

Abstract
Giant plasma membrane vesicles (GPMVs) are isolated directly from living cells and provide an alternative to vesicles constructed of synthetic or purified lipids as an experimental model system for use in a wide range of assays. GPMVs capture much of the compositional protein and lipid complexity of intact cell plasma membranes, are filled with cytoplasm, and are free from contamination with membranes from internal organelles. GPMVs often exhibit a miscibility transition below the growth temperature of their parent cells. GPMVs labeled with a fluorescent protein or lipid analog appear uniform on the micron-scale when imaged above the miscibility transition temperature, and separate into coexisting liquid domains with differing membrane compositions and physical properties below this temperature. The presence of this miscibility transition in isolated GPMVs suggests that a similar phase-like heterogeneity occurs in intact plasma membranes under growth conditions, albeit on smaller length scales. In this context, GPMVs provide a simple and controlled experimental system to explore how drugs and other environmental conditions alter the composition and stability of phase-like domains in intact cell membranes. This chapter describes methods to generate and isolate GPMVs from adherent mammalian cells and to interrogate their miscibility transition temperatures using fluorescence microscopy.
1. Introduction
It has long been known that general anesthetics, themselves hydrophobic molecules, partition into lipid membranes and influence their physical properties (Meyer 1899; Mullins 1954; Seeman 1972). Much of the past work in this area has focused on simple, single component membranes. These membranes undergo a main chain melting transition between a liquid phase and gel or solid phases, whose properties have been extensively studied by a variety of experimental methods. In all cases, incorporating hydrophobic general anesthetics into single component membranes lowers the main chain transition temperature (TM) and disorders lipid chains (makes their motions more isotropic) at fixed temperature (Shieh, Ueda, Lin and Eyring 1976; Kamaya, Ueda, Moore and Eyring 1979; Miller, Firestone, Alifimoff and Streicher 1989; Heimburg and Jackson 2007; Lorincz, Mihaly, Nemeth, Wacha and Bota 2015). These molecules also can impact the lateral pressure profiles, bending rigidity, and water permeability of simple membranes (Trudell, Payan, Chin and Cohen 1975; Mountcastle, Biltonen and Halsey 1978; Gruner and Shyamsunder 1991; Jorgensen, Ipsen, Mouritsen, Bennett and Zuckermann 1991; Jorgensen, Ipsen, Mouritsen and Zuckermann 1993). Over the years, it has been argued that these types of physical effects are unlikely to contribute to the molecular mechanisms of general anesthetics, as they tend to produce small perturbations at physiologically relevant concentrations (e.g. ΔTM<0.5°C) (Franks and Lieb 1982; Franks and Lieb 1986; Franks and Lieb 1990). In fact, many observations can be simply explained through the general lens of ‘freezing point depression’ where any hydrophobic impurity will have a qualitatively similar effect on membrane properties that simply depend on TM. However, many hydrophobic molecules are not general anesthetics, though they do impact the main chain transition temperature of simple membranes (Franks 1986; Franks and Lieb 1991; Krasowski, Jenkins, Flood, Kung, Hopfinger and Harrison 2001). Since membrane physical properties that are simply related to a changing TM can be produced by both anesthetics and non-anesthetics, it is unlikely that they contribute to the molecular mechanisms of general anesthetic function.
In addition to the main chain transition, multicomponent membranes that incorporate a sterol such as cholesterol can experience a miscibility transition between a single and two coexisting liquid phases (Veatch and Keller 2005) (Figure 1). One of these liquid phases, referred to as liquid-disordered or Ld, closely resembles the liquid crystalline phase present in a single component membrane above TM. The second liquid, called liquid-ordered or Lo, has chain order resembling the solid phase but with individual molecules retaining a high level of translational and rotational mobility (Ipsen, Karlstrom, Mouritsen, Wennerstrom and Zuckermann 1987). This miscibility transition occurs at a temperature called Tmix, and was first observed experimentally in giant vesicles and black lipid membranes in 2001 (Dietrich, Bagatolli, Volovyk, Thompson, Levi, Jacobson et al. 2001; Samsonov, Mihalyov and Cohen 2001), and is also observed in extracted and isolated biological membranes (Dietrich 2001; Baumgart, Hammond, Sengupta, Hess, Holowka, Baird et al. 2007) (Figure 1). While all hydrophobic impurities are expected to lower the main chain transition temperature to roughly the same extent, the magnitude and sign of an impurity’s effect on the miscibility transition temperature depends more strongly on the membrane composition and the detailed molecular structure of the impurity (Allender and Schick 2017; Cornell, McCarthy, Levental, Levental, Brooks and Keller 2017). We recently found that several anesthetic n-alcohols lower Tmix in vesicles isolated from RBL-2H3 membranes, but two non-anesthetic n-alcohols do not (Gray, Karslake, Machta and Veatch 2013; Machta, Gray, Nouri, McCarthy, Gray, Miller et al. 2016). This suggests that, at least in principle, membrane physical properties that depend on Tmix could contribute to the molecular mechanisms of n-alcohol general anesthesia.
Figure 1: Giant plasma membrane vesicles (GPMVs) can contain two coexisting liquid phases.
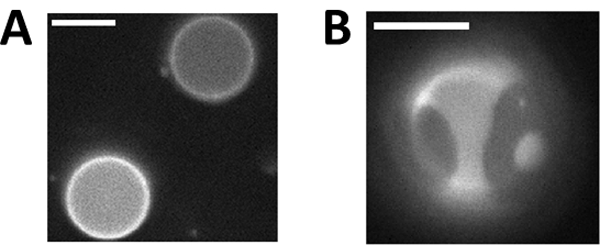
(A) GPMVs imaged above their miscibility transition temperature (Tmix) are in a single liquid phase, as indicated by the uniform distribution of a fluorescent lipid analog across the vesicle surface. (B) GPMVs separate into coexisting liquid-ordered (Lo) and liquid-disordered (Ld) phases at a temperature below Tmix. The fluorescent dye added to the GPMVs incorporates into the Ld phase, facilitating easy distinction between the phases. Scale bars are 10μm.
