

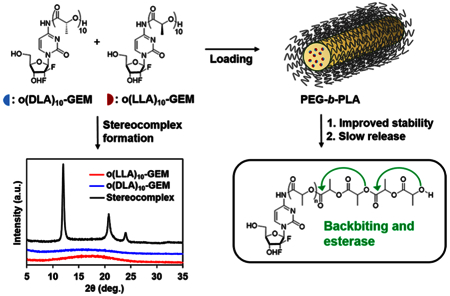
Keywords: oligo(lactic acid)n (o(LA)n); stereocomplex; gemcitabine; prodrug; backbiting conversion; poly(ethylene glycol)-block-poly(D,L-lactic acid) (PEG-b-PLA); polymeric micelles
Abstract
Herein we demonstrate the formation of stereo-complex prodrugs of oligo(L-lactic acid)n-gemcitabine (o(LLA)n-GEM) and oligo(D-lactic acid)n-gemcitabine (o(DLA)n-GEM) for stable incorporation in poly(ethylene glycol)-block-poly(D,L-lactic acid) (PEG-b-PLA) micelles. O(LLA)n or o(DLA)n was attached at the amino group (4-(N)) of GEM via an amide linkage. When n = 10, a 1:1 mixture of o(LLA)10-GEM and o(DLA)10-GEM (o(L+DLA)10-GEM) was able to form a stereocomplex with a distinctive crystalline pattern. Degradation of o(L+DLA)10-GEM was driven by both backbiting conversion and esterase contribution, generating primarily o(L+DLA)1-GEM and GEM. O(L+DLA)10-GEM stably loaded in PEG-b-PLA micelles in the size range of 140−200 nm with an unexpected elongated morphology. The resulting micelles showed improved physical stability in aqueous media and inhibited backbiting conversion of o(L+DLA)10-GEM within micelles. Release of o(L+DLA)10-GEM from micelles was relatively slow, with a t1/2 at ca. 60 h. Furthermore, weekly administration of o(L+DLA)10-GEM micelles i.v. displayed potent antitumor activity in an A549 human non-small-cell lung carcinoma xenograft model. Thus, stereocomplexation of isotactic o(LLA)n and o(DLA)n acts as a potential prodrug strategy for improved stability and sustained drug release in PEG-b-PLA micelles.
Stereocomplexation of isotactic poly(lactic acid) (PLA) has gained increasing attention over the last decades because of its ability to impart improved physical and chemical stability to drug formulations.1 Ikada et al. first reported the formation of a stereocomplex by blending a 1:1 ratio of poly(L-lactide) (PLLA) and poly(D-lactide) (PDLA), forming a distinctive crystalline structure with a different melting temperature and a different diffraction pattern from the homopolymer.2 Due to the ease of scale-up and biodegradability of PLA, a large research effort has described stereocomplex formation between enantiomeric PLA in hydrogels and polymeric micelles to enhance kinetic stability and control drug release.3 Hennink et al. demonstrated stable dextran hydrogels cross-linked by stereocomplex lactic acid oligomers acted as a controlled release matrix for pharmaceutical proteins.4 Leroux’s research group showed enhanced physical stability of poly(ethylene glycol)-block-poly(L-lactic acid) (PEG-b-PLLA) and poly(ethylene glycol)-block-poly(D-lactic acid) (PEG-b-PDLA) micelles by introducing stereo-complex PLLA and PDLA blocks in the core.5 In addition, PLA stereocomplexation was also applied to the delivery of luminescent molecules in polymeric micelles. Kersey et al. introduced a PLLA moiety on a dual-emissive boron dye, which was then loaded in PEG-b-PDLA micelles, forming stable stereocomplex micelles ranging from 80 to 120 nm.6 While the PLA stereocomplexation approach has been widely implemented on polymer-based drug delivery systems, no prior research has studied PLA stereocomplexation as a pro-moiety in prodrug design.
