan of the whole mouse was first acquired using the following parameters: X-ray tube current 177 μA, X-ray tube voltage 45 kVp, 90 s and 180 frames per rotation, pitch 1. The helical SPECT scan was then acquired from head to toe using multi-purpose pinhole collimators (APT1). The energy window width was 20% centred symmetrically over the energy peak of 99mTc at 140 keV. Twenty-four projections (200 s per projection) were used, allowing the acquisition of at least 50 kilocounts/projection.
SPECT images were reconstructed iteratively and filtered using the software package (HiSPECT v1.4.1876, SciVis GmbH, Goettingen, Germany) and the manufacturer’s algorithm (3 subsets, 9 iterations, 35% post-filtering, 128 × 128 matrix, zoom 1, 30 × 20 mm transaxial field of view, resulting in a pixel size of 0.3 mm). CT images were reconstructed using CTReco version r1.146, with a standard filtered back projection algorithm (Exact Cone Beam) and post-filtered (RamLak, 100% frequency cutoff), resulting in a pixel size of 0.2 mm. Co-registered images were visualised in the three orthogonal planes and as maximal intensity projection with InVivoScope v1.43 (Bioscan Inc.).
Data analysis
Statistical analysis was performed using an unpaired two-tailed t test with Prism software (Prism 5.01, September 2007, GraphPad Software Inc.). Differences at the 95% confidence level (P < 0.05) were considered significant.
Results
Synthesis, radiolabelling, and distribution coefficients (log D)
All conjugates (Fig. 1) were synthesised with the maximum yield of 30–40% and purity ≥ 97%. The conjugates were characterised by analytical reversed phase HPLC and ESI-MS (Table 1, Additional file 1).
Fig. 1.
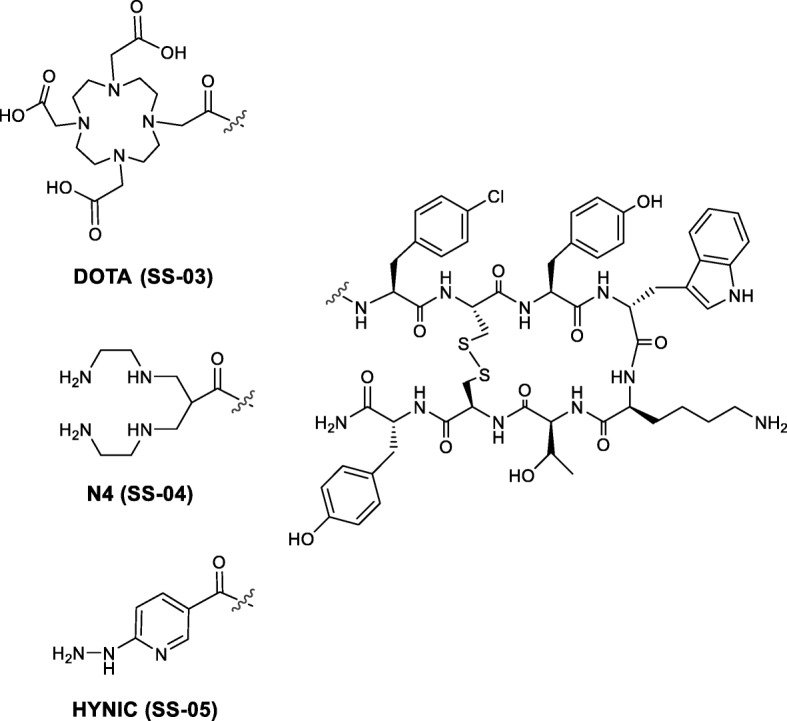
Structures of the somatostatin receptor subtype 2 antagonist conjugated to different chelators
Table 1.
Analytical data of the purified chelator-peptide conjugates
| Compound | Sequence | Molecular weight (g/mol) | m/z (calc.) | MS (ESI):(m/z) | Purity (%) |
|---|---|---|---|---|---|
| SS-03 | DOTA-(4-Cl)-Phe-Cyclo(D-Cys-Tyr-D-Trp-Lys-Thr-Cys)-D-Tyr-NH2 | 1531.15 | 1529.59 | 767.2 [M+2H]++ | 99.5 |
| 1531.8 [M+H]+ | |||||
| SS-04 | N4-(4-Cl)-Phe-Cyclo(D-Cys-Tyr-D-Trp-Lys-Thr-Cys)-D-Tyr-NH2 | 1331.01 | 1329.56 | 667.7 [M+2H]++ | 98 |
| 1332 [M+H]+ | |||||
| SS-05 | HYNIC-(4-Cl)-Phe-Cyclo(D-Cys-Tyr-D-Trp-Lys-Thr-Cys)-D-Tyr-NH2 | 1279.88 | 1278.45 | 642 [M+2H]++ | 97 |
| 1280.8 [M+H]+ |
SS-03 was labelled with 177Lu with labelling yields of > 97% at a maximum specific activity of 50 GBq/μmol. The conjugates SS-04 and SS-05 were labelled with 99mTc at room temperature (30 min) and elevated temperature (95 °C, 10 min), respectively. Tin(II)chloride was used as the reducing agent and citrate as an intermediate supporting Tc(V) ligand for the labelling of SS-04. For SS-05, 99mTc labelling was performed using edda (ethylenediamine, N,N′-diacetic acid) as coligand. The radiolabelling yields of both 99mTc-SS-04 and 99mTc-SS-05 were > 97% at a specific activity of approximately 100 GBq/μmol.
The distribution coefficients (log D) were determined using the shake-flask method (see Additional file 1). All radiopeptides showed high hydrophilicity (99mTc-SS-04, log D = − 2.49 ± 0.34, 177Lu-SS-03, log D = − 2.35 ± 0.22 and 99mTc-SS-05, log D = − 2.03 ± 0.27).