Since the early observations of coexisting liquid phases in synthetic and isolated biological membranes (Dietrich 2001; Veatch 2005; Baumgart 2007), there has been much speculation that this phase transition is in some way related to the submicron lipid raft domains hypothesized to exist in intact cell plasma membranes (Simons and Ikonen 1997; Lingwood and Simons 2010). In fact, the original proposal of the ‘raft’ hypothesis (Simons 1997) was in part based on even earlier observations of submicron heterogeneities (Shimshick and McConnell 1973; Vist and Davis 1990; Sankaram and Thompson 1991) and detergent resistant membrane fractions in purified membranes (Schroeder, London and Brown 1994), both of which are now thought to be related to the presence of a near-by miscibility transition (Heerklotz 2002; Honerkamp-Smith, Veatch and Keller 2009). We have proposed that lipid raft domains are critical composition fluctuations that occur within a single phase membrane that is biologically tuned to be close to a miscibility transition under physiological conditions (Veatch, Cicuta, Sengupta, Honerkamp-Smith, Holowka and Baird 2008; Machta, Papanikolaou, Sethna and Veatch 2011; Machta, Veatch and Sethna 2012; Zhao, Wu and Veatch 2013; Burns, Wisser, Wu, Levental and Veatch 2017). If so, changes in Tmix are expected to impact the properties of these domains as well as the functions that they promote. Past work has shown that the ligand gated ion channels thought to be responsible for general anesthesia are functionally modulated by other perturbations of lipid raft structure (Sooksawate and Simmonds 2001; Allen, Halverson-Tamboli and Rasenick 2007; Li, Serwanski, Miralles, Bahr and De Blas 2007). Ongoing work is more directly probing the connections between the immiscibility transition in membranes and lipid mediated heterogeneity in intact cells in a wide range of cellular and signaling systems (Zhou, Maxwell, Sezgin, Lu, Liang, Hancock et al. 2013; Levental and Veatch 2016; Stone, Shelby, Nunez, Wisser and Veatch 2017).
The goal of this chapter is to describe methods used to generate and isolate giant plasma membrane vesicles (GPMVs) from intact cells. These vesicles closely mimic the protein and lipid compositions from the intact plasma membranes from which they were derived, but lack polymerized cytoskeletal elements (Holowka and Baird 1983; Fridriksson, Shipkova, Sheets, Holowka, Baird and McLafferty 1999). These vesicles experience a miscibility transition below growth temperatures, and microscopic liquid-ordered and liquid-disordered domains are easily visualized on the vesicle surface when membranes are doped with fluorescent lipid analogs and imaged using conventional epi-fluorescence or confocal microscopy. The described GPMV isolation method produces vesicles with a distribution of compositions and miscibility transition temperatures. The distribution of transition temperatures can shift to lower or higher values in the presence of drugs or upon the application of hydrostatic pressure (Gray 2013; Raghunathan, Ahsan, Ray, Nyati and Veatch 2015; Machta 2016). Methods are described to quantify the average miscibility transition temperatures over a population of vesicles.
2. GPMV formation
Below, a procedure is described for isolating giant plasma membrane vesicles (GPMVs) that is built upon protocols described previously (Scott 1976; Holowka 1983; Baumgart 2007; Sezgin, Kaiser, Baumgart, Schwille, Simons and Levental 2012). GPMV formation and isolation can be accomplished in roughly two hours, and it is recommended that vesicles be used within several hours of preparation. This is only one of several possible methods available to form GPMVs. Some other methods described involve long incubations in nutrient poor buffers (Lingwood, Ries, Schwille and Simons 2008), or incubation of cells in buffers of different salts and osmotic pressures (Sarabipour, Chan, Zhou, Di Paolo and Hristova 2015). Overall, we find that the method outlined below, sometimes along with suggested modifications, provides the fastest and most robust method to produce GPMVs. Typically, each adherent cell present produces one to several 5–20 μm diameter vesicles that are filled with cytoplasm, lack internal membranes, and whose membranes are rich in protein and lipid components associated with the plasma membrane. The methods described below are most effective on adherent cells. GPMVs can also be made from cells in suspension, but yields are frequently lower, most vesicles remain attached to cells, and it is challenging to isolate GPMVs from cells after their formation. We use rat basophilic leukemia (RBL-2H3) cells for most studies, but have had success with the described techniques in other cell types, including the mammalian cell lines CHO, HELA, HEK, CH27 B cells, as well as cell lines from fish (ZF4), amphibian (XtC-2), and fly (SF9).
2.1. Equipment
37°C Incubator with shaker
Cell culture flasks and dishes
2.2. Buffers and Reagents
Vesiculation buffer: 150 mM NaCl, 2 mM CaCl2, and 20 mM HEPES in water, pH 7.4
- Fluorescent lipid Labeling Buffer: vesiculation buffer containing 1% (v/v) methanol, 2 μg/mL of a fluorescently labeled lipid probe such as DiI-C12 which is available from ThermoFisher Scientific (Prepare on day of GPMV formation)
- Dilute concentrated fluorophore stock in methanol to a final concentration of 200 μg/ml. For example, add 1 μL of a 10 mg/ml DiI-C12 solution to 49 μL methanol). If fluorophore is suspended in chloroform, evaporate solvent prior to resuspension in methanol.
- Add 10 μL of solution (a) to 1 mL of vesiculation buffer resulting in a final concentration of 2 μg/mL of fluorophore in vesiculation buffer. Alternate concentrations of probe can be used as needed, but it is recommend to remain below 10 μg/ml and to maintain at least 1% methanol to facilitate transfer of the lipid probe into the membrane.
- Sometimes it is beneficial to include a lipid transporter protein (such as BSA) in the labeling buffer. This is useful for more hydrophobic lipid probes (e.g. ones with long and saturated hydrocarbon chains). Labeling with DiI-C12 does not require BSA for efficient incorporation.
- Active Vesiculation Buffer: 1.9 mM DTT, 27.6 mM formaldehyde (HCHO), in vesiculation buffer (make on day of GPMV formation).
- Dissolve 15 mg of dithiothreitol (DTT) in 500 μL of vesiculation buffer. (This step is included to avoid measuring only 3mg of DTT, since this quantity is difficult to measure accurately on most laboratory scales.)
- Add 100 μL of (a) to 10 mL of vesiculation buffer.
- Add 20.3 μL of 37% formaldehyde to (b).
Flask or dish of adherent cells.
2.3. Procedure
The cells used for GPMV isolation should be prepared so that they stably adhere to the surface. GPMV composition and properties such as transition temperature will be somewhat dependent on growth conditions (serum concentration, cell density, etc.) (Gray, Diaz-Vazquez and Veatch 2015) therefore it is beneficial to maintain and/or characterize these variables throughout all trials of an experiment. Typically, we prepare cells such that they are 50–80% confluent and have adhered to the substrate at least overnight, and try to maintain details of cell maintenance (number of cells plated, time of incubation) throughout a given measurement.
Gently rinse cells twice with 1 mL of vesiculation buffer to remove dead cells and other cellular debris.
If labeling with a lipid fluorophore, add 1 mL of labeling buffer to the cell culture dish while gently shaking or rocking for 5–15 minutes, allowing the dye to incorporate into the cell membrane. This can be accomplished at room temperature on a benchtop rocker, at 37°C inside a shaking incubator (100 rev/min), or on a benchtop or incubator with occasional agitation. Labeled cells can be imaged prior to vesiculation to optimize fluorophore incorporation or to control for growth density. If needed, cells can be labeled more than once via this procedure.
To increase efficiency, the active buffer can be prepared while the cells are being labeled.
