

Abstract
Background: Omipalisib has been found to affect the viability of cancer cells. However, its effect on clonogenicity – a feature of cancer stem cells, is not clear. Cells isolated from neurocutaneous melanocytosis (NCM) patients’ lesions grow clonogenically. The aim of this study was to investigate the effect of omipalisib treatment on clonogenic growth of NCM cells in vitro. Materials and Methods: Clonogenic growth efficiency was evaluated by colony formation assays with or without specific growth factors. Activation of MEK and Akt was determined by immunoblots. Colony formation and cell viability were assessed upon pharmacological inhibition of MEK, Akt and mToR. Results: Clonogenicity appeared to depend on bFGF and IGF1signaling through ERK and Akt. Omipalisib treatment prevented colony formation and induced autophagic cell death. Conclusion: Signaling through Akt is important for survival of clonogenic cells in NCM, and omipalisib treatment as a monotherapy or in combination with MEK162 could be an effective therapeutic strategy to inhibit clonogenic growth.
Keywords: Clonogenicity, neurocutaneous melanocytosis, MEK162, omipalisib
Neurocutaneous melanocytosis (NCM) is a congenital neoplastic disease characterized by melanocytic proliferations in the brain and skin (1). Although patients with asymptomatic NCM may live without much problems for extended periods, the onset of symptoms in NCM generally indicates the beginning of an aggressive clinical course, with a poor prognosis and frequently death occurring within 2-3 years of diagnosis (2). There is little information on the cellular and molecular characteristics of the NCM cells, and no effective chemotherapy is available. Radiation and conventional broad- spectrum chemotherapy with Vincristine, Cyclophosphamide, Dacarbazine and Etoposide have been ineffective (3). Recently, post-natal treatment with MEK162 has been found to be effective in a murine model of NCM (4).
Omipalisib, also known as GSK2126458 is a new investigational Akt inihibitor drug by Glaxo SmithKline, which dually targets direct phosphorylation of Akt by PI3Kinase and reverse phosphorylation of Akt by mToR, thereby completely deactivating Akt. This drug has already passed human phase-1 trials for idiopathic pulmonary fibrosis and solid tumors making it a potential candidate to be tested in other diseases (5). Recent studies have suggested that Insulin like growth factor receptor (IGF-IR) signaling via Akt could be a general therapeutic target in NCM, due to the sensitivity of NCM cells to IGF-IR blockers (6) in general, irrespective of their oncogenic status (7,8). However, the specific role that Akt plays in the survival of NCM cells is not clearly understood. Recent evidence supports the emerging role of Akt in the maintenance of stemness in cancer stem cells (9-12). Insulin-like growth factors (IGFs) and signaling through Akt have been implicated in cancer stem cell self-renewal and tumorigenicity in several cancers, including solid tumors of breast (13), lung (14), colon (15) and hepatocellular carcinoma (16).
Herein, we report the role of Akt activation by IGF1 and bFGF in the maintenance of clonogenicity of cells isolated from NCM lesions. We recently reported an in vitro system for sustainable culture of clonogenic nevo-melanocytes from (NCM) lesions as Nevospheres (17). In this communication, we report the role of omipalisib (GSK2126458) in preventing clonogenic colony formation in vitro and induction of autophagic cell death in clonogenically growing cells from NCM lesions.
Materials and Methods
NCM patients. Following a standardized protocol, clinical data and melanocytic tissue lesions were prospectively collected from 3 NCM patients enrolled into the Gavin Bailey Tissue Repository for Neural Crest Disorders at the Children’s Hospital of Pittsburgh of UPMC. Informed written consent was obtained from parents in all cases, and the study was approved by the institutional review board of the University of Pittsburgh (IRB-PRO10030357). The characteristics of neoplastic cells used in this study were described in more detail in a previous report(1) including clinical and pathological features. The clinical features of patient C42N are included in Table I.
Table I. Clinical characteristics of patient C42N.

Reagents, drugs and antibodies. MEK inhibitor MEK162 and dual PI3K-mToR inhibitor omipalisib were purchased from Selleck Chemicals LLC, Houston, TX, USA. 3-methyl Adenine (3-MA) was purchased from Santa Cruz Biotechnology, Dallas, TX, USA. Geltrex® matrix was obtained from Thermo Fisher Scientific, Waltham, MA, USA and used according to manufacturer’s instructions. All Antibodies were from Cell Signaling Technology, Danvers, MA, USA. Anti β-actin was from Millipore-Sigma St. Louis, MO, USA.