Binding affinity and immunofluorescence microscopy
Table 2 summarises the IC50 values of SS-03 and SS-04 for the five somatostatin receptor subtypes (sst1-sst5). Both SS-03 and SS-04 show high selectivity and affinity to sst2. The affinity of SS-03 for the sst2 subtype is threefold higher than SS-04. In comparison with natural somatostatin-28 (SS-28) and the potent sst2 antagonist DOTA-sst2-ANT (DOTA-pNO2Phe-cyclo[D-Cys-Tyr-D-Trp-Lys-Thr-Cys]-D-Tyr-NH2), both SS-03 and SS-04 retained high affinity to sst2.
Table 2.
Binding affinities (IC50, nM) of chelator-peptide conjugates
| Compound | IC50 (nM)* | ||||
|---|---|---|---|---|---|
| sst1 | sst2 | sst3 | sst4 | sst5 | |
| SS-03 | > 1000 | 1.7 ± 0.06 | > 1000 | 404 ± 92 | 564 ± 174 |
| SS-04 | > 1000 | 5.3 ± 0.17 | 720 ± 74 | 171 ± 35 | 228 ± 73 |
| DOTA-sst2-ANT [22] | > 1000 | 1.5 ± 0.4 | > 1000 | 287 ± 27 | > 1000 |
| SS-28 | 5.2 ± 0.3 | 2.7 ± 0.38 | 7.7 ± 0.9 | 5.6 ± 0.4 | 4.0 ± 0.3 |
*Values are IC50 in nM (mean ± SEM; n ≥ 3)
Immunofluorescence-based internalisation was performed using HEK-sst2 cells to demonstrate the antagonistic property of the conjugates. Figure 2 illustrates that 10 nM of the agonist [Tyr3]octreotide (TOC) triggers massive receptor internalisation, whereas SS-03 or SS-04 at the much higher concentration of 1000 nM does not stimulate receptor internalisation. However, at a concentration of 1 μM together with 10 nM of TOC, the conjugates were able to prevent the agonist-induced receptor internalisation.
Fig. 2.
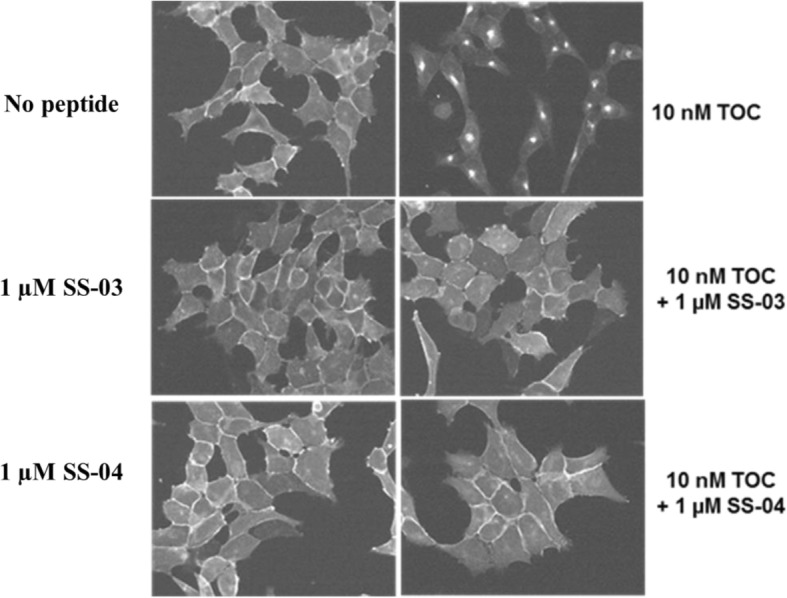
Immunofluorescence microscopy-based internalisation assay on HEK-sst2 cells. Immunofluorescence microscopy-based internalisation assay with HEK-sst2 cells showing the sst2 internalisation induced by [Tyr3]octreotide (TOC) is efficiently antagonised by SS-03 and SS-04. Control experiment showing membrane bound sst2-receptor in the absence of peptide. The agonist, TOC, triggered massive sst2-receptor internalisation at a concentration of 10 nM. The antagonists SS-03 and SS-04 failed to induce sst2 internalisation even at a concentration of 1 μM. Further, both SS-03 and SS-04 at a concentration of 1 μM efficiently blocked the agonist (TOC, 10 nM)-mediated sst2-receptor internalisation
Receptor-binding, internalisation, and dissociation kinetics
Figure 3 shows the cellular uptake profiles of the radioligands as measured with HEK-rsst2 cells. 177Lu-SS-03 and 99mTc-SS-04 showed high uptake and blocking studies demonstrated that the uptake was receptor-mediated. Within 2 h of incubation, the specifically bound fraction levelled off at 48%, demonstrating rapid binding of 177Lu-SS-03 and 99mTc-SS-04 to the receptors. The internalised fraction was about 8–10% and increased very slowly. Surprisingly, 99mTc-SS-05 showed very low cellular uptake of < 1% even after 4 h of incubation.
Fig. 3.