Rinse the cells several times with vesiculation buffer to remove excess fluorophores and once more with 1 mL of active buffer. Remove any remaining active buffer in the dish with a pipette, and add enough active buffer to cover cells completely. We find that 200–600uL of buffer is sufficient for a 35 mm dish and 1ml for a 25 cm2 flask. (See note 2).
Incubate cells in buffer for 60–120 minutes at growth temperature with gentle agitation. This can be accomplished in a 37°C shaking incubator set at 100 revolutions per min, in a 37°C shaking water bath on a low setting, or in a stationary incubator, water bath, or hot plate with occasional manual agitation. During this time, cells vesiculate and GPMVs separate from cells.
Progress can be monitored by imaging the dish or flask. Under phase contrast, GPMVs appear dark and can be seen attached to cells or flowing over cells upon gentle agitation. GPMVs can be hard to image at this stage with fluorescence excitation as cells often appear much brighter than vesicles, since the fluorescent probe is often trafficked into internal membranes during the incubation (Figure 2).
To harvest GPMVs, lightly rap the bottom of the dish to release additional cell-attached GPMVs. Decant the liquid containing active buffer and GPMVs into an appropriately sized tube.
If needed, GPMVs can be further separated from detached cells through a brief spin (we spin at 500 x g for 5 minutes). GPMVs remain in solution while detached cells will tend to pellet. To concentrate the sample of GPMVs (e.g. for lipidomics analysis), spin at 25 to 50k x g for 20 minutes. It may be beneficial to wash the pellet and then repeat the spinning for another 20 minutes.
Chemicals used for GPMV generation can be dialyzed from vesicles using cassettes or dialysis tubing and multiple buffer exchanges. We used 10,000 MWCO tubing and 5 × 200 mL exchanges. (See note 8.)
Figure 2: Phase contrast (A) and fluorescent (B,C) images of cell attached GPMVs.
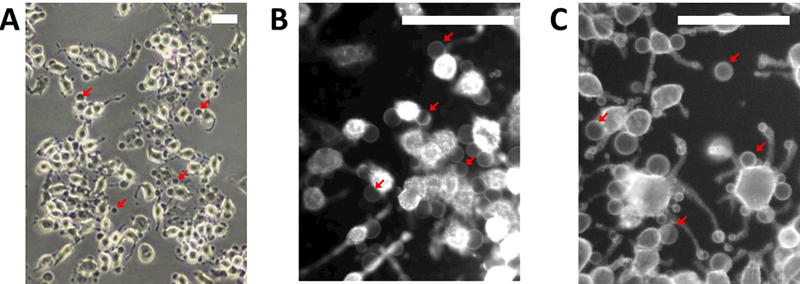
(A) GPMVs appear dark in phase contrast images and can be seen attached to cells or flowing above the adherent cell layer (not shown). (B) Cell attached GPMVs are difficult to visualize when cells membranes are fluorescently tagged prior to incubation with active vesiculation buffer due to trafficking of fluorophores to internal membranes. (C) GPMVs and their parent cell membranes have roughly the same fluorescent intensity when cells are labeled after incubation with active vesiculation buffer. Red arrows point to GPMVs in all panels. Scale bars are 100μm.
2.4. Notes
DiI-C12 is a fluorescent dye which labels the liquid-disordered phase of a lipid membrane and is excited maximally by 549 nm light. Other fluorescent dyes which selectively partition into either the Lo or the Ld phase may be used.
A minimal volume of active buffer should be used in order to increase the final density of prepared vesicles. A volume should be used that ensures no parts of the dish or flask surface will dry over the course of the incubation.
Sometimes it is beneficial to dilute the vesiculation buffer, either to match the osmotic strength of the growth media of the given cell type (as is the case in Xtc2 cells), or to apply an osmotic gradient that favors vesiculation (as we found useful for ZF4 cells).
We and others find that most vesicles will form within 1 hour of incubation with active buffer at 37°C, although in some cases longer incubations increase vesicle yields.
GPMVs can be fluorescently labeled post isolation with lipid dyes, lipid binding proteins, or antibodies. The advantage of pre-labeling with lipid fluorophores is that it enables washing of unbound probes, which is difficult to accomplish once vesicles are prepared. A disadvantage of pre-labeling is that some probes will be trafficked to internal membranes during the incubation, leading to lower final concentrations of the probe in the isolated vesicles. Examples of pre and post labeled cell attached GPMVs are shown in Figure 2 B,C.
Our standard protocol uses 2 mM DTT and 25 mM HCHO in the vesiculation buffer to optimize the density of the vesicles. Table 1 shows a titration of DTT and HCHO, highlighting the necessity of HCHO in the vesiculation process.
DTT can be replaced with other reducing agents such as β-Mercaptoethylamine (BME) or glutathione (GT) without significant modification to the described protocol. We recommend matching the reducing potential to achieve comparable yields (e.g. replace 2 mM DTT with 4 mM GT). Vesicle sizes are often smaller and yields lower when alternate reducing agents are used. It should be noted that DTT is membrane permeable, and can chemically modify lipids or remove lipid modifications on proteins (Levental, Grzybek and Simons 2011; Sezgin 2012).
GPMVs can also be prepared through a 5min incubation in DTT (or another reducing agent) and HCHO containing buffer, followed by a wash and longer incubation in vesiculation buffer containing only HCHO. This method achieves similar yields of GPMVs.
This protocol utilizes a minimal vesiculation buffer but other solutions can be used, as long as they are compatible with the cells being investigated. GPMVs have been successfully prepared with different buffering agents (e.g. phosphate buffer) or in solutions containing serum.
Table 1:
GPMV yields in from RBL-2H3 cells as a function of DTT and HCHO concentration in the active vesiculation buffer.
| 0 mM DTT | 1 mM DTT | 2 mM DTT | |
|---|---|---|---|
| 0 mM HCHO | none | none | none |
| 12.5 mM HCHO | low | medium | low |
| 25 mM HCHO | medium | high | high |
| 50 mM HCHO | medium | very high | very high |
3. Measuring GPMV Miscibility Transition Temperatures
Miscibility transition temperatures of single GPMVs or the average transition temperature of a population of GPMVs can be accomplished by imaging vesicles over a range of temperatures using a fluorescence microscope equipped with a temperature controlled stage. Vesicles should be examined on the same day that they are prepared to minimize the degradation of vesicle components.
3.1. Equipment
Temperature-controlled microscope stage. The stage should have a flat surface that is thermally isolated from the microscope base where the sample can be adhered using a thermal compound. It is helpful to measure temperature close to the sample, for example with a thermistor fastened to the stage surface. GPMVs prepared as described in the previous section often have transition temperatures between 0 and 30°C, therefore the stage would ideally be able to achieve temperatures between −10° and 40°C. This temperature range is well within accessible with a peltier heating/cooling element. Our stage is assembled primarily from commercial components and is displayed in Figure 3. Fully assembled stages and control units with similar functionality can be obtained commercially. (E.g. the Heating Cooling Thermal Stages available from Brook Industries.)