Previously, we have demonstrated that oligo(lactic acid)n (o(LA)n) can act as a pro-moiety and form an ester prodrug of paclitaxel (o(LA)n-PTX) that enhances its compatibility in the PLA core of PEG-b-PLA micelles and undergoes backbiting after release.7 As a result, stable prodrug micelles were formed with a high drug loading content and a sustained drug release profile, thereby enhancing antitumor efficacy.7 In this study, we report the use of enantiomeric oligo(L-lactic acid)n (o(LLA)n) and oligo(D-lactic acid)n (o(DLA)n) as pro-moieties for prodrug design, utilizing PLA stereocomplexation between prodrugs for stable incorporation in poly(ethylene glycol)-block-poly(D,L-lactic acid) (PEG-b-PLA) micelles. To demonstrate the additional benefits of prodrug stereocomplexation, gemcitabine (GEM) was used as a model drug due to its hydrophilic nature attenuating hydrophobic core loading in polymeric micelles.
GEM (2′,2′-difluoro-2′-deoxycytidine) is a potent anti-cancer agent that acts against a wide spectrum of solid tumors.8 It is highly water-soluble (>15 mg/mL) and can be directly reconstituted with 0.9% sodium chloride without preservatives before infusion into humans.9 However, due to its extensive metabolism at the amino group (4-(N)) of GEM by cytidine deaminase during circulation (plasma half-life <20 min), a high dose of weekly GEM infusion is often required (1000−1250 mg/m2).10 Consequently, undesired pharmaco-kinetic profiles such as an exceedingly high initial plasma concentration of GEM can induce severe toxicity.11 In addition, its rapid metabolic inactivation can vastly limit the delivery of GEM to the tumor site.
To improve the metabolic stability and delivery of GEM, lipophilic prodrugs of GEM have been synthesized for liposomal or polymeric micellar carriers. Cattel’s research group showed that a stearoyl prodrug of GEM (GemC18) at the 4-(N)-position was more stable in plasma than GEM.12 Incorporation of GemC18 in liposomes significantly improved exposure and reduced clearance versus GEM, resulting in increased drug accumulation in the tumor and enhanced antitumor efficacy in mice grafted with HT-29 human colorectal adenocarcinoma and KB 396p nasopharyngeal carcinoma.13 Tan’s research team successfully incorporated GemC18 in a mixed micelle system consisting of PEG-distearoyl-phosphatidyl-ethanolamine (DSPE) and tocopheryl polyethylene glycol 1000 succinate (TPGS), which prolonged the circulation time of GEM and improved GEM concentration in tumors by 3-fold.14 In addition, Couvreur’s team demonstrated that squalenoylation of GEM at the 4-(N)-position allowed formation of stable nanoassemblies, which significantly inhibited tumor growth and prolonged survival in mice bearing subcutaneous pancreatic tumors.15,16 Thus, lipophilic prodrugs of GEM have been shown to improve metabolic stability and therapeutic efficacy of GEM.
In this study, we note three key features of the GEM prodrug micelle platform using PLA stereocomplexation: (1) extensive metabolism of GEM in plasma is reduced because of enantiomeric o(LA)n linkage via amide conjugation at the 4-(N)-position of GEM; (2) degradation of the stereocomplex o(LA)n pro-moiety after release is driven by backbiting conversion and potential esterase contribution, cleaving nontoxic lactic acid units as side products; (3) improved micelle stability can be obtained through the formation of stable stereocomplex prodrugs between o(LLA)n-GEM and o(DLA)n-GEM (o(L+DLA)n-GEM). We hypothesize that o(L+DLA)n-GEM can form a distinctive stereocomplex crystal and hinder backbiting conversion after incorporation in PEG-b-PLA micelles, resulting in improved physical stability and sustained drug release (Figure 1). Therefore, stable incorpo ration of o(L+DLA)n-GEM in PEG-b-PLA micelles can show the versatility of the PLA stereocomplexation of isotactic o(LLA)n and o(DLA)n as a potential prodrug design for prodrug micelle delivery systems.
Figure 1.