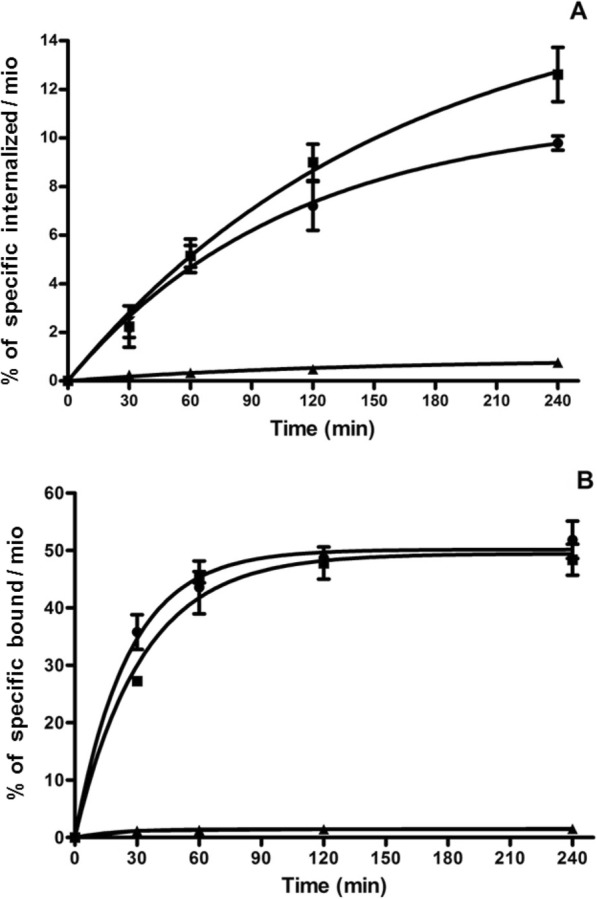
In vitro cellular uptake profile of the radioligands on HEK293-rsst2 cells. Cellular uptake profile of 177Lu-SS-03 (circle), 99mTc-SS-04 (square), and 99mTc-SS-05 (triangle) as measured with HEK293-rsst2 cells. a The amount of specifically internalised and b radiopeptides specifically bound to the membrane. Values and standard deviations are the result of two independent experiments with triplicates in each experiment
The dissociation kinetics of 177Lu-SS-03 and 99mTc-SS-04 were studied by a temperature shift experiment and showed a plateau at 2 h, indicating that a steady state had been achieved (Fig. 4). Both conjugates bind strongly to the receptor, displaying only 30 and 48% of dissociation, respectively, after 4 h at 37 °C.
Fig. 4.
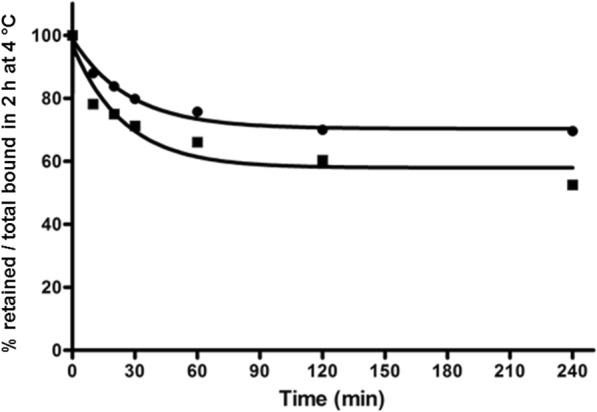
Dissociation kinetics of the cell surface bound radioligands on HEK293-rsst2 cells. Dissociation kinetics of membrane bound 177Lu-SS-03 (circle) and 99mTc-SS-04 (square) as measured with HEK293-rsst2 cells. The profile illustrates the varying degree of dissociation kinetics exhibited by 177Lu-SS-03 (koff = 0.037 min-1 and t1/2 = 18.66 min) and 99mTc-SS-04 (koff = 0.040 min-1 and t1/2 = 17.24 min). Values and standard deviations are the result of two independent experiments with triplicates in each experiment. The rate constants were determined by fitting to a pseudofirst order reaction using GraphPad Prism
Biodistribution studies and SPECT imaging
Table 3 summarises the biodistribution of 177Lu-SS-03 and 99mTc-SS-04 evaluated at 1, 4, and 24 h after injection in HEK-rsst2 xenografts. At 1 h, 99mTc-SS-04 showed high uptake in the liver (7.18 ± 0.40 %IA/g) and kidneys (49.82 ± 4.31 %IA/g) and higher blood pool activity (1.85 ± 0.41 %IA/g) than 177Lu-SS-03 (liver 3.00 ± 0.66 %IA/g, kidneys 15.15 ± 1.86 %IA/g, and blood 0.57 ± 0.04 %IA/g). 99mTc-SS-04 exhibits twofold higher tumour uptake compared to 177Lu-SS-03. Uptake in other sst2 receptor-positive organs such as the stomach and pancreas is lower for 99mTc-SS-04 (11.1 ± 2.9 and 15.6 ± 0.8 %IA/g, respectively) than 177Lu-SS-03 (48.7 ± 13.1 and 72.6 ± 13.8 %IA/g, respectively). At 4 h, the tumour uptake of 177Lu-SS-03 increased to 31.68 ± 4.00 %IA/g, whereas the tumour uptake of 99mTc-SS-04 remained constant. Tumour uptake of 177Lu-SS-03 and 99mTc-SS-04 was significantly reduced by pre-injection of 20 nmol of unlabelled peptide. At 24 h, both conjugates showed clearance from the blood pool (< 0.1 %IA/g) and significant wash-out from the liver (177Lu-SS-03: 0.47 ± 0.07 %IA/g and 99mTc-SS-04: 1.87 ± 0.33 %IA/g) and kidneys (177Lu-SS-03: 7.3 ± 1.6 %IA/g and 99mTc-SS-04: 6.3 ± 1.8 %IA/g). 99mTc-SS-04 showed higher tumour uptake at all time points including 24 h p.i. (32.5 ± 0.78 %IA/g) compared to 177Lu-SS-03 (26.32 ± 4.42 %IA/g). The tumour-to-normal tissue ratios of 177Lu-SS-03 and 99mTc-SS-04 at each time point are illustrated in Table 4. At 1 h, 177Lu-SS-03 shows higher tumour-to-normal tissue ratios compared to 99mTc-SS-04. At 4 h, both radiopeptides showed similar ratios except 177Lu-SS-03 had a twofold higher tumour-to-liver ratio. However, at 24 h, 99mTc-SS-04 exhibited excellent biodistribution with a 4.5-fold higher tumour-to-blood ratio, approximately threefold higher tumour-to-kidney ratio, and approximately twofold higher tumour-to-liver ratio compared to 177Lu-SS-03. All relevant tumour-to-normal organ ratios increased with time and were > 4 at 24 h.
Table 3.