Fluorescence microscope with epifluorescence light source, filter cube matching the fluorophore used, an objective (20–60x), and camera appropriate for low light fluorescence imaging. (See notes 1 and 2)
Figure 3: Temperature can be controlled using a simple, home-built temperature stage.

Our temperature stage consists of a water-circulating heat sink and peltier thermoelectric device (Custom Thermoelectric, Bishopville, MD) and a PID-type controller unit (Oven Industries, Mechanicsburg, PA). Water is circulated through the heat sink using a garden fountain pump (available at hardware stores) submerged in a bucket. Temperature is measured with a thermistor probe calibrated to work with the controller mounted on the copper plate close to the sample. The sample, placed between two coverslips, is adhered to the copper plate near the thermistor and the assembly is placed, sample down, on a standard Olympus inverted microscope stage and is fastened with clips to prevent drift. We have also constructed stages where the plexiglass thermal insulator was replaced by rubber or cork simply adhered to a metal plate.
3.2. Procedure
GPMV samples are easily imaged when placed between two coverslips. This sample can be prepared by 1) lining the edges of a glass coverslip with a thin layer of Dow Corning high vacuum grease or similar compound; 2) Pipetting an appropriate volume of GPMV suspension (20–40uL) onto this coverslip; and 3) Covering with a second coverslip. The goal is to generate a thin layer of vesicles and fluid that is fully encapsulated within the chamber. The thin liquid layer greatly suppresses convective currents that lead to vesicle movement while imaging. When forming the chamber, avoid allowing liquid to leak out of the sealed area, as these channels frequently remain open and can lead to currents within the chamber that disrupt imaging. We use 22×22mm number 1.5 coverslips for imaging.
When preparing a sample containing freshly prepared GPMVs mixed with a chemical perturbation such as an n-alcohol anesthetic, we recommend first mixing vesicles with chemicals in a separate container (e.g. a microcentrifuge tube) prior to assembling the sample. This ensures mixing and allows the system to equilibrate prior to imaging. Care should be taken when mixing hydrophobic chemicals with GPMVs (see note 3). We recommend maintaining concentrations of chemicals used to make GPMVs if still present in GPMV solutions while imaging, as some of these (namely DTT) can independently impact GPMV miscibility transition temperatures (see note 4).
Secure the sealed coverslips to the temperature controller stage using a thermally conductive paste such as Arctic Silver polysynthetic thermal compound. The goal is to secure the coverslips to the stage both mechanically and thermally without compromising the sample integrity. Position the apparatus on the microscope in the proper orientation for viewing. Because GPMVs are filled with cytoplasm which has a higher density than the buffers used, they will settle to the bottom coverslip within roughly 5–10 minutes of becoming properly oriented.
Focus the objective lens on the vesicles settled at the bottom coverslip. Take care to not run the objective into the sample, as this could compromise its integrity. The most information rich images often are focused on the bottom vesicle surfaces, where even vesicles of different sizes are coplanar. Images can also be taken of vesicle midplanes, which show the circumference, or the top surface of vesicles. For transition temperature measurements, the focus should be set such that the user can distinguish vesicles in which the probe is uniformly distributed from those containing domains. Sometimes it is useful to acquire images at multiple focal planes for more robust identification. Some examples of phase-separated GPMVs are shown in Figure 4.
The acquisition time for the images should be set between 0.05 to 2 seconds, depending on the fluorescence intensity. Acquisition time or excitation light intensity may need to be adjusted as the temperature decreases. This is because the quantum yield of most fluorophores increases with decreasing temperature and because the probe will become concentrated within a subset of the vesicle surface when they separate into coexisting phases.
In order to measure the average transition temperature of a population of vesicles, start by acquiring images at a temperature where all vesicles are in the 1-phase state. Acquire images at a fixed temperature over several visual fields. It is recommended that at least 100 GPMVs be imaged per temperature. Lower the temperature in increments of 2–5°C and repeat the image acquisition procedure after allowing the sample to equilibrate for 2–5 min (or until the sample stops changing). If possible, continue lowering temperature until all or nearly all vesicles are in the 2-phase state. Use smaller increments when the temperature nears the transition temperature. Even though, the transition is fully reversible in most cases, we recommend imaging vesicles from high to low temperature for the reasons described in Note 5. We recommend blocking the excitation source when not acquiring images to avoid probe photo-bleaching and membrane photo-oxidation, which itself can impact transition temperatures in purified membranes (Veatch and Keller 2003; Veatch 2005; Zhao, Wu, Shao, Kong, Jain, Hunt et al. 2007), although in our experience this is less of a concern in isolated GPMVs.
-
Following image acquisition, average transition temperatures can be quantified by tabulating the fraction of vesicles containing two phases at each temperature investigated, then fitting these points to a sigmoidal curve to extrapolate the temperature where 50% of vesicles contain two coexisting liquid phases. We accomplish this analysis using software written in Matlab in which a user identifies if a given vesicle contains one or two phases, and this assignment is stored in memory as the user cycles through all vesicles imaged (see note 6). Once all vesicles in a given measurement have been assigned, the fraction of phase separated vesicles is computed at each temperature and fit to the following sigmoidal form:
The best fit value of the parameter Tmix is the extrapolated temperature where 50% of vesicles contain two phases and the parameter B is a measure of the width of the transition.
Vesicles with a single liquid phase will have equal fluorescence across the plane when focused on the top or bottom vesicle surface, or equal intensity across the circumference when focused at the midplane. Vesicles in a two liquid state will present as having light and dark parts of the vesicles, as the dye selectively incorporates into the Ld phase. Typically, vesicles containing two liquid phases will have roughly equal surface fractions of the vesicle surface occupied by either phase. In some cases, liquid domains will remain dispersed or bud from or into the vesicle, deforming the vesicle surface. These vesicles are also categorized as containing two liquid phases. Sometimes, uniform vesicles appear heterogeneous because they are attached to smaller vesicles or probe aggregates. These should be categorized as uniform or skipped in the identification process if phase state is ambiguous. In some cases, GPMVs can present with coexisting gel and liquid phases, or even coexisting gel with multiple liquid phases. This is most common when cells or vesicles are treated in a way that reduces their cholesterol content. Gel domains tend to be non-circular and rotate as ridged bodies. In some cases, gel domains can form rigid networks that span the vesicle surface. Examples of GPMVs with different domain morphologies are shown in Figure 5.
Errors in the measurement of the fraction of phase-separated vesicles at a given temperature can be determined by tabulating confidence intervals using binomial errors: and , where LB and UB are the lower and upper bounds of the confidence interval, respectively, n is the total number of GPMVs investigated at that temperature, k is average value for the number of GPMVs phase separated at that temperature, and βInv is the β-inverse cumulative distribution function. The error in determining the average transition temperature is evaluated by computing the confidence intervals on the Tmix parameter in the nonlinear least squares fit. Typically, the same sample will produce equivalent Tmix values within error bounds when the same GPMV preparation is examined on the same day, but absolute measurements of Tmix can vary between different GPMV preparations. For this reason, we often report the impact of a biochemical perturbation in terms of the shift of Tmix from a control (ΔTmix) instead of absolute values of Tmix.