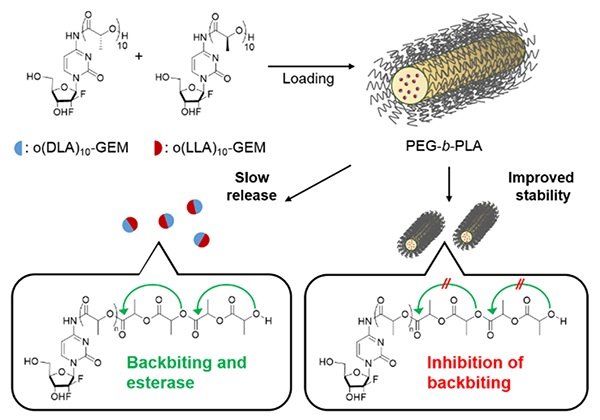
Stereocomplex prodrugs of o(LLA)n-GEM and o(DLA)n-GEM for PEG-b-PLA micelles: loading, stability, and prodrug conversion by backbiting and esterase after release.
RESULTS AND DISCUSSION
Monodisperse o(LLA)n or o(DLA)n was synthesized by a ring-opening polymerization (ROP) of either cyclic L-lactide or cyclic D-lactide using benzyl alcohol as initiator and tin(II) ethylhexanoate (Sn(Oct)2) as catalyst as reported previously, followed by fractionation and direct hydrogenation.17,18 Coupling of o(LLA)n or o(DLA)n with GEM at the 4-(N)-position via amide linkage was mediated by 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDCI) and N-hydroxysuccinimide (NHS), resulting in o(LLA)n-GEM and o-(DLA)n-GEM prodrugs (n = 6 or 10) (Scheme 1). The chemical structures were supported by 1H NMR spectroscopy and electrospray mass spectroscopy analysis (Supporting Information).
Scheme 1.
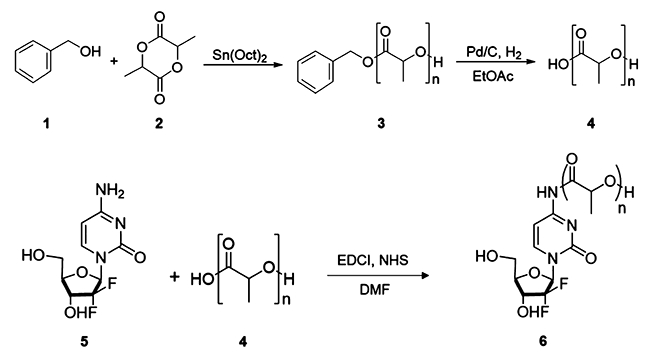
Synthetic scheme for o(LLA)n-GEM and o(DLA)n-GEM. (1) Benzyl alcohol, (2) cyclic-L-lactide or cyclic-D-lactide, (3) benzyl-oligo(L-lactic acid)n (Bn-o(LLA)n) or benzyl-oligo(D-lactic acid)n (Bn-o(DLA)n), (4) oligo(L-lactic acid)n (o(LLA)n) or oligo(D-lactic acid)n (o(DLA)n), (5) gemcitabine (GEM), (6) oligo(L-lactic acid)n-gemcitabine (o(LLA)n-GEM) or oligo(D-lactic acid)n-gemcitabine (o(DLA)n-GEM).
To explore the stereocomplex formation of o(LLA)n-GEM and o(DLA)n-GEM, both prodrugs were mixed in an equal mass ratio in a glass vial before dissolving in acetonitrile (CH3CN). The resulting solution was then placed in a vacuum oven at 70 °C for 12 h to allow complete solvent removal. Powder X-ray diffraction (PXRD) and differential scanning calorimetry (DSC) were used to confirm the formation of a crystalline stereocomplex through the identification of different diffraction patterns and melting transition (Tm) as compared to each enantiomeric form of the prodrug (Figure 2A and B). When n = 10, both o(LLA)10-GEM and o(DLA)10-GEM are amorphous, as evidenced by the broad diffraction patterns and the absence of a DSC melting endotherm. By contrast, a 1:1 mixture of o(LLA)10-GEM and o(DLA)10-GEM shows three distinct crystalline peaks at 12.0°, 20.8°, and 23.9°, which is the unique diffraction signature of the formation of a crystalline PLA stereocomplex.2 The presence of the crystalline stereo-complex was further proven by a melting endotherm at 117.8 °C. Interestingly, when n = 6, no crystalline stereocomplex can be formed after blending an equal mass ratio of o(LLA)6-GEM and o(DLA)6-GEM, as evidenced by a broad diffraction peak and the absence of a melting endotherm (Figure S1). These findings corroborate previously published results reporting that a minimum of seven o(LA) was required for crystalline stereocomplex formation.17
Figure 2.