Biodistribution results of 177Lu-SS-03 and 99mTc-SS-04 in nude mice bearing HEK293-rsst2 tumour xenografts. Data expressed as %IA/g (percentage of injected activity per gram) and presented as mean ± SD (n = 3–5)
| Organs | 1 h | 4 h | 4 h blocking* | 24 h |
|---|---|---|---|---|
| 177Lu-SS-03 | ||||
| Blood | 0.57 ± 0.04 | 0.21 ± 0.04 | 0.07 ± 0.02 | 0.07 ± 0.04 |
| Heart | 0.69 ± 0.12 | 0.38 ± 0.07 | 0.11 ± 0.01 | 0.11 ± 0.04 |
| Liver | 3.00 ± 0.66 | 1.75 ± 0.22 | 1.04 ± 0.09 | 0.47 ± 0.07 |
| Spleen | 1.51 ± 0.19 | 1.42 ± 0.77 | 0.28 ± 0.04 | 0.33 ± 0.06 |
| Lung | 13.24 ± 4.49 | 6.71 ± 1.37 | 0.59 ± 0.11 | 0.74 ± 0.22 |
| Kidney | 15.15 ± 1.86 | 17.04 ± 1.81 | 20.24 ± 4.30 | 7.35 ± 1.66 |
| Stomach | 48.74 ± 13.11 | 30.21 ± 4.25 | 0.37 ± 0.06 | 6.02 ± 1.40 |
| Intestine | 3.13 ± 1.16 | 2.28 ± 0.31 | 0.24 ± 0.03 | 0.27 ± 0.06 |
| Adrenal | 3.70 ± 0.63 | 2.90 ± 0.46 | 0.06 ± 0.03 | 1.16 ± 0.46 |
| Pancreas | 72.61 ± 13.77 | 54.49 ± 7.28 | 0.24 ± 0.07 | 3.03 ± 0.62 |
| Muscle | 0.22 ± 0.06 | 0.12 ± 0.02 | 0.06 ± 0.00 | 0.07 ± 0.02 |
| Bone | 4.11 ± 0.53 | 2.77 ± 1.06 | 0.12 ± 0.03 | 1.25 ± 0.16 |
| Tumour | 23.64 ± 1.28 | 31.68 ± 4.00 | 11.15 ± 1.93 | 26.32 ± 4.42 |
| 99mTc-SS-04 | ||||
| Blood | 1.85 ± 0.41 | 0.22 ± 0.03 | 0.22 ± 0.01 | 0.03 ± 0.01 |
| Heart | 1.42 ± 0.30 | 0.25 ± 0.06 | 0.23 ± 0.02 | 0.09 ± 0.01 |
| Liver | 7.18 ± 0.40 | 5.13 ± 0.51 | 4.07 ± 0.65 | 1.87 ± 0.33 |
| Spleen | 2.09 ± 0.34 | 0.85 ± 0.08 | 0.63 ± 0.02 | 0.41 ± 0.11 |
| Lung | 15.93 ± 2.69 | 2.03 ± 0.14 | 2.00 ± 0.37 | 0.54 ± 0.10 |
| Kidney | 49.82 ± 4.31 | 25.56 ± 1.01 | 18.48 ± 3.66 | 6.31 ± 1.82 |
| Stomach | 11.13 ± 2.98 | 1.99 ± 0.39 | 0.56 ± 0.03 | 0.62 ± 0.14 |
| Intestine | 2.12 ± 0.57 | 0.57 ± 0.06 | 0.42 ± 0.03 | 0.16 ± 0.02 |
| Adrenal | 3.17 ± 0.69 | 0.99 ± 0.20 | 0.37 ± 0.10 | 0.50 ± 0.13 |
| Pancreas | 15.58 ± 0.82 | 1.16 ± 0.86 | 0.33 ± 0.06 | 0.31 ± 0.11 |
| Muscle | 0.64 ± 0.13 | 0.12 ± 0.02 | 0.12 ± 0.02 | 0.05 ± 0.03 |
| Bone | 3.15 ± 0.72 | 0.92 ± 0.15 | 0.40 ± 0.01 | 0.44 ± 0.23 |
| Tumour | 47.14 ± 7.23 | 47.24 ± 7.96 | 14.17 ± 1.69 | 32.51 ± 0.78 |
*Pre-injection with 20 nmol of unlabelled peptide (SS-03 or SS-04)
Table 4.
Tumour-to-normal tissue ratios of 177Lu-SS-03 and 99mTc-SS-04 after 1, 4, and 24 h of administration in nude mice bearing HEK293-rsst2 tumour xenografts
| 177Lu-SS-03 | 99mTc-SS-04 | |||||
|---|---|---|---|---|---|---|
| 1 h | 4 h | 24 h | 1 h | 4 h | 24 h | |
| Tumour/blood | 41.3 | 150.5 | 360.9 | 25.5 | 218.1 | 981.3 |
| Tumour/liver | 7.9 | 18.1 | 55.8 | 6.6 | 9.2 | 17.4 |
| Tumour/kidney | 1.56 | 1.86 | 3.58 | 0.95 | 1.85 | 5.16 |
| Tumour/muscles | 109.5 | 259.3 | 372.1 | 73.6 | 382.1 | 616.7 |
| Tumour/bone | 5.7 | 11.5 | 21.1 | 14.9 | 51.3 | 74.1 |
The SPECT/CT images (Fig. 5) were acquired at 4 h after intravenous injection of 99mTc-SS-04. The highest uptake was visible in the tumour and the kidneys. To a lesser extent, uptake was also visible in the abdomen due to tracer accumulation in the sst2-expressing organs.
Fig. 5.