In order to measure the transition temperature of a single vesicle, first conduct a rough determination of the transition temperature by taking the large (2–5°C) temperature steps while visualizing the vesicle surface. This could be done in a separate sample to avoid additional temperature cycling. Then, set the temperature to be ~2°C above the transition and repeat the above procedure at smaller temperature increments, e.g. 0.5, 0.2 or 0.1°C until a temperature is reach where two distinct phases are clearly visible. In many instances, flickering fluctuations are visible on the vesicle surface above the transition and undulating boundaries are visible below the transition (Veatch 2008) (REF). These fluctuations can be quantified to precisely identify the transition using analytical methods described elsewhere (Honerkamp-Smith, Cicuta, Collins, Veatch, den Nijs, Schick et al. 2008; Veatch 2008; Honerkamp-Smith 2009).
Similar procedures can be followed for measuring transition temperatures for vesicles held at different hydrostatic pressures. This requires a specialized microscope stage that can stably maintain and manipulate both temperature and pressure, which has been described previously (McCarthy, Ces, Law, Seddon and Brooks 2015; Purushothaman, Cicuta, Ces and Brooks 2015).
Figure 4: assigning phase state of individual vesicles within a population imaged at fixed temperature.
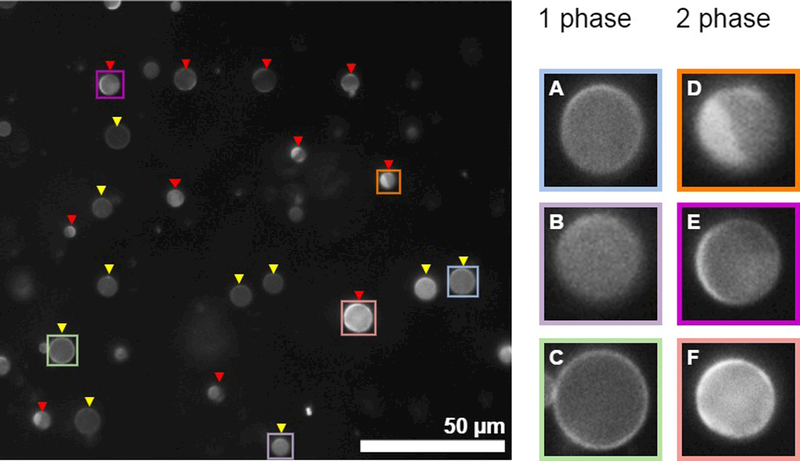
(Left) An example image showing a field of GPMVs in which most vesicles can be unambiguously assigned to contain either 1 or liquid 2 phases. Yellow triangles point towards 1 phase vesicles while red triangles point towards 2 phase vesicles. (right) isolated view of selected vesicles shown at the Left. GPMVs shown in A-C contain a single phase. A and B show GPMVs where the image is focused on a vesicle surface, and the brightness of the fluorophore is uniform across this surface. The GPMV in C is in focus along the midplane of the vesicle, and the brightness of fluorophore is uniform around the circumference. GPMVs shown in D and E contain coexisting liquid phases. The vesicle in D where the image is focused on the vesicle surface and a clear phase boundary is observed. The vesicle in E is focused close to the midplane, and the fluorophore intensity varies along the perimeter. F is difficult to assign. However, both the surface and the circumference vary in brightness so it was classified as containing two phases. In this example, some GPMVs in the field were not labeled because they are too small or too out of focus. In general, it is most important to remain consistent regarding assignments and the characteristics of vesicles that the investigator chooses not to assign. One reason to image at least 100 vesicles is to reduce the possible contributions of user bias in assigning vesicles.
Figure 5: Images of vesicles highlighting the range of possible shapes and domain morphologies.
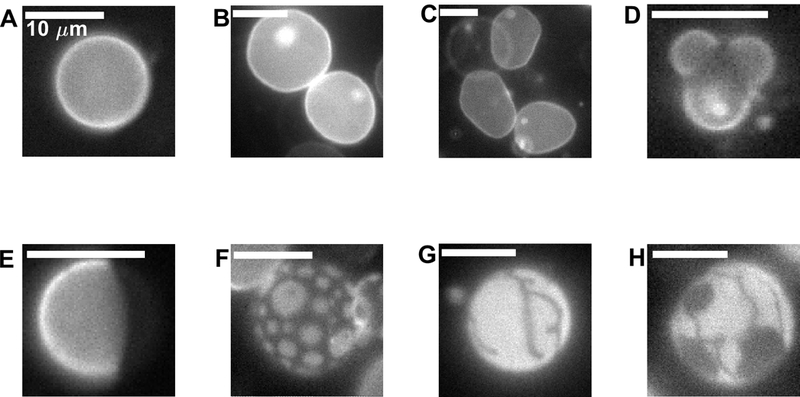
(A) Spherical vesicle with a single liquid phase. (B) Two single phase vesicles that each contain internal smaller vesicles (bright spots) . (C) Non-spherical vesicles imaged above Tmix. Vesicle edges undulate when vesicles are imaged in time (not shown). (D) Vesicle with bulging liquid-disordered domains. (E) Spherical vesicle exhibiting normal separation of 2 liquid phases where domains have fully coarsened through coalescence. (F) Liquid domains dispersed on the vesicle surface. In some cases domains do not coalesce, or do not coalesce on the time-scale of the measurement. (G) Vesicle that contains a gel phase domain. (H) Apparent coexistence of three phases within a single vesicle. Both gel and liquid-disordered phases exclude the fluorescent probe. The vesicle in D was imaged in the presence of detergent which increased membrane permeability, enabling the dramatic shape observed. Vesicles in G and H are GPMVs isolated from cells incubated with methyl beta cyclodextrin to reduce their cholesterol content.
3.3. Notes
Water or oil emersion objectives are not recommended when accurate temperature control is required, as they establish thermal contact between the sample and the objective or microscope base and can promote significant temperature gradients across the sample.
We use a 40X air objective on an IX81 inverted microscope (OLYMPUS) with a mercury lamp excitation source. A SCMOS camera (ANDOR) is used to collect images for analysis. DiI-C12 imaging is accomplished using a cy3 filter cube (Chroma).
When incubating GPMVs with hydrophobic compounds, it is possible to generate equilibrated or super-saturated solutions when using co-solvents such as DMSO or ethanol. To make equilibrated solutions, first add a small volume of a concentrated stock of the compound in solvent into the GPMV solution. This will often cause the compound to precipitate out of solution. Then, add enough co-solvent to re-dissolve the compound. It is recommended that the exact volumes of co-solvent required for resuspension of the compound be determined in a vesicle free solution, since small sample volumes prevent careful inspection for precipitates. To make super-saturated solutions, first dilute the stock solution in co-solvent so that when added to GPMVs, the desired final concentration of both compound and co-solvent is achieved. Then, add this diluted compound directly to GPMVs.