Cell culture, cytotoxicity, viability and clonogenicity assays. Culture of clonogenic cells from NCM patients’ lesions and nevosphere colony formation has been described by us in detail previously (1). Clonogenicity was estimated by measuring colony formation on Geltrex® matrix (ThermoFisher Scientific, Waltham, MA, USA cat. no. A1314201) following the procedure described by Franken et al. (18). All drugs were dissolved in DMSO. A maximum dose of 0.1% DMSO was never exceeded. All experiments were done in the presence of a vehicle control. Cells were seeded onto a 96-well plate at a density of 5000 cells per well and treated with increasing concentrations of inhibitor drugs in triplicates. Corresponding dilutions of Dimethyl sulphoxide (DMSO) were used as untreated vehicle controls. For cytotoxicity assays, incubation was stopped after 48 h, unless mentioned otherwise and lactate dehydrogenase activity estimated from the culture media of the treated cells using Pierce™ LDH Cytotoxicity Assay Kit from ThermoFisher Scientific, Waltham, MA, USA following manufacturer’s instructions. For viability assays, after 96 h of incubation unless indicated otherwise, cell viability was quantitatively determined by MTT assay using Cell Titer 96 Non-Radioactive Cell Proliferation Assay kit (Promega Corporation, Madison WI, USA) according to manufacturer’s instructions.
Mutation analysis. The presence of mutation was detected by Sanger Sequencing method. PCR products were sequenced in sense and antisense directions using the BigDye Terminator v3.1 cycle sequencing kit on ABI 3130 (Applied Biosystems, Inc., Foster City, CA, USA) according to the manufacturer’s instructions. The sequences were analyzed using Mutation Surveyor software (SoftGenetics, LLC., State College, PA, USA).
Immunoblotting. Cells were lysed in ice-cold lysis buffer containing 50 mM Tris-HCl, pH7.4, 5mM EDTA, 0.1% SDS, 1% Triton X-100, 0.5% sodium deoxycholate with phosphatase and protease inhibitors (Roche Diagnostics GmBH, Mannheim, Germany) for protein extraction. Protein was estimated using Bradford reagent (Bio-Rad Laboratories Inc., Hercules, CA, USA). For immunoblots with nevospheres, prior to lysis, nevospheres were detached from Geltrex matrix by incubating in ice-cold PBS/EDTA with gentle shaking until the gel matrix is completely dissolved. Nevospheres were centrifuged at ~115 × g for 5 min, supernatant aspirated and cells were lysed in cold lysis buffer for protein extraction. Lysis buffer contained 50mM Tris-HCl, pH 7.4, 5mM EDTA, 0.1% SDS, 1% Triton X-100, 0.5% sodium deoxycholate with phosphatase and protease inhibitors (Roche). Protein was estimated using Bradford reagent (Bio-Rad). Equal amount of protein was loaded and electrophoretically separated on a 4-15% SDS-PAGE gel and transferred onto PVDF membrane (Bio-Rad). The membranes were blocked for 1 h at room temperature with 5% BSA (Fisher Scientific) in Tris buffered saline containing 0.1% Tween-20 (Bio-Rad). The blots were incubated with indicated primary antibodies overnight at 4˚C, washed and probed with appropriate HRP-conjugated secondary antibodies. The blots were developed by exposure to x-ray film (Bioexpress Corporation, Kaysville, UT, USA) after incubation with a luminol-based substrate (Millipore-Sigma, St. Louis, MO, USA).
Results
Colony-forming efficiency of nevomelanocytes is dependent on both IGF1 and bFGF. Clinical characteristics of patients C76N and PD1N have been reported in our previous publication (1). Nevomelanocytic cells from both patients carried oncogenic mutation NRAS Q61K. In this study, and in order to have a different genetic background as a reference, we included nevo-melanocytes isolated from patient C42N, who had a large/giant congenital nevus with CNS involvement and a BRAF V600E mutation. Cells grown with the full complement of growth factors (HMGS) formed 20-40 colonies per square millimeter as reported earlier (1), with C42N forming less colonies under the same conditions (Figure 1). Cells seeded without any of the required growth factors failed to grow any observable colonies. However, addition of IGF1 in the absence of other growth factors promoted formation of small colonies although in lower numbers compared to those growing in HMGS. The same observation was made with bFGF in all the cell types described. Figure 1 illustrates a representative observation from cells derived from our patient C76N. It was noted that media without growth factors (-GF) but containing PMA, hydrocortisone and bovine pituitary extract did not produce colonies, indicating that these accessory mitogens do not contribute to colony growth. Only addition of IGF1 and bFGF directly induced colony formation. Addition of fetal bovine serum rescued the colony formation to a similar extent as with IGF1 and bFGF added individually. However, addition of both growth factors together at the optimum concentration completely rescued colony formation to an extent comparable to HMGS. Colony formation efficiency was measured in terms of number of colonies per square millimeter and the diameter of colonies.