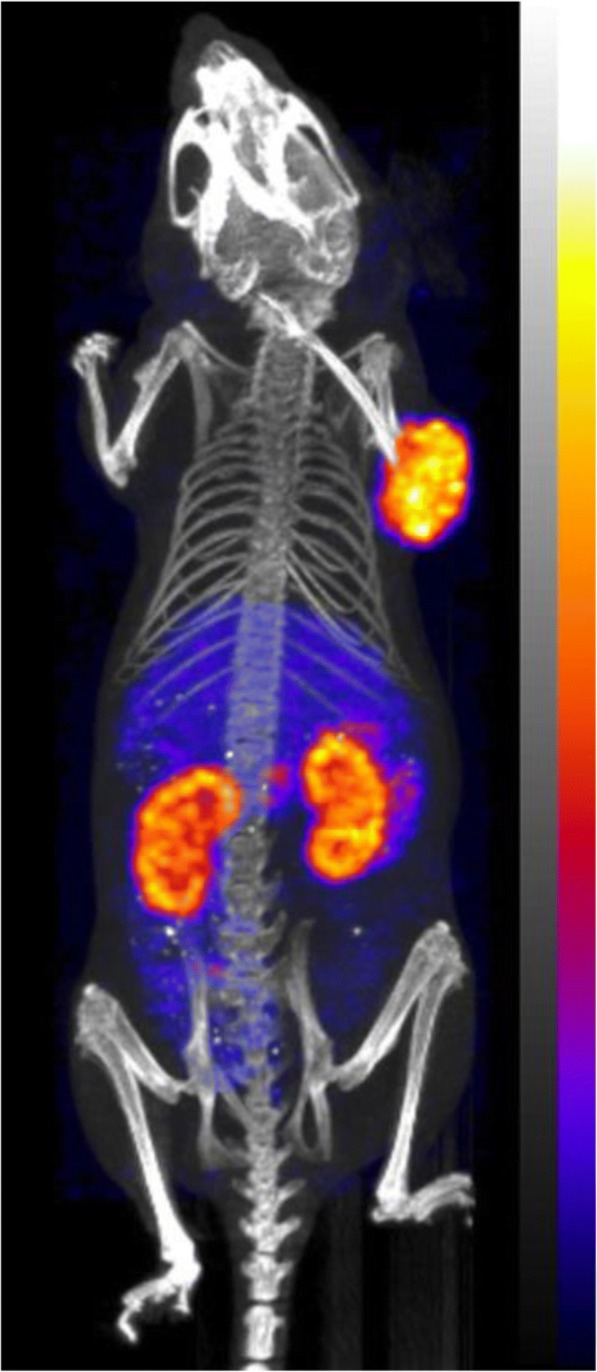
SPECT/CT imaging of 99mTc-SS-04 on HEK-rsst2-xenograft-bearing mouse. Maximum intensity projection (MIP) of the nanoSPECT/CT image acquired 4 h after injection of 15 MBq (150 pmol peptide) 99mTc-SS-04 in a HEK-rsst2-xenograft-bearing mouse. The colour bar corresponds to 0–90%IA/g
Discussion
Radiolabelled peptides targeting G-protein-coupled receptors have been an important focus in the radiopharmaceutical and nuclear oncology fields over the last 20 years [27–29]. The clinically employed somatostatin receptor-targeting peptides are agonists, which exhibit nanomolar binding affinities and fast receptor-mediated internalisation in vitro and in vivo via endocytosis. More recently, sst2 antagonists were studied in animal models [6–8, 18] and in humans [9–12]. Surprisingly, the antagonists showed high and long-lasting tumour uptake in most cases. However, antagonist superiority cannot be generalised, as shown by Wadas et al. [18] and Dude et al. [19].
To date, many peptides labelled with gamma-, positron-, and beta-emitters (111In, 64Cu, 68Ga, and 177Lu) have been described, but only one 99mTc-labelled antagonist has been reported to date [21].
We selected two of the more popular and successful chelators for 99mTc: HYNIC with the co-ligand edda (ethylenediamine-N,N′-diacetic acid) and the bifunctional tetraamine N4. The HYNIC core has been widely used for the labelling of octreotide-based somatostatin receptor-targeting peptides, and the [HYNIC, Tyr3]octreotide/edda kit (HYNIC-TOC) is registered in some European countries. The [O=Tc=O]+ core is formed with linear and macrocyclic tetraamines such as 1,4,8,11-tetraazaundecane and cyclam-14. This core exhibits high kinetic stability, can be labelled at room temperature, is highly hydrophilic, and with different functions in 6-position, such as carboxylic acid can be easily coupled to the N-termini of biomolecules [30]. We have conjugated these two chelators to a new antagonist, p-Cl-Phe-cyclo(D-Cys-Tyr-D-Trp-Lys-Thr-Cys)D-TyrNH2. As mentioned earlier, we chose the most easily accessible amino acids leading to antagonistic peptides. Our new antagonist is based on a modification of the first radiolabelled sst2 antagonists (DOTA-BASS) [5, 9, 31], where the p-NO2-Phe has been replaced by p-Cl-Phe. We also conjugated DOTA to this antagonist and labelled the conjugate with 177Lu for comparative in vitro and in vivo studies.
The antagonistic properties of SS-03 and SS-04 were investigated using immunofluorescence microscopy; the two compounds inhibit receptor internalisation triggered by the potent agonist [Tyr3]octreotide but do not trigger internalisation on their own. Somewhat surprisingly, but as previously reported [6, 14, 17], we observed low but significant cell internalisation of 177Lu-SS-03 and 99mTc-SS-04 (around 10% of the added radiopeptide activity per one million cells at 4 h). This uptake can be blocked by excess unlabelled peptide. The internalisation rate is significantly lower than that of somatostatin-based radiolabelled agonists [14, 22, 32]. The reason for the difference between the two assays is unclear.
A very important outcome of this work is that [99mTc-HYNIC/edda]-SS-01 shows practically no cell binding or internalisation in our HEK-rsst2 cell assay. This is contrary to HYNIC conjugated to somatostatin-based agonistic octapeptides such as [Tyr3]octreotide (TOC) and [Tyr3,Thr8]octreotide (TATE). These conjugates showed impressive preclinical [3] and clinical pharmacokinetics [33–35]. The influence of chelator and even radiometal on the pharmacologic properties was also reported for DOTA- and NODAGA-conjugated sst2 antagonists [6, 7].