We find that DTT strongly impacts GPMV transition temperatures. GPMVs viewed in the absence of DTT often have transition temperatures 10–20°C below vesicles examined in the presence of 2 mM DTT.
GPMV membranes tend to reduce in size as temperature is lowered. This occurs because the average area per lipid in membranes is temperature dependent, and because the average area per lipid is further reduced upon the formation of the more condensed liquid-ordered phase. When temperature is lowered on freshly prepared vesicles for the first time, vesicle membranes become tense and then slowly relax by squeezing fluid from the vesicle interior in a way that retains their integrity and spherical shape. Upon raising temperature, the vesicle membrane expands but typically the volume of fluid enclosed within the vesicle remains constant, giving the membrane excess area and allowing the vesicle to take on non-spherical geometries. We typically view vesicles from high to low temperature to avoid complications introduced by this geometric flexibility. Even so, we find that the miscibility transition temperature is typically reversible, although vesicles imaged after temperature cycling tend to be non-spherical above Tmix and contain bulging domains below Tmix (Figure 5).
Due to the necessity of tabulating the fraction of vesicles containing two phases by hand, user bias is present at some degree, although in our experience this effect does not significantly impact transition temperature determination. To investigate the effects of user bias, we have had multiple researchers analyze the same set of data, and have randomized the images of GPMVs with the researcher blind to the temperature. We find that researchers that are more experienced in identifying vesicle states tend to generate sharper transitions because they are less likely to mis-identify individual vesicles, but the midpoint of the transition is not significantly shifted when large numbers of vesicles are analyzed.
4. Summary and Conclusion
Giant plasma membrane vesicles are simple to prepare and are easily visualized using wide field or confocal microscopy methods. Also, GPMV membranes can be concentrated and biochemically analyzed for their lipid and protein content (Holowka 1983; Fridriksson 1999; Levental, Lorent, Lin, Skinkle, Surma, Stockenbojer et al. 2016; Burns 2017), probed via spectroscopic methods to assay molecular scale structures (Ge, Gidwani, Brown, Holowka, Baird and Freed 2003), or deposited onto surfaces to make supported bilayers (Richards, Hsia, Singh, Haider, Kumpf, Kawate et al. 2016). Notably, most GPMV preparations contain two coexisting liquid phases when imaged at low temperature (Baumgart 2007), which is remarkable given their biological origin and complex composition. The protocols detailed in the previous sections will lead to consistent yields of vesicles that have reproducible miscibility transition temperatures. These vesicles are model membranes that retain much of the compositional complexity of the intact cell plasma membranes from which they are derived; although have important differences that should be considered when interpreting results. For example, they are no longer coupled to cytoskeleton, and do not generally contain cytosleleton associated plasma membrane proteins. They also are depleted of PI(4,5)P2 (Keller, Lorizate and Schwille 2009), and have some signatures of loss of asymmetry, as indicated by PS lipids being exposed on the extracellular face (Baumgart 2007). Even with these important differences, GPMVs are membrane models that much more closely reproduce intact cell plasma membranes than do purified giant unimalellar vesicles made of purified components. Understanding the similarities (Honerkamp-Smith 2009) and differences (Cornell 2017) between these two model membrane systems could shed light on the molecular mechanisms of membrane organization in cells.
Figure 6: Measurement of phase transition temperatures from GPMV images.
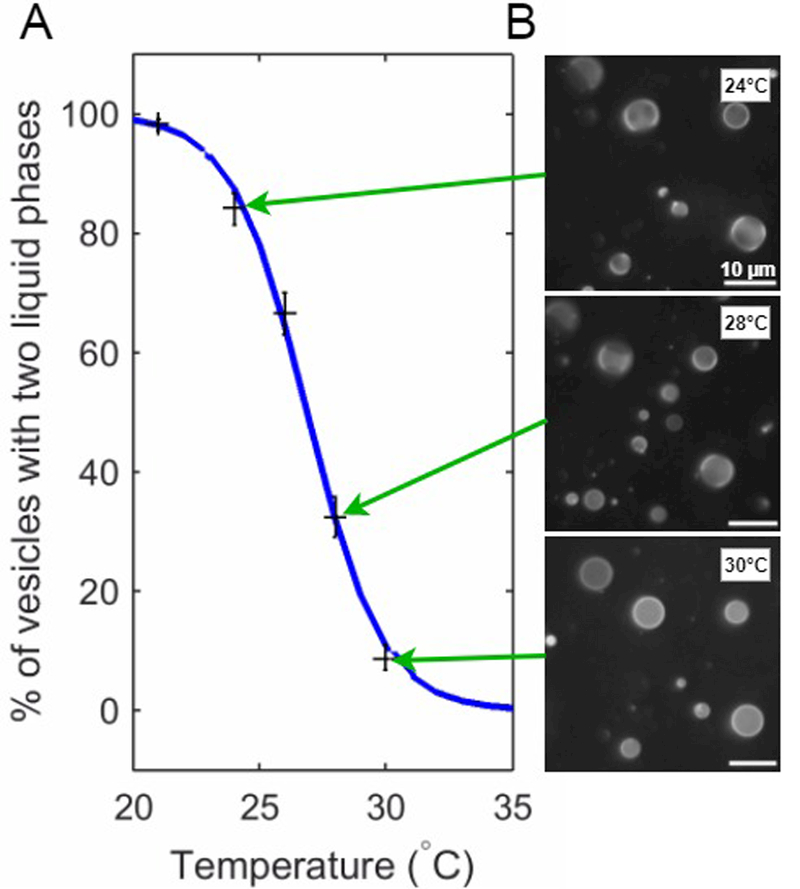
(A) Plot summarizing the fraction of GPMVs assigned to contain two liquid phases as a function of temperature (black crosses). The blue curve is a fit to these points by the sigmoid function stated in the main text. For this example, the best fit value for the average transition temperature Tmix is 26.9°C. (B) A small subset of the GPMV images that were assigned and used to obtain the data points shown in part A.
CITED REFERENCES
- Allen JA, Halverson-Tamboli RA and Rasenick MM (2007). “Lipid raft microdomains and neurotransmitter signalling.” Nature Reviews Neuroscience 8(2): 128–140. [DOI] [PubMed] [Google Scholar]
- Allender DW and Schick M (2017). “The Effect of Solutes on the Temperature of Miscibility Transitions in Multi-component Membranes.” Biophysical Journal: in perss. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart T, Hammond AT, Sengupta P, Hess ST, Holowka DA, Baird BA and Webb WW (2007). “Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles.” Proc Natl Acad Sci U S A 104(9): 3165–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M, Wisser K, Wu J, Levental I and Veatch SL (2017). “Miscibility Transition Temperature Scales with Growth Temperature in a Zebrafish Cell Line.” Biophys J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell CE, McCarthy NLC, Levental KR, Levental I, Brooks NJ and Keller SL (2017). “n-Alcohol Length Governs Shift in Lo-Ld Mixing Temperatures in Synthetic and Cell-Derived Membranes.” Biophys J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich C, Bagatolli LA, Volovyk ZN, Thompson NL, Levi M, Jacobson K and Gratton E (2001). “Lipid rafts reconstituted in model membranes.” Biophys J 80(3): 1417–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP and Lieb WR (1982). “Molecular mechanisms of general anaesthesia.” Nature 300(5892): 487–493. [DOI] [PubMed] [Google Scholar]
- Franks NP and Lieb WR (1986). “Partitioning of long-chain alcohols into lipid bilayers: implications for mechanisms of general anesthesia.” Proc Natl Acad Sci U S A 83(14): 5116–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP and Lieb WR (1990). “Mechanisms of general anesthesia.” Environ Health Perspect 87: 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP and Lieb WR (1991). “Stereospecific effects of inhalational general anesthetic optical isomers on nerve ion channels.” Science 254(5030): 427–430. [DOI] [PubMed] [Google Scholar]
- Fridriksson EK, Shipkova PA, Sheets ED, Holowka D, Baird B and McLafferty FW (1999). “Quantitative analysis of phospholipids in functionally important membrane domains from RBL-2H3 mast cells using tandem high-resolution mass spectrometry.” Biochemistry 38(25): 8056–8063. [DOI] [PubMed] [Google Scholar]
- Ge M, Gidwani A, Brown HA, Holowka D, Baird B and Freed JH (2003). “Ordered and disordered phases coexist in plasma membrane vesicles of RBL-2H3 mast cells. An ESR study.” Biophys J 85(2): 1278–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray E, Karslake J, Machta BB and Veatch SL (2013). “Liquid general anesthetics lower critical temperatures in plasma membrane vesicles.” Biophys J 105(12): 2751–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EM, Diaz-Vazquez G and Veatch SL (2015). “Growth Conditions and Cell Cycle Phase Modulate Phase Transition Temperatures in RBL-2H3 Derived Plasma Membrane Vesicles.” PLoS One 10(9): e0137741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruner SM and Shyamsunder E (1991). “Is the mechanism of general anesthesia related to lipid membrane spontaneous curvature?” Ann N Y Acad Sci 625: 685–697. [DOI] [PubMed] [Google Scholar]
- Heerklotz H (2002). “Triton promotes domain formation in lipid raft mixtures.” Biophys J 83(5): 2693–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimburg T and Jackson AD (2007). “The thermodynamics of general anesthesia.” Biophys J 92(9): 3159–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowka D and Baird B (1983). “Structural studies on the membrane-bound immunoglobulin E-receptor complex. 1. Characterization of large plasma membrane vesicles from rat basophilic leukemia cells and insertion of amphipathic fluorescent probes.” Biochemistry 22(14): 3466–3474. [DOI] [PubMed] [Google Scholar]
- Honerkamp-Smith AR, Cicuta P, Collins MD, Veatch SL, den Nijs M, Schick M and Keller SL (2008). “Line tensions, correlation lengths, and critical exponents in lipid membranes near critical points.” Biophys J 95(1): 236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honerkamp-Smith AR, Veatch SL and Keller SL (2009). “An introduction to critical points for biophysicists; observations of compositional heterogeneity in lipid membranes.” Biochim Biophys Acta 1788(1): 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipsen JH, Karlstrom G, Mouritsen OG, Wennerstrom H and Zuckermann MJ (1987). “Phase equilibria in the phosphatidylcholine-cholesterol system.” Biochim Biophys Acta 905(1): 162–172. [DOI] [PubMed] [Google Scholar]
- Jorgensen K, Ipsen JH, Mouritsen OG, Bennett D and Zuckermann MJ (1991). “Anesthetics affect membrane heterogeneity.” Ann N Y Acad Sci 625: 747–750. [DOI] [PubMed] [Google Scholar]
- Jorgensen K, Ipsen JH, Mouritsen OG and Zuckermann MJ (1993). “The effect of anaesthetics on the dynamic heterogeneity of lipid membranes.” Chem Phys Lipids 65(3): 205–216. [DOI] [PubMed] [Google Scholar]
- Kamaya H, Ueda I, Moore PS and Eyring H (1979). “Antagonism between high pressure and anesthetics in the thermal phase-transition of dipalmitoyl phosphatidylcholine bilayer.” Biochim Biophys Acta 550(1): 131–137. [DOI] [PubMed] [Google Scholar]
- Keller H, Lorizate M and Schwille P (2009). “PI(4,5)P2 degradation promotes the formation of cytoskeleton-free model membrane systems.” Chemphyschem 10(16): 2805–2812. [DOI] [PubMed] [Google Scholar]
- Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ and Harrison NL (2001). “General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of gamma-aminobutyric acid (GABA) current at the GABA(A) receptor but not with lipid solubility.” J Pharmacol Exp Ther 297(1): 338–351. [PubMed] [Google Scholar]
- Levental I, Grzybek M and Simons K (2011). “Raft domains of variable properties and compositions in plasma membrane vesicles.” Proc Natl Acad Sci U S A 108(28): 11411–11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental I and Veatch S (2016). “The Continuing Mystery of Lipid Rafts.” J Mol Biol 428(24 Pt A): 4749–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental KR, Lorent JH, Lin X, Skinkle AD, Surma MA, Stockenbojer EA, Gorfe AA and Levental I (2016). “Polyunsaturated Lipids Regulate Membrane Domain Stability by Tuning Membrane Order.” Biophys J 110(8): 1800–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Serwanski DR, Miralles CP, Bahr BA and De Blas AL (2007). “Two pools of Triton X-100-insoluble GABA(A) receptors are present in the brain, one associated to lipid rafts and another one to the post-synaptic GABAergic complex.” Journal of Neurochemistry 102(4): 1329–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D, Ries J, Schwille P and Simons K (2008). “Plasma membranes are poised for activation of raft phase coalescence at physiological temperature.” Proc Natl Acad Sci U S A 105(29): 10005–10010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D and Simons K (2010). “Lipid rafts as a membrane-organizing principle.” Science 327(5961): 46–50. [DOI] [PubMed] [Google Scholar]
- Lorincz A, Mihaly J, Nemeth C, Wacha A and Bota A (2015). “Effects of ursolic acid on the structural and morphological behaviours of dipalmitoyl lecithin vesicles.” Biochim Biophys Acta 1848(5): 1092–1098. [DOI] [PubMed] [Google Scholar]
- Machta BB, Gray E, Nouri M, McCarthy NLC, Gray EM, Miller AL, Brooks NJ and Veatch SL (2016). “Conditions that Stabilize Membrane Domains Also Antagonize n-Alcohol Anesthesia.” Biophys J 111(3): 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machta BB, Papanikolaou S, Sethna JP and Veatch SL (2011). “Minimal model of plasma membrane heterogeneity requires coupling cortical actin to criticality.” Biophys J 100(7): 1668–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machta BB, Veatch SL and Sethna JP (2012). “Critical Casimir forces in cellular membranes.” Phys Rev Lett 109(13): 138101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy NL, Ces O, Law RV, Seddon JM and Brooks NJ (2015). “Separation of liquid domains in model membranes induced with high hydrostatic pressure.” Chem Commun (Camb) 51(41): 8675–8678. [DOI] [PubMed] [Google Scholar]
- Meyer H (1899). “Zur Theorie der Alkoholnarkose.” Archiv für experimentelle Pathologie und Pharmakologie 42(2–4): 109–118. [Google Scholar]
- Miller KW, Firestone LL, Alifimoff JK and Streicher P (1989). “Nonanesthetic alcohols dissolve in synaptic membranes without perturbing their lipids.” Proc Natl Acad Sci U S A 86(3): 1084–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountcastle DB, Biltonen RL and Halsey MJ (1978). “Effect of Anesthetics and Pressure on Thermotropic Behavior of Multilamellar Dipalmitoylphosphatidylcholine Liposomes.” Proceedings of the National Academy of Sciences of the United States of America 75(10): 4906–4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins LJ (1954). “Some Physical Mechanisms in Narcosis.” Chemical Reviews 54(2): 289–323. [Google Scholar]
- Purushothaman S, Cicuta P, Ces O and Brooks NJ (2015). “Influence of High Pressure on the Bending Rigidity of Model Membranes.” J Phys Chem B 119(30): 9805–9810. [DOI] [PubMed] [Google Scholar]
- Raghunathan K, Ahsan A, Ray D, Nyati MK and Veatch SL (2015). “Membrane Transition Temperature Determines Cisplatin Response.” PLoS One 10(10): e0140925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards MJ, Hsia CY, Singh RR, Haider H, Kumpf J, Kawate T and Daniel S (2016). “Membrane Protein Mobility and Orientation Preserved in Supported Bilayers Created Directly from Cell Plasma Membrane Blebs.” Langmuir 32(12): 2963–2974. [DOI] [PubMed] [Google Scholar]
- Samsonov AV, Mihalyov I and Cohen FS (2001). “Characterization of cholesterol-sphingomyelin domains and their dynamics in bilayer membranes.” Biophys J 81(3): 1486–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaram MB and Thompson TE (1991). “Cholesterol-induced fluid-phase immiscibility in membranes.” Proc Natl Acad Sci U S A 88(19): 8686–8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarabipour S, Chan RB, Zhou B, Di Paolo G and Hristova K (2015). “Analytical characterization of plasma membrane-derived vesicles produced via osmotic and chemical vesiculation.” Biochim Biophys Acta 1848(7): 1591–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder R, London E and Brown D (1994). “Interactions between saturated acyl chains confer detergent resistance on lipids and glycosylphosphatidylinositol (GPI)-anchored proteins: GPI-anchored proteins in liposomes and cells show similar behavior.” Proc Natl Acad Sci U S A 91(25): 12130–12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RE (1976). “Plasma membrane vesiculation: a new technique for isolation of plasma membranes.” Science 194(4266): 743–745. [DOI] [PubMed] [Google Scholar]
- Seeman P (1972). “The membrane actions of anesthetics and tranquilizers.” Pharmacol Rev 24(4): 583–655. [PubMed] [Google Scholar]
- Sezgin E, Kaiser HJ, Baumgart T, Schwille P, Simons K and Levental I (2012). “Elucidating membrane structure and protein behavior using giant plasma membrane vesicles.” Nat Protoc 7(6): 1042–1051. [DOI] [PubMed] [Google Scholar]
- Shieh DD, Ueda I, Lin H and Eyring H (1976). “Nuclear magnetic resonance studies of the interaction of general anesthetics with 1,2-dihexadecyl-sn-glycero-3-phosphorylcholine bilayer.” Proc Natl Acad Sci U S A 73(11): 3999–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimshick EJ and McConnell HM (1973). “Lateral phase separation in phospholipid membranes.” Biochemistry 12(12): 2351–2360. [DOI] [PubMed] [Google Scholar]
- Simons K and Ikonen E (1997). “Functional rafts in cell membranes.” Nature 387(6633): 569–572. [DOI] [PubMed] [Google Scholar]
- Sooksawate T and Simmonds MA (2001). “Effects of membrane cholesterol on the sensitivity of the GABA(A) receptor to GABA in acutely dissociated rat hippocampal neurones.” Neuropharmacology 40(2): 178–184. [DOI] [PubMed] [Google Scholar]
- Stone MB, Shelby SA, Nunez MF, Wisser K and Veatch SL (2017). “Protein sorting by lipid phase-like domains supports emergent signaling function in B lymphocyte plasma membranes.” Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudell JR, Payan DG, Chin JH and Cohen EN (1975). “Antagonistic Effect of an Inhalation Anesthetic and High-Pressure on Phase-Diagram of Mixed Dipalmitoyl-Dimyristoylphosphatidylcholine Bilayers.” Proceedings of the National Academy of Sciences of the United States of America 72(1): 210–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch SL, Cicuta P, Sengupta P, Honerkamp-Smith A, Holowka D and Baird B (2008). “Critical fluctuations in plasma membrane vesicles.” ACS Chem Biol 3(5): 287–293. [DOI] [PubMed] [Google Scholar]
- Veatch SL and Keller SL (2003). “Separation of liquid phases in giant vesicles of ternary mixtures of phospholipids and cholesterol.” Biophys J 85(5): 3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch SL and Keller SL (2005). “Seeing spots: complex phase behavior in simple membranes.” Biochim Biophys Acta 1746(3): 172–185. [DOI] [PubMed] [Google Scholar]
- Vist MR and Davis JH (1990). “Phase equilibria of cholesterol/dipalmitoylphosphatidylcholine mixtures: 2H nuclear magnetic resonance and differential scanning calorimetry.” Biochemistry 29(2): 451–464. [DOI] [PubMed] [Google Scholar]
- Zhao J, Wu J, Shao H, Kong F, Jain N, Hunt G and Feigenson G (2007). “Phase studies of model biomembranes: macroscopic coexistence of Lalpha+Lbeta, with light-induced coexistence of Lalpha+Lo Phases.” Biochim Biophys Acta 1768(11): 2777–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Wu J and Veatch SL (2013). “Adhesion stabilizes robust lipid heterogeneity in supercritical membranes at physiological temperature.” Biophys J 104(4): 825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Maxwell KN, Sezgin E, Lu M, Liang H, Hancock JF, Dial EJ, Lichtenberger LM and Levental I (2013). “Bile acids modulate signaling by functional perturbation of plasma membrane domains.” J Biol Chem 288(50): 35660–35670. [DOI] [PMC free article] [PubMed] [Google Scholar]