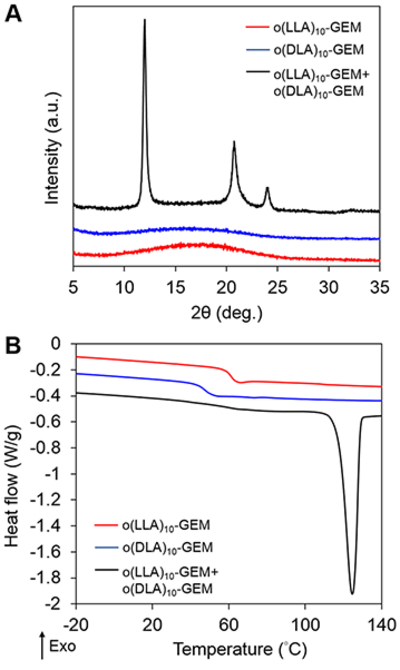
(A) Powder XRD profiles and (B) DSC thermograms of o(LLA)10-GEM, o(DLA)10-GEM, and the stereocomplex mixture of both prodrugs after evaporation from CH3CN.
The incorporation of o(LLA)6-GEM, o(LLA)10-GEM, or o(L+DLA)10-GEM in PEG-b-PLA micelles was achieved by lyophilization from a tert-butanol/water mixture. As shown in Table 1, both o(LLA)6-GEM and o(LLA)10-GEM were successfully incorporated in PEG-b-PLA, forming micelles with an average hydrodynamic diameter at ca. 30 nm and ca. 7% drug loading. Although it is hypothesized that introducing o(LLA)n to GEM as pro-moiety can enhance the compatibility of GEM in the PLA core of PEG-b-PLA micelles, loading of o(LLA)6-GEM or o(LLA)10-GEM within micelles did not show improved physical stability, precipitating in <24 h or <1 h, respectively. By contrast, PEG-b-PLA micelles containing o(L+DLA)10-GEM demonstrated improved physical stability, with no substantial change in particle size and no drug precipitation at >168 h. Interestingly, the incorporation of o(L+DLA)10-GEM in PEG-b-PLA micelles at 5.4% and 15.3% increased the hydrodynamic diameter from 30 nm to ca. 140 nm and ca. 200 nm, respectively, compared to their noncomplexed counterparts. The morphology of o(L+DLA)10-GEM micelles was further investigated using atomic force microscopy (AFM). Unexpectedly, an elongated structure of o(L+DLA)10-GEM micelles was observed (Figure 3A,B). According to Portinha et al., a mixture of poly(ε-caprolactone)-b-PLLA and poly(ε-caprolactone)-b-PDLA formed nanoparticles with a cylindrical morphology.19 However, a recent study reported the formation of stereo-complex spherical micelles of a diblock copolymer containing PLLA and PDLA blocks.20 Nonetheless, one distinct difference of our system is that o(L+DLA)10-GEM prodrugs are loaded in PEG-b-PLA micelles, instead of direct stereocomplexation between the core blocks of the copolymer. Although the formation of a spherical morphology for PEG-b-PLA micelles is commonly reported in the literature after self-assembly,21 detailed physical study is required to further understand the morphological change of PEG-b-PLA micelles for stable incorporation of stereocomplex prodrugs. Release of o-(LLA)6-GEM and o(LLA)10-GEM from PEG-b-PLA micelles was relatively rapid in vitro, with t1/2 at ca. 0.8 h and ca. 4 h, respectively (Figure 4). It may be attributed to the hydrophilic nature of GEM, resulting in reduced partition of the drug for the hydrophobic micelle core of PEG-b-PLA micelles and therefore faster drug release. By contrast, o(L+DLA)10-GEM was gradually released from PEG-b-PLA micelles at 5.4% and 15.3%, both with t1/2 at ca. 60 h in the first 3 days, followed by a much slower release rate thereafter throughout 3 weeks (Figure 4). Overall, o(L+DLA)10-GEM was stably loaded in PEG-b-PLA micelles with an unexpected cylindrical morphology, demonstrating improved physical stability and sustained drug release.