In contrast, the N4-conjugated and 99mTc-labelled radiopeptide showed superior properties, with very high cell uptake in vitro and about the highest tumour uptake at 1 and 4 h of any somatostatin-based radiopeptide studied to date in this xenograft model [6, 7]. The higher tumour uptake of 99mTc-SS-04, compared with 177Lu-SS-03, may be attributed to the somewhat slower blood clearance of 99mTc-SS-04, even though at 4 h p.i, the blood values are at the same level for both radiopeptides. In addition, it shows very good tumour retention, no change between 1 and 4 h, and only about 30% release between 1 and 24 h p.i. The target-to-relevant organ ratios are > 5 at 24 h, reflected in excellent SPECT/CT images. For both radiotracers, the kidney uptake is somewhat high but the tumour-to-kidney ratios are still within a reasonable range when compared to potent radioagonists. We are currently studying somewhat different peptide motifs coupled to tetraamine chelators for 99mTc-labelling to improve the overall pharmacokinetics. The hypothesised improvement is based on the properties of 177Lu-SS-03, which exhibits pharmacokinetics somewhat inferior to the previously published 111In-DOTA and 177Lu-DOTA conjugated antagonists [31]. As previously mentioned, Radford et al. studied a somewhat different sst2 antagonist peptide labelled using the 99mTc tricarbonyl strategy and an NSN-type chelator [21]. The Re-complexed congener showed reasonably good receptor affinity indicating that this strategy, other than the HYNIC strategy, retains binding affinity [21]. The problem with the radioligand is its high abdominal uptake and low tumour-to-normal organ ratios, which is below 1 for some relevant tissues. As the authors pointed out, this is due to the high lipophilic character of the radioligand. The new 99mTc-based sst2 antagonist 99mTc-SS-04 is hydrophilic, as indicated by its log D value. Consequently, it has limited accumulation in the abdomen and rather high renal excretion. The biodistribution profile of 99mTc-SS-04, together with its very high tumour uptake, favours this tracer for clinical translation.
Conclusions
Sst2 antagonists were labelled with 99mTc using two common chelators. Surprisingly, the widely used 99mTc ligand HYNIC along with the co-ligand edda resulted in almost complete loss of sst2 binding affinity, whereas 99mTc-SS-04 demonstrated impressive tumour uptake and high tumour-to-normal organ ratios. Therefore, 99mTc-SS-04 appears to be an excellent candidate for SPECT imaging of sst2-positive tumours and is a promising option for further modification of the peptide motif. The pharmacokinetics of 99mTc-SS-04 demonstrate once again that the 99mTcO2(N4) core offers favourable pharmacokinetic features for small peptides and may potentially be the 99mTc-labelling strategy of choice.
Additional file
Materials and methods. (PDF 484 kb)
Acknowledgements
The authors wish to thank Leah Bassity for her editorial assistance.
Funding
This work was supported by the Swiss National Science Foundation.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
Abbreviations
- %IA/g
Injected activity per gram
- DIC
N,N′-Diisopropylcarbodiimide
- DMF
N,N-Dimethylformamide
- edda
Ethylenediamine,N,N′-diacetic acid
- Fmoc
Fluorenylmethyloxycarbonyl
- GPCR
G-protein coupled receptors
- GRP
Gastrin-releasing peptide
- HEK
Human embryonic kidney
- HOBt
Hydroxybenzotriazole
- HPLC
High-performance liquid chromatography
- HYNIC
6-hydrazinonicotinic acid
- IC50
Half maximal inhibitory concentration
- NETs
Neuroendocrine tumours
- NMP
N-methylpyrrolidinone
- p.i.
Post-injection
- PBS
Phosphate-buffered saline
- s.c.
Subcutaneously
- SPECT/CT
Single-photon emission computed tomography/computed tomography
- sst
Somatostatin receptor
- sst2
Somatostatin receptor subtype 2
- TATE
[Tyr3,Thr8]octreotide
- TOC
[Tyr3]octreotide
- CB-TE2A
4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane
- NODAGA
1,4,7-triazacyclononane,1-glutaric acid-4,7-acetic acid
- DOTA
1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid
- N4
6-carboxy-1,4,8,11-tetraazaundecane
Authors’ contributions
HM and JCR conceived and designed the study and wrote the manuscript. KA, MF, SU, and GN performed the experiments, analysed the data, and wrote the manuscript. MLT and BW performed the experiments and analysed the data. SNR and ECC analysed the data and wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval
All animal studies were performed in accordance with institutional and national guidelines regarding animal care and approved by the ethics committee of the University Hospital Basel (Approval #789).
This article does not contain any studies with human participants performed by any of the authors.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Keelara Abiraj, Email: abiraj.keelara@roche.com.
Samer Ursillo, Email: samer.ursillo@gmail.com.
Maria Luisa Tamma, Email: marialuisa.tamma@virgilio.it.
Svetlana N. Rylova, Email: rylova@hotmail.com
Beatrice Waser, Email: waserpatho@rubigen.ch.
Edwin C. Constable, Email: edwin.constable@unibas.ch
Melpomeni Fani, Email: melpomeni.fani@usb.ch.
Guillaume P. Nicolas, Email: guillaume.nicolas@usb.ch
Jean Claude Reubi, Email: reubi@pathology.unibe.ch.