Figure 1. Role of bFGF and IGF1 in maintaining clonogenic growth. (A) Colonies from C76N cells on Geltrex® matrix supplemented with indicated growth factors. HMGS, Human melanocytic growth supplement; GF, Media devoid of growth factors but supplemented with hydrocortisone (0.18 μg/ml), phorbol 12-myristate 13-acetate (PMA) 10 ng/ml, bovine pituitary extract (0.2% v/v), bovine transferrin (5 μg/ml) and heparin (3 μg/ml). GF wells were supplemented with growth factors (IGF1 and bFGF and serum as indicated. Scale bar represents 200 μm. (B) Number of colonies formed from each cell line after seeding ~50,000 cells per well in a 24-well plate and treated with indicated growth factors. (C) Average size of colonies (y-axis represents diameter in μm) formed from each cell line under indicated treatments after 72 h of growth on Geltrex™ matrix. IGF1 and bFGF together can reconstitute the colony formation observed in HMGS. One-way ANOVA analysis shows significant difference between control and treatments at p<0.05 (denoted by asterisk). No significance was observed at p<0.05 between HMGS and bFGF+IGF1 treatments in Figure 1B (denoted by double asterisk).

bFGF activates both ERK and Akt. In all three cell lines studied, bFGF treatment was able to activate both ERK and Akt, albeit to different degrees. bFGF treatment primarily induced robust phosphorylation of ERK on residue tyr204 in a dose-dependent manner, while at higher doses also stimulated low levels of phosphorylation of Akt on residue Ser473 (Figure 2A, top panel). This was observed in C76N and C42N cells but not the case in PD1N. The overall total levels of ERK and Akt proteins remained unaffected. The residual ERK activity noted in the untreated control was significantly higher in C42N than in C76N and PD1N. This could be the result of constitutively active BRAF in C42N. On the other hand, IGF1 could only activate Akt and not ERK in all cell lines studied. There was no observable ERK activation and the residual level of ERK activity in C42N stayed fairly constant over the range of doses studied. It was interesting to note that the residual ERK phosphorylation observed in the untreated controls of C76N and PD1N decreased with IGF1 treatment, and was undetectable concomitant to increase in pAkt. This suggests the redundancy in NRAS signaling and the ability of NRAS being a membrane bound GTPase to switch cellular signaling pathways depending on the nature of growth factors it is exposed to.
Figure 2. Effect of bFGF and IGF1 on downstream signaling mediators in NCM cells. (A) Cells from C76N, PD1N and C42N were treated or not with increasing concentrations of bFGF (upper panel) and IGF1 (lower panel). Cell lysates were prepared in the presence of protease and phosphatase inhibitors, and equivalent protein was loaded onto each well. Western blots were probed with phosphor-specific antibodies targeted to phospho-ERK1/2, phospho-Akt, total ERK1/2, total Akt and cyclin D. β-actin was used as loading control. (B) Densitometric analysis of relative band intensities using Image J and plotted as percent densitometric units assuming control (HMGS) as one hundred percent.

Suppression of Akt phosphorylation, not MEK leads to loss of clonogenicity. We wanted to investigate, which one of these signaling pathways is indispensable to maintaining clonogenicity. To address this question, we used MEK162, a pharmacological inhibitor of MEK, an upstream effector of (ERK 1/2) to block the ERK pathway. To block Akt activation, we used omipalisib, which inhibits PI3-kinase and mToR, two upstream effectors of Akt. Cells were grown under adherent conditions and pre-treated with the inhibitors at a lower than EC50 dose before being trypsinized and seeded onto Geltrex® for colony formation (Figure 3A). All the cell types failed to form any colonies when Akt was suppressed by omipalisib. The surviving fraction was significantly lower than control without affecting cell viability significantly. In contrast, MEK inhibition did not affect colony formation significantly in any of the cell types studied, including BRAF mutated C42N. The surviving fraction remained almost unaltered upon MEK162 treatment and there was no loss of cell viability (Figure 3B and C). Next, we asked if Akt suppression would lead to activation of cell death pathways, such as apoptosis or autophagy.
Figure 3. Omipalisib has a superior impact on the clonogenicity of NCM cells. (A) Scheme of experiment. (B) Representative pictures of colony formation 5 days post seeding. Scale bar represents 100 μm. (C) Graphs showing relative impact of each drug compared to vehicle on colony formation. Total number of colonies, surviving fraction as well as viability was estimated. *p<0.001 (one-way ANOVA). Plating efficiency and surviving fraction was calculated according to Franken et al. (23).