Table 1.
Physicochemical Characterizations of o(LLA)6-GEM, o(LLA)10-GEM, and o(L+DLA)10-GEM Loaded PEG-b-PLA Micelles (Mean ± SD, n = 3)
| drug | particle size (nm) | PDI | loading efficiency (%) | loading content (%) | stability (h)d) |
|---|---|---|---|---|---|
| o(LLA)6-GEMa | 27.4 ± 0.3 | 0.23 ± 0.01 | 67.5 ± 11.3 | 6.3 ± 1.1 | <24 |
| o(LLA)10-GEMa | 29.4 ± 0.1 | 0.13 ± 0.03 | 82.1 ± 11.4 | 7.6 ± 1.1 | <1 |
| o(L+DLA)10-GEMb | 139.5 ± 24.9 | 0.35 ± 0.08 | 56.7 ± 4.3 | 5.4 ± 0.4 | >168 |
| o(L+DLA)10-GEMc | 202.3 ± 19.0 | 0.33 ± 0.07 | 90.3 ± 4.4 | 15.3 ± 0.9 | >168 |
1.0 mg of o(LLA)6-GEM or o(LLA)10-GEM and 10.0 mg of PEG4000-b-PLA2200 were used.
1.0 mg of o(LLA)10-GEM, 1.0 mg of o(DLA)10-GEM, and 20.0 mg of PEG4000-b-PLA2200 were used.
1.0 mg of o(LLA)10-GEM, 1.0 mg of o(DLA)10-GEM, and 10.0 mg of PEG4000-b-PLA2200 were used.
Formulations were dispersed in Milli-Qwater at 25 °C.
Figure 3.

AFM images of two representative regions of o(L+DLA)10-GEM-loaded PEG-b-PLA micelles.
Figure 4.
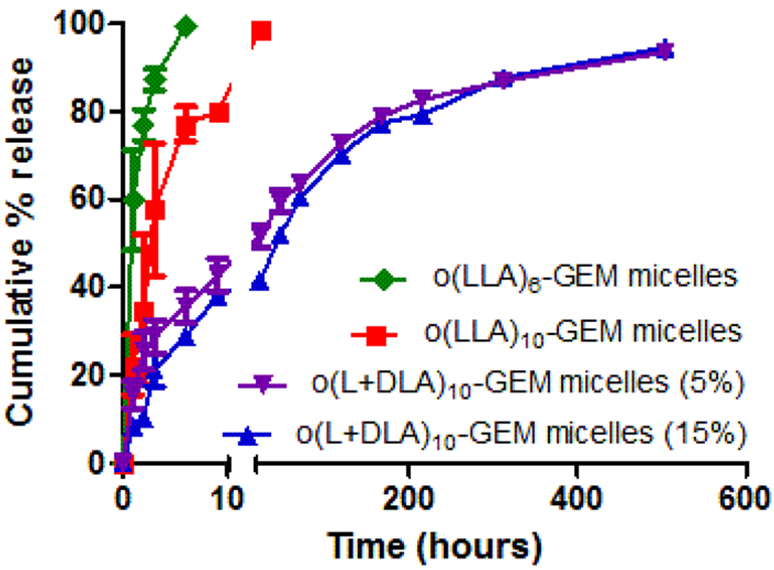
In vitro release of o(LLA)6-GEM, o(LLA)10-GEM, or o(L+DLA)10-GEM from PEG-b-PLA micelles at 5% and 15% loading (mean ± SEM, n = 3, 4).
Previously, we have demonstrated that the degradation of o(LA)n-PTX was driven by an intramolecular backbiting reaction of the terminal hydroxyl group on the penultimate ester bond of o(LA)n in a mixture of acetonitrile and phosphate-buffered saline (1:1 v/v CH3CN/PBS), cleaving lactoyl lactate in a stepwise chain end scission.7 For o(L+DLA)10-GEM, the backbiting kinetics is expected to be slower due to the strong interaction of the PLA stereocomplex. However, conversion of o(L+DLA)10-GEM (λmax = 300 nm) was found to have a similar backbiting kinetics in 1:1 v/v CH3CN/PBS, with t1/2 at ca. 6.6 h versus ca. 7.3 h of o(LA)8-PTX (Figure 5C).7 A series of well-defined peaks with shorter elution times was produced upon cleavage of lactoyl lactate in a stepwise manner, producing an even number of degradation intermediates: o(L+DLA)8-GEM, o(L+DLA)6-GEM, and o(L+DLA)4-GEM (Figure 5A). Interestingly, both o(L+DLA)1-GEM and GEM (λmax = 270 nm) emerged as major degradation products as early as 1 h after backbiting initiation, indicating amide hydrolysis and surprising ester hydrolysis at the ester bond adjacent to the amide (Figure 5A and B). Similarly, o(LLA)6-GEM and o(LLA)10-GEM followed back-biting conversion and amide hydrolysis, producing o(LLA)1-GEM and GEM as major degradation species (Figures S3–S8). On the contrary, degradation of o(L+DLA)10-GEM in PEG-b-PLA micelles was relatively slow, with t1/2 at ca. 630 h (Figure 5F). Notably, only GEM was observed as major degradation species and there was no appearance of backbiting degradation intermediates (Figure 5D and E). The improved stability of o(L+DLA)10-GEM in PEG-b-PLA micelles could be ascribed to the strong interaction between o(LLA)10 and o(DLA)10 pro-moieties in PEG-b-PLA micelles, which potentially inhibited backbiting. However, when o(L+DLA)10-GEM was dissolved in the CH3CN/PBS mixture, no stereocomplexation was formed due to complete solubilization of o(L+DLA)10-GEM, and therefore backbiting conversion followed.
Figure 5.
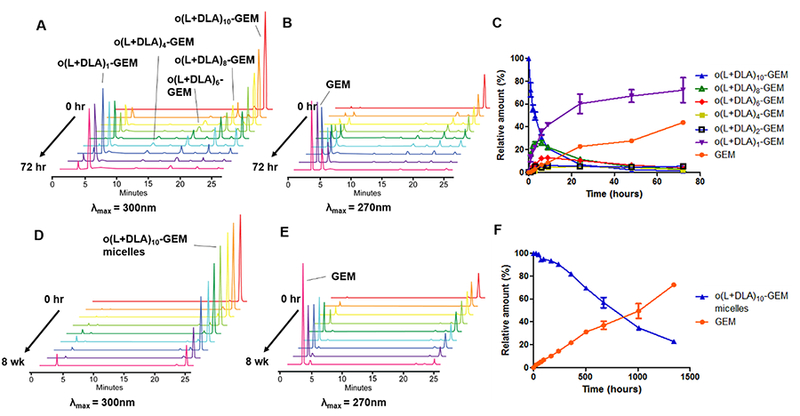
Representative reverse-phase HPLC chromatograms with (A) λmax = 300 nm, (B) λmax = 270 nm, and (C) relative amount of a physical mixture of o(L+DLA)10-GEM and its backbiting conversion products after incubation in 1:1 v/v CH3CN/10 mM PBS at 37 °C, pH 7.4, from 0 to 72 h (mean ± SD, n = 3). Representative reverse-phase HPLC chromatograms with (D) λmax = 300 nm, (E) λmax = 270 nm, and (F) relative amount of o(L+DLA)10-GEM-loaded PEG-b-PLA micelles at 15% loading and its conversion product after incubation in Milli-Q water at 25 °C from 0 to 8 weeks (mean ± SD, n = 3).
As alluded to earlier, GEM is susceptible to deamination in plasma by metabolic enzymes such as cytidine deaminase, rendering it pharmacologically inactive. The attachment of a pro-moiety at the 4-(N)-position of GEM is hypothesized to obscure the deamination site and therefore improve the metabolic stability of GEM. Consequently, the metabolic stability of o(L+DLA)10-GEM in PEG-b-PLA micelles and o(L+DLA)10-GEM in comparison to GEM was investigated in rat plasma. As expected, GEM was not stable in plasma, with less than 50% of intact GEM detected after 24 h of incubation in plasma at 37 °C (Figure 6A). Surprisingly, degradation of o(L+DLA)10-GEM was extremely rapid in plasma, generating a random distribution of degradation species and ca. 20% GEM during the first sampling time point, suggesting a fragile amide linkage and contribution of esterases in addition to backbiting (Figure 6B). When o(L+DLA)10-GEM was loaded in PEG-b-PLA micelles, o(L+DLA)10-GEM gained extra stability, generating GEM as the only major degradation species in plasma over 24 h (Figure 6C). Notably, no degradation intermediates were generated throughout the study, indicating stable o(L+DLA)10-GEM in PEG-b-PLA micelles could potentially prevent esterase degradation of o(LLA)n and o(DLA)n.
Figure 6.
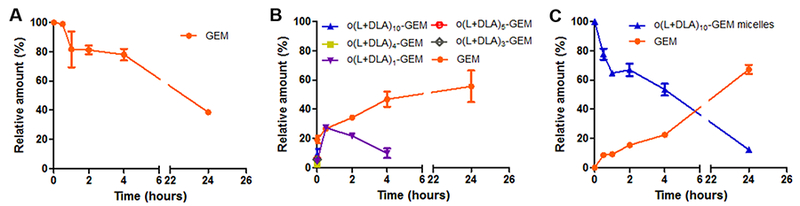
Relative amount of (A) GEM, (B) o(L+DLA)10-GEM, and (C) o(L+DLA)10-GEM prodrugs in PEG-b-PLA micelles at 15% loading and its conversion product GEM in rat plasma after incubation at 37 °C from 0 to 24 h (mean ± SEM, n = 3).
The bioactivity of o(L+DLA)10-GEM micelles was evaluated by an in vitro cell viability assay to test the half-maximal inhibitory concentration (IC50) in comparison to GEM. Since GEM was used as the first-line treatment for non-small-cell lung cancer (NSCLC) and pancreatic cancer,22,23 the human A549 NSCLC cell line and the PANC-1 pancreatic cancer cell line were investigated using a CellTiter-Blue assay. As shown in Figure 7A and B, GEM had a relatively low IC50 value against A549 and PANC-1 cells, at ca. 1.1 μM and ca. 9.2 μM, respectively. In contrast, o(L+DLA)10-GEM micelles were less cytotoxic than GEM in A549 and PANC-1 cells after 72 h of incubation, with an IC50 value of ca. 12.7 μM and ca. 97.9 μM, respectively. The in vitro cytotoxicity data provided further supportive evidence of high stability of o(L+DLA)10-GEM in PEG-b-PLA micelles and a relatively slow prodrug release from micelles, which led to decreased cytotoxicity.
Figure 7.
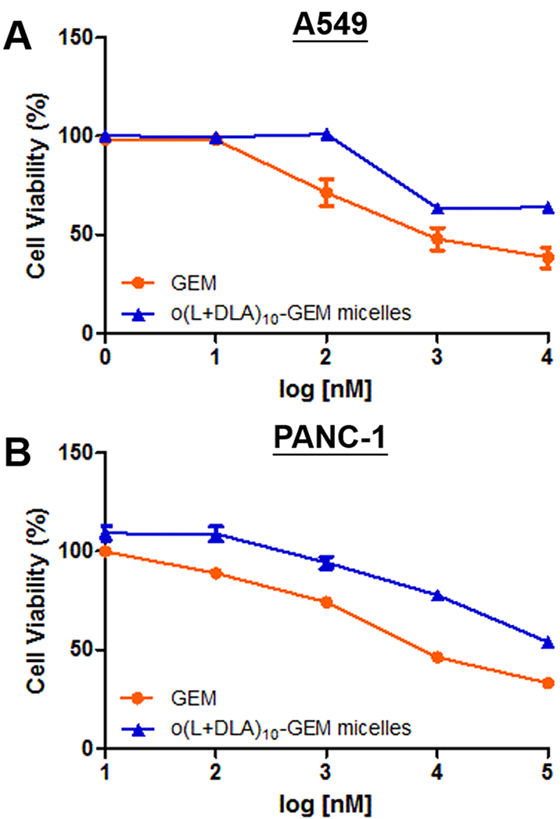
In vitro cytotoxicity of GEM and o(L+DLA)10-GEM micelles at 15% loading against (A) human A549 non-small-cell lung cancer cells and (B) PANC-1 pancreatic cancer cells (mean of quintuplicate determinations ± SEM).
To verify the antitumor efficacy of o(L+DLA)10-GEM micelles in vivo, mice bearing subcutaneous A549 xenografts were treated with normal saline, GEM, or o(L+DLA)10-GEM micelles via weekly i.v. injections for 3 weeks at 10 mg/kg equivalent of GEM. Notably, administration of o(L+DLA)10-GEM micelles showed significant tumor growth inhibition compared to GEM or the saline control (Figure 8A). Interestingly, tumors in mice that were treated with GEM grew at a faster rate than the saline control, indicating GEM was not effective (Figure 8A). A slight decrease of body weight was recorded after the first dose of o(L+DLA)10-GEM micelles, but no significant difference was observed among all the treatment groups (Figure 8B). Delivery of GEM at 10 mg/kg is generally ineffective against subcutaneous tumors in mice. The literature reports that 100 mg/kg of GEM is required to inhibit tumor growth in mice bearing subcutaneous tumors due to its metabolic instability.24 Furthermore, other reports indicate that a lipophilic linker attached at the 4-(N)-position of GEM improves the pharmacokinetic properties of GEM and thus alters its toxicity. Couvreur’s team reported that the maximum tolerated dose of the squalenoyl GEM nanoassemblies in mice (20 mg/kg) was 5-fold lower than that of GEM (100 mg/kg).25 This study’s in vivo efficacy results are supported by these findings. Although a relatively low dose of GEM was used (10 mg/kg), o(L+DLA)10-GEM micelles displayed a potent antitumor effect in vivo, suggesting stable o(L+DLA)10-GEM in PEG-b-PLA micelles could effectively deliver GEM to tumor sites for drug action.
Figure 8.
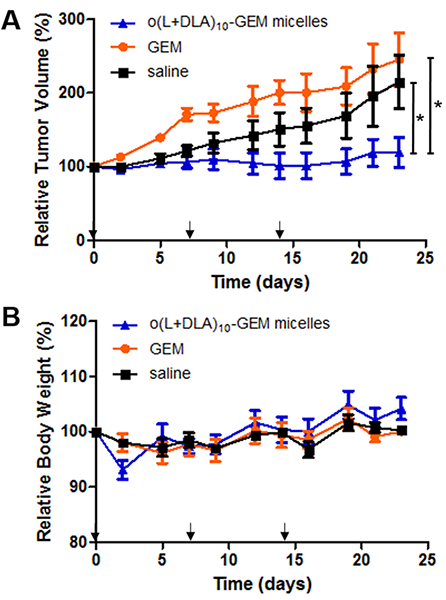
In vivo antitumor efficacies of GEM and o(L+DLA)10-GEM micelles in an A549 non-small-cell lung cancer xenograft model. Mice were administered i.v. weekly for 3 weeks (as indicated by the arrows) with GEM (10 mg/kg) or o(L+DLA)10-GEM micelles (5% loading, 10 mg/kg GEM equivalent). Results are