Helmut R. Maecke, Email: helmut.maecke@uniklinik-freiburg.de
References
- 1.Deupi X. Molecular dynamics: a stitch in time. Nat Chem. 2014;6(1):7–8. doi: 10.1038/nchem.1832. [DOI] [PubMed] [Google Scholar]
- 2.Maecke HR, Reubi JC. Somatostatin receptors as targets for nuclear medicine imaging and radionuclide treatment. J Nucl Med. 2011;52(6):841–844. doi: 10.2967/jnumed.110.084236. [DOI] [PubMed] [Google Scholar]
- 3.Storch D, Behe M, Walter MA, Chen J, Powell P, Mikolajczak R, et al. Evaluation of [99mTc/EDDA/HYNIC0]octreotide derivatives compared with [111In-DOTA0,Tyr3, Thr8]octreotide and [111In-DTPA0]octreotide: does tumor or pancreas uptake correlate with the rate of internalization? J Nucl Med. 2005;46(9):1561–9. [PubMed] [Google Scholar]
- 4.Sprague JE, Peng Y, Sun X, Weisman GR, Wong EH, Achilefu S, et al. Preparation and biological evaluation of copper-64-labeled tyr3-octreotate using a cross-bridged macrocyclic chelator. Clin Cancer Res. 2004;10(24):8674–8682. doi: 10.1158/1078-0432.CCR-04-1084. [DOI] [PubMed] [Google Scholar]
- 5.Ginj M, Zhang H, Waser B, Cescato R, Wild D, Wang X, et al. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc Natl Acad Sci U S A. 2006;103(44):16436–16441. doi: 10.1073/pnas.0607761103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fani M, Del Pozzo L, Abiraj K, Mansi R, Tamma ML, Cescato R, et al. PET of somatostatin receptor-positive tumors using 64Cu- and 68Ga-somatostatin antagonists: the chelate makes the difference. J Nucl Med. 2011;52(7):1110–1118. doi: 10.2967/jnumed.111.087999. [DOI] [PubMed] [Google Scholar]
- 7.Fani M, Braun F, Waser B, Beetschen K, Cescato R, Erchegyi J, et al. Unexpected sensitivity of sst2 antagonists to N-terminal radiometal modifications. J Nucl Med. 2012;53(9):1481–1489. doi: 10.2967/jnumed.112.102764. [DOI] [PubMed] [Google Scholar]
- 8.Nicolas GP, Mansi R, McDougall L, Kaufmann J, Bouterfa H, Wild D, et al. Biodistribution, pharmacokinetics, and dosimetry of (177)Lu-, (90)Y-, and (111)In-labeled somatostatin receptor antagonist OPS201 in comparison to the agonist (177)Lu-DOTATATE: the mass effect. J Nucl Med. 2017;58(9):1435–1441. doi: 10.2967/jnumed.117.191684. [DOI] [PubMed] [Google Scholar]
- 9.Wild D, Fani M, Behe M, Brink I, Rivier JE, Reubi JC, et al. First clinical evidence that imaging with somatostatin receptor antagonists is feasible. J Nucl Med. 2011;52(9):1412–1417. doi: 10.2967/jnumed.111.088922. [DOI] [PubMed] [Google Scholar]
- 10.Wild D, Fani M, Fischer R, Del Pozzo L, Kaul F, Krebs S, et al. Comparison of somatostatin receptor agonist and antagonist for peptide receptor radionuclide therapy: a pilot study. J Nucl Med. 2014;55(8):1248–1252. doi: 10.2967/jnumed.114.138834. [DOI] [PubMed] [Google Scholar]
- 11.Nicolas GP, Beykan S, Bouterfa H, Kaufmann J, Bauman A, Lassmann M, et al. Safety, biodistribution, and radiation dosimetry of (68)Ga-OPS202 in patients with gastroenteropancreatic neuroendocrine tumors: a prospective phase I imaging study. J Nucl Med. 2018;59(6):909–914. doi: 10.2967/jnumed.117.199737. [DOI] [PubMed] [Google Scholar]
- 12.Nicolas GP, Schreiter N, Kaul F, Uiters J, Bouterfa H, Kaufmann J, et al. Sensitivity comparison of (68)Ga-OPS202 and (68)Ga-DOTATOC PET/CT in patients with gastroenteropancreatic neuroendocrine tumors: a prospective phase II imaging study. J Nucl Med. 2018;59(6):915–921. doi: 10.2967/jnumed.117.199760. [DOI] [PubMed] [Google Scholar]
- 13.Abiraj K, Mansi R, Tamma ML, Fani M, Forrer F, Nicolas G, et al. Bombesin antagonist-based radioligands for translational nuclear imaging of gastrin-releasing peptide receptor-positive tumors. J Nucl Med. 2011;52(12):1970–1978. doi: 10.2967/jnumed.111.094375. [DOI] [PubMed] [Google Scholar]
- 14.Cescato R, Maina T, Nock B, Nikolopoulou A, Charalambidis D, Piccand V, et al. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J Nucl Med. 2008;49(2):318–326. doi: 10.2967/jnumed.107.045054. [DOI] [PubMed] [Google Scholar]
- 15.Dalm SU, Bakker IL, de Blois E, Doeswijk GN, Konijnenberg MW, Orlandi F, et al. 68Ga/177Lu-NeoBOMB1, a novel radiolabeled GRPR antagonist for theranostic use in oncology. J Nucl Med. 2017;58(2):293–299. doi: 10.2967/jnumed.116.176636. [DOI] [PubMed] [Google Scholar]
- 16.Abd-Elgaliel WR, Gallazzi F, Garrison JC, Rold TL, Sieckman GL, Figueroa SD, et al. Design, synthesis, and biological evaluation of an antagonist-bombesin analogue as targeting vector. Bioconjug Chem. 2008;19(10):2040–2048. doi: 10.1021/bc800290c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wieser G, Mansi R, Grosu AL, Schultze-Seemann W, Dumont-Walter RA, Meyer PT, et al. Positron emission tomography (PET) imaging of prostate cancer with a gastrin releasing peptide receptor antagonist--from mice to men. Theranostics. 2014;4(4):412–419. doi: 10.7150/thno.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wadas TJ, Eiblmaier M, Zheleznyak A, Sherman CD, Ferdani R, Liang K, et al. Preparation and biological evaluation of 64Cu-CB-TE2A-sst2-ANT, a somatostatin antagonist for PET imaging of somatostatin receptor-positive tumors. J Nucl Med. 2008;49(11):1819–1827. doi: 10.2967/jnumed.108.054502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dude I, Zhang Z, Rousseau J, Hundal-Jabal N, Colpo N, Merkens H, et al. Evaluation of agonist and antagonist radioligands for somatostatin receptor imaging of breast cancer using positron emission tomography. EJNMMI Radiopharm Chem. 2017;2(1):4. doi: 10.1186/s41181-017-0023-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reubi JC, Waser B, Macke H, Rivier J. Highly increased 125I-JR11 antagonist binding in vitro reveals novel indications for sst2 targeting in human cancers. J Nucl Med. 2017;58(2):300–306. doi: 10.2967/jnumed.116.177733. [DOI] [PubMed] [Google Scholar]