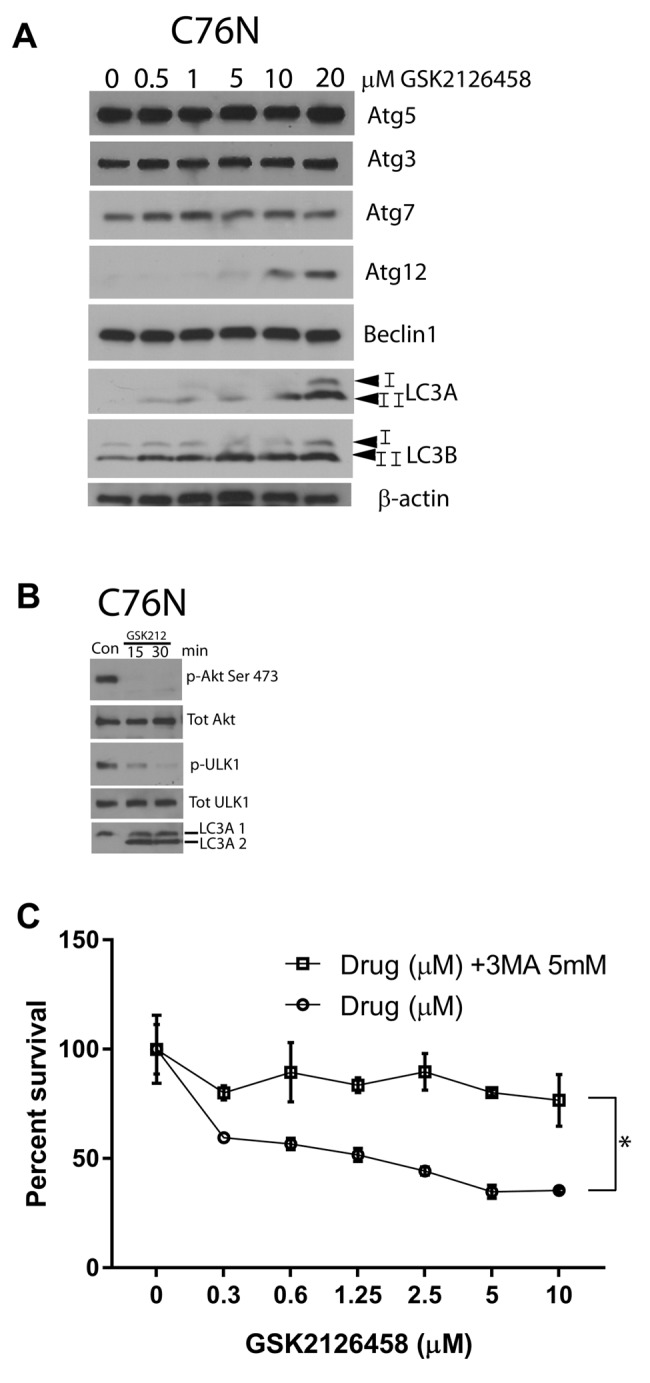
Akt inhibition by omipalisib induces cytotoxicity and autophagic cell death. Figure 4A shows the dose-dependent increase in cytotoxicity in omipalisib treated cells compared to MEK162 which did not cause detectable cytotoxicity. This was similar in all cell lines studied, but the extent of cell death was noted to be higher in C76N compared to the others. Cell death was observed at 0.5 μM dose of GSK212645, and it increased further in a dose-dependent manner (Figure 4B). We then assessed whether the mechanism of cell death inflicted by omipalisib is through apoptosis or autophagy. We ran a panel of apoptotic markers and did not detect differences between treated and untreated cells (data not shown). Therefore, we then studied autophagy induction by this drug. A representative immunoblot analysis of autophagic proteins from one of the cell lines is shown in Figure 5A. The other two cell lines showed similar results (data not shown). Among a panel of autophagic marker proteins, increase was detected in Atg12, LC3A and LC3B. Notably, a marked increase was detected in the fast migrating band of LC3 (LC3A-II and LC3B-II). Drug treatment possibly caused an upregulation of the process of autophagy, as indicated by the increase in LC3 (lower band) that is known to participate in the formation of autophagic vesicles. LC3A-2 appeared within 15 min of treatment with GSK2126458, which coincided with the loss of inhibitory phosphorylation on ULK1, hinting to a possible mechanism of autophagy induction (Figure 5B). ULK1 is known to be the initiator of autophagy and is kept inactive by mToR phosphorylation at Ser757 therefore; it is likely that inhibition of mToR releases ULK1 from inactivity, triggering autophagic vesicle formation leading to cell death. To confirm that drug induced cell death is indeed mediated through autophagy, we pre-treated cells with 3-methyl adenine (3MA) at a dose known to inhibit autophagy in most cell types. Five mM 3MA successfully reduced omipalisib mediated cell death, confirming that autophagy was responsible for the drug-induced cell death (Figure 5C). Based on this paradigm, we hypothesized that combined inhibition of pathways induced by bFGF and IGF1 would be more efficacious in reducing clonogenicity in these cells.
Figure 4. MEK162 has a lower cytotoxicity than omipalisib. (A) Graphs (mean±S.E.M.) showing percentage of cytotoxicity in C76N (left), PD1N (middle) and C42N (right) cells calculated as a function of LDH activity according to manufacturer’s instructions. (B) Pictures showing C76N cells treated with vehicle or omipalisib at the indicated concentrations. Cells rounded up and detached from the surface. Less cells are seen attached at the higher dose. Scale bar represents 100 μm.

Figure 5. Omipalisib treatment induces autophagy. (A) Cells from C76N treated with indicated doses of omipalisib were lysed and electrophoresced through a 4-15% gradient gel and immunoblotted using the indicated antibodies. Dose-dependent increase in LC3 A and B (lower band) is noted. (B) Cells were treated with 1 μM omipalisib and harvested at the indicated timepoints and immunoblotted against the indicated antibodies. Phospho-Akt was undetectable after 15 min, phospho-ULK1 gradually disappeared by 30 min. LC3A lower band could be detected after 15 min of treatment. (C) Cells pre-treated or not with 5 mM 3MA were subjected to increasing doses of omipalisib and cell viability measured after 48 h by MTT assay. Cells without omipalisib treatment were used as control and considered as 100% viable. 3MA pre-treated cells remained significantly more viable compared to the untreated ones in response to omipalisib treatment. Significant at *p<0.05 (one-way ANOVA).
Combined inhibition of MEK and Akt leads to lower cell viability. Cells were treated with increased doses of MEK162 with or without a fixed dose (<EC50) of omipalisib (1μM). Cell viability went down significantly when combined with omipalisib compared to MEK162 alone for all the doses studied in all the cell lines (Figure 6A-C, left panel). A reverse experimental approach with a fixed dose (<EC50) of MEK162 when combined with increased doses of omipalisib reduced viability significantly in all the doses studied in all the cell lines (Figure 6A-C, right panel). This indicated that both signaling pathways are synergistic and need to be suppressed in order to suppress cell viability and clonogenicity. This also suggests the redundancy in signaling pathways affecting clonogenicity in nevomelanocytic cells and that bFGF and IGF1 pathways are equally important for maintenance of clonogenicity in NRAS and BRAF mutated cells alike.
Figure 6. Omipalisib augments MEK162 effect in combined administration and vice-versa. (A, B, C, left panel) Treatment with increased doses of MEK162 alone or in combination with 1μM omipalisib of C76N, PD1N and C42N cells. (A, B, C, right panel) Increasing doses of omipalisib alone was compared to combination with 1μM MEK162 in C76N, PD1N and C42N cells. Viability was measured by MTT assay after 72 h of treatment. Untreated controls were considered 100% viable. Curves were fitted according to the non-linear regression analysis using Graphpad PRISM.

Discussion
Akt inhibition by omipalisib (GSK2126458) has been recently identified as a therapeutic strategy in Rhabdomyosarcoma (19) and KRAS mutant non-small cell lung cancer (20). We have previously shown that Akt inhibition by omipalisib affects viability of NCM cells in a three-dimensional cell culture system. However, detailed mechanism of action of omipalisib on NCM cells is not known. In this communication, we show that omipalisib prevents colony formation and induces autophagic cell death in NRAS mutant NCM cells that depend on IGF1 and bFGF for clonogenic growth.
Neurocutaneous melanocytosis is a disease without an effective chemotherapy treatment, and once it become symptomatic, it frequently follows an aggressive and lethal progress. The rarity of the disease and scarcity of human samples make it difficult to carry on research on it and improve our understanding of its pathophysiology. Recently we have shown its clonogenic cellular properties, which appear to contribute to the growth of its lesions (17). Under in vitro conditions, these cells exhibit a slow growth and can form anchorage-independent colonies. However, the factors contributing to the clonogenic growth of these cells are largely unknown. Here we have shown that bFGF and IGF1, two growth factors abundant in the microenvironment of these lesions are necessary for the clonogenic growth of NCM cells in vitro. In our hands, neither of these growth factors alone could successfully rescue the colony-formation efficiency of NCM cells; however, the presence of both growth factors eliminated the necessity of any other component in the culture medium to restore normal clonogenic efficiency. This shows the absolute dependence of these cells on both bFGF and IGF1 in order to maintain their clonogenic growth ability.
Activated NRAS is known to signal through both ERK and Akt intermediates affecting downstream targets involved in cell survival and proliferation. Interestingly, IGF1 did not affect the ERK pathway at all and only induced Akt in these cells. Surprisingly, the residual ERK activity found in untreated cells disappeared with increasing IGF1 treatment only in NRAS mutated cells suggesting a switch in NRAS-mediated signaling. This is not the case with BRAF mutated cells which maintained their residual activation of ERK. This can be due to the fact that BRAF, being a kinase instead of a GTPase, does not associate with any growth factor receptor and therefore its activity is independent of the nature of growth factor present in the environment. We observed that colony formation was not inhibited by pre-treatment with MEK162, while Akt inhibition by omipalisib completely stopped colony formation in the presence of both growth factors (Figure 3). This suggests that signaling through Akt is necessary for both growth factors to be able to induce clonogenic growth, while ERK pathway induction in itself is not sufficient. This supports our idea that Akt could be a major nodal point of survival signaling in NCM (7). The fact that omipalisib treatment and not MEK162 affected cell viability (Figure 3C) suggests the induction of a death response concomitant to Akt inhibition. Omipalisib treatment produced significant cytotoxicity in these cells (Figure 4). We investigated apoptotic and autophagic mediators to determine the nature of cell death and we found that omipalisib treatment could induce autophagic cell death in a dose-dependent manner, which can be prevented by 5 mM 3-methyl-adenine (3MA), an autophagy inhibitor, confirming the role of autophagy in omipalisib mediated cell death. Autophagic marker LC3 appears within 15 min of GSK212645 treatment in vitro. This could be due to the release of inhibition of Unc like kinase 1(ULK1) by mToR. Akt is a common substrate for PI3-kinase and mToR, each responding to different signaling cues. Omipalisib inhibits PI3-kinase and mToR simultaneously stopping Akt activation from all possible kinases. It is possible that mToR inhibition in this case resulted in the activation of autophagy, otherwise held in check by inhibitory phosphorylation of ULK1 (one of the initiating kinases of autophagic process) by mToR. This idea follows from the observation that omipalisib caused disappearance of ULK inhibitory phosphorylation in a time-dependent manner coincidentally with the appearance of autophagic marker LC3. These observations led us to hypothesize that combined targeting of ERK and Akt would be more effective.
Combined targeting of MAPK and Akt pathways has been proposed in cutaneous melanoma (21) and more recently in conjunctival melanoma (22). New successful drug combinations with MAPK pathway inhibitors have been proposed recently in KRAS-mutated colorectal cancer cell lines (23) and melanoma (24,25) to improve the outcome of MAPK inhibitors. To prove our hypothesis, we treated NCM cells with increasing doses of one drug combined with a fixed dose (<EC50) of the other drug. Cell viability studies showed that the combination always led to less viability than monotherapy. However, dose dependent increase in omipalisib combined with a fixed dose of MEK162 was more effective than the treatment with increasing doses of MEK162 combined with a fixed dose of omipalisib.
These data point to an important possibility in NCM therapy. Omipalisib is in clinical trial for solid tumors and idiopathic pulmonary fibrosis (5) and could be considered a new candidate to be used alone or in a combination therapy along with MEK162 on NCM patients. Given the lack of effective pharmacological treatment for congenital melanocytic proliferations, including L/GCMN and NCM, our findings represent a step toward developing alternatives to surgical treatments, which are the current approaches in clinic in trying to help patients with these conditions, although their effectiveness is limited, and their complications are not insignificant.
Acknowledgements
The Authors acknowledge the support of patients donating tissues for the Gavin Bailey Tissue Repository for Neural Crest Disorders.
Funding
The Marjorie K. Harmer endowment for Research in Pediatric Pathology (MRM); A generous gift from Mr. and Mrs. Travis Bailey and Nevus Outreach, Inc. (MRM); and Grant # I#0048403 from the Morgan Family Foundation through the Silicon Valley Community Foundation (CMS).
Conflicts of Interest
The Authors declare no conflicts of interest.
References
- 1.Reyes-Múgica M, Beckwith M, Etchevers HC. Etiology of congenital melanocytic nevi and related conditions. In: Nevogenesis (Practical Clinical Medicine series) (ed. Marghoob, A.A.). Berlin, Heidelberg. Springer Berlin Heidelberg. 2012;24 [Google Scholar]
- 2.Kadonaga JN, Frieden IJ. Neurocutaneous melanosis: Definition and review of the literature. J Am Acad Dermatol. 1991;24:747–755. doi: 10.1016/0190-9622(91)70115-i. [DOI] [PubMed] [Google Scholar]
- 3.Makin GWJ, Eden OB, Lashford LS, Moppett J, Gerrard MP, Davies HA, Powell CVE, Campbell AN, Child HF. Leptomeningeal melanoma in childhood. Cancer. 1999;86:878–886. [PubMed] [Google Scholar]
- 4.Pawlikowski JS, Brock C, Chen SC, Al-Olabi L, Nixon C, McGregor F, Paine S, Chanudet E, Lambie W, Holmes WM, Mullin JM, Richmond A, Wu H, Blyth K, King A, Kinsler VA, Adams PD. Acute inhibition of MEK suppresses congenital melanocytic nevus syndrome in a murine model driven by activated NRAS and Wnt signaling. J Invest Dermatol. 2015;135:2093–2101. doi: 10.1038/jid.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munster P, Aggarwal R, Hong D, Schellens JHM, van der Noll R, Specht J, Witteveen PO, Werner TL, Dees EC, Bergsland E, Agarwal N, Kleha JF, Durante M, Adams L, Smith DA, Lampkin TA, Morris SR, Kurzrock R. First-in-human phase I study of GSK2126458, an oral pan-class I phosphatidylinositol-3-kinase inhibitor, in patients with advanced solid tumor malignancies. Clin Cancer Res. 2016;22:1932–1939. doi: 10.1158/1078-0432.CCR-15-1665. [DOI] [PubMed] [Google Scholar]
- 6.Ruan Y, Kovalchuk A, Jayanthan A, Lun X, Nagashima Y, Kovalchuk O, Wright JR, Jr, Pinto A, Kirton A, Anderson R, Narendran A. Druggable targets in pediatric neurocutaneous melanocytosis: Molecular and drug sensitivity studies in xenograft and ex vivo tumor cell culture to identify agents for therapy. Neuro Oncol. 2015;17:822–831. doi: 10.1093/neuonc/nou310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel J, Salgado CM, Múgica MR, Basu D. Insulin-like growth factor 1 receptor signaling via Akt: a general therapeutic target in neurocutaneous melanocytosis. Neuro Oncol. 2016;18:142–143. doi: 10.1093/neuonc/nov271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruan Y, Narendran A. Response to “Insulin-like growth factor 1 receptor signaling via Akt: a general therapeutic target in neurocutaneous melanocytosis?”. Neuro Oncol. 2016;18:143–144. doi: 10.1093/neuonc/nov290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bendall SC, Stewart MH, Menendez P, George D, Vijayaragavan K, Werbowetski-Ogilvie T, Ramos-Mejia V, Rouleau A, Yang J, Bosse M, Lajoie G, Bhatia M. IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature. 2007;448:1015–1021. doi: 10.1038/nature06027. [DOI] [PubMed] [Google Scholar]
- 10.Malaguarnera R, Belfiore A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front Endocrinol. 2014;5:10. doi: 10.3389/fendo.2014.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malaguarnera R, Frasca F, Garozzo A, Gianì F, Pandini G, Vella V, Vigneri R, Belfiore A. insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J Clin Endocr Metab. 2011;96:766–774. doi: 10.1210/jc.2010-1255. [DOI] [PubMed] [Google Scholar]
- 12.Shan J, Shen J, Liu L, Xia F, Xu C, Duan G, Xu Y, Ma Q, Yang Z, Zhang Q, Ma L, Liu J, Xu S, Yan X, Bie P, Cui Y, Bian X-W, Qian C. Nanog regulates self-renewal of cancer stem cells through the insulin-like growth factor pathway in human hepatocellular carcinoma. Hepatology. 2012;56:1004–1014. doi: 10.1002/hep.25745. [DOI] [PubMed] [Google Scholar]
- 13.Chang W-W, Lin R-J, Yu J, Chang W-Y, Fu C-H, Lai AC-Y, Yu J-C, Yu AL. The expression and significance of insulin-like growth factor-1 receptor and its pathway on breast cancer stem/progenitors. Breast Cancer Res. 2013;15:1–16. doi: 10.1186/bcr3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu C, Xie D, Yu S-C, Yang X-J, He L-R, Yang J, Ping Y-F, Wang B, Yang L, Xu S-L, Cui W, Wang Q-L, Fu W-J, Liu Q, Qian C, Cui Y-H, Rich JN, Kung H-F, Zhang X, Bian X-W. β-Catenin/POU5F1/SOX2 transcription factor complex mediates IGF-I receptor signaling and predicts poor prognosis in lung adenocarcinoma. Cancer Res. 2013;73:3181–3189. doi: 10.1158/0008-5472.CAN-12-4403. [DOI] [PubMed] [Google Scholar]
- 15.Dallas NA, Xia L, Fan F, Gray MJ, Gaur P, van Buren G, Samuel S, Kim MP, Lim SJ, Ellis LM. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009;69(5):1951–1957. doi: 10.1158/0008-5472.CAN-08-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bodzin AS, Wei Z, Hurtt R, Gu T, Doria C. Gefitinib resistance in HCC mahlavu cells: Upregulation of CD133 expression, activation of IGF-1R signaling pathway, and enhancement of IGF-1R nuclear translocation. J Cell Physiol. 2012;227:2947–2952. doi: 10.1002/jcp.23041. [DOI] [PubMed] [Google Scholar]
- 17.Basu D, Salgado CM, Bauer BS, Johnson D, Rundell V, Nikiforova M, Khakoo Y, Gunwaldt LJ, Panigrahy A, Reyes-Múgica M. Nevospheres from neurocutaneous melanocytosis cells show reduced viability when treated with specific inhibitors of NRAS signaling pathway. Neuro Oncol. 2016;18:528–537. doi: 10.1093/neuonc/nov184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J, Aguila JR, Alipio Z, Lai R, Fink LM, Ma Y. Enhanced Self-Renewal of Hematopoietic Stem/Progenitor Cells Mediated by the Stem Cell Gene Sall4. J Hematol Oncol. 2011;4:38. doi: 10.1186/1756-8722-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKinnon T, Venier R, Yohe M, Sindiri S, Gryder BE, Shern JF, Kabaroff L, Dickson B, Schleicher K, Chouinard-Pelletier G, Menezes S, Gupta A, Zhang X, Guha R, Ferrer M, Thomas CJ, Wei Y, Davani D, Guidos CJ, Khan J, Gladdy RA. Functional screening of FGFR4-driven tumorigenesis identifies PI3K/mTOR inhibition as a therapeutic strategy in rhabdomyosarcoma. Oncogene. 2018;37:2630–2644. doi: 10.1038/s41388-017-0122-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park K-S, Yang H, Choi J, Seo S, Kim D, Lee CH, Jeon H, Kim S-W, Lee DH. The HSP90 inhibitor, NVP-AUY922, attenuates intrinsic PI3K inhibitor resistance in KRAS-mutant non-small cell lung cancer. Cancer Lett. 2017;406:47–53. doi: 10.1016/j.canlet.2017.07.028. [DOI] [PubMed] [Google Scholar]
- 21.Meier F, Busch S, Lasithiotakis K, Kulms D, Garbe C, Maczey E, Herlyn M, Schittek B. Combined targeting of MAPK and AKT signalling pathways is a promising strategy for melanoma treatment. Br J Dermatol. 2007;156:1204–1213. doi: 10.1111/j.1365-2133.2007.07821.x. [DOI] [PubMed] [Google Scholar]
- 22.Cao J, Heijkants RC, Jochemsen AG, Dogrusöz M, de Lange MJ, van der Velden PA, van der Burg SH, Jager MJ, Verdijk RM. Targeting of the MAPK and AKT pathways in conjunctival melanoma shows potential synergy. Oncotarget. 2017;8:58021–58036. doi: 10.18632/oncotarget.10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong J, Chen Y, Yang L, Pillai R, Shirasawa S, Fakih M. MEK162 enhances antitumor activity of 5-fluorouracil and trifluridine in KRAS-mutated human colorectal cancer cell lines. Anticancer Res. 2017;37:2831–2838. doi: 10.21873/anticanres.11634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Najem A, Krayem M, Perdrix A, Kerger J, Awada A, Journe F, Ghanem G. new drug combination strategies in melanoma: current status and future directions. Anticancer Res. 2017;37:5941–5953. doi: 10.21873/anticanres.12041. [DOI] [PubMed] [Google Scholar]
- 25.Luo H, Umebayashi M, Doi K, Morisaki T, Shirasawa S, Tsunoda T. Resveratrol Overcomes Cellular Resistance to Vemurafenib Through Dephosphorylation of AKT in BRAF-mutated Melanoma Cells. Anticancer Res. 2016;36:3585–3589. [PubMed] [Google Scholar]