- 21.Radford L, Gallazzi F, Watkinson L, Carmack T, Berendzen A, Lewis MR, et al. Synthesis and evaluation of a (99m)Tc tricarbonyl-labeled somatostatin receptor-targeting antagonist peptide for imaging of neuroendocrine tumors. Nucl Med Biol. 2017;47:4–9. doi: 10.1016/j.nucmedbio.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Cescato R, Erchegyi J, Waser B, Piccand V, Maecke HR, Rivier JE, et al. Design and in vitro characterization of highly sst2-selective somatostatin antagonists suitable for radiotargeting. J Med Chem. 2008;51(13):4030–4037. doi: 10.1021/jm701618q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bass RT, Buckwalter BL, Patel BP, Pausch MH, Price LA, Strnad J, et al. Identification and characterization of novel somatostatin antagonists. Mol Pharmacol. 1996;50(4):709–715. [PubMed] [Google Scholar]
- 24.Reubi JC, Schar JC, Waser B, Wenger S, Heppeler A, Schmitt JS, et al. Affinity profiles for human somatostatin receptor subtypes SST1-SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur J Nucl Med. 2000;27(3):273–282. doi: 10.1007/s002590050034. [DOI] [PubMed] [Google Scholar]
- 25.Cescato R, Schulz S, Waser B, Eltschinger V, Rivier JE, Wester HJ, et al. Internalization of sst2, sst3, and sst5 receptors: effects of somatostatin agonists and antagonists. J Nucl Med. 2006;47(3):502–511. [PubMed] [Google Scholar]
- 26.Gu YZ, Schonbrunn A. Coupling specificity between somatostatin receptor sst2A and G proteins: isolation of the receptor-G protein complex with a receptor antibody. Mol Endocrinol. 1997;11(5):527–537. doi: 10.1210/mend.11.5.9926. [DOI] [PubMed] [Google Scholar]
- 27.Fani M, Maecke HR. Radiopharmaceutical development of radiolabelled peptides. Eur J Nucl Med Mol Imaging. 2012;39(Suppl 1):S11–S30. doi: 10.1007/s00259-011-2001-z. [DOI] [PubMed] [Google Scholar]
- 28.Fani M, Maecke HR, Okarvi SM. Radiolabeled peptides: valuable tools for the detection and treatment of cancer. Theranostics. 2012;2(5):481–501. doi: 10.7150/thno.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schottelius M, Wester HJ. Molecular imaging targeting peptide receptors. Methods. 2009;48(2):161–177. doi: 10.1016/j.ymeth.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 30.Nock B, Nikolopoulou A, Chiotellis E, Loudos G, Maintas D, Reubi JC, et al. [99mTc]Demobesin 1, a novel potent bombesin analogue for GRP receptor-targeted tumour imaging. Eur J Nucl Med Mol Imaging. 2003;30(2):247–258. doi: 10.1007/s00259-002-1040-x. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Fani M, Schulz S, Rivier J, Reubi JC, Maecke HR. Comprehensive evaluation of a somatostatin-based radiolabelled antagonist for diagnostic imaging and radionuclide therapy. Eur J Nucl Med Mol Imaging. 2012;39(12):1876–1885. doi: 10.1007/s00259-012-2231-8. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen K, Parry JJ, Rogers BE, Anderson CJ. Evaluation of copper-64-labeled somatostatin agonists and antagonist in SSTr2-transfected cell lines that are positive and negative for p53: implications for cancer therapy. Nucl Med Biol. 2012;39(2):187–197. doi: 10.1016/j.nucmedbio.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Decristoforo C, Mather SJ, Cholewinski W, Donnemiller E, Riccabona G, Moncayo R. 99mTc-EDDA/HYNIC-TOC: a new 99mTc-labelled radiopharmaceutical for imaging somatostatin receptor-positive tumours; first clinical results and intra-patient comparison with 111In-labelled octreotide derivatives. Eur J Nucl Med. 2000;27(9):1318–1325. doi: 10.1007/s002590000289. [DOI] [PubMed] [Google Scholar]
- 34.Cwikla JB, Mikolajczak R, Pawlak D, Buscombe JR, Nasierowska-Guttmejer A, Bator A, et al. Initial direct comparison of 99mTc-TOC and 99mTc-TATE in identifying sites of disease in patients with proven GEP NETs. J Nucl Med. 2008;49(7):1060–1065. doi: 10.2967/jnumed.107.046961. [DOI] [PubMed] [Google Scholar]
- 35.Hubalewska-Dydejczyk A, Fross-Baron K, Mikolajczak R, Maecke HR, Huszno B, Pach D, et al. 99mTc-EDDA/HYNIC-octreotate scintigraphy, an efficient method for the detection and staging of carcinoid tumours: results of 3 years’ experience. Eur J Nucl Med Mol Imaging. 2006;33(10):1123–1133. doi: 10.1007/s00259-006-0113-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and methods. (PDF 484 kb)
Data Availability Statement
All data generated or analysed during this study are included in this published article and its supplementary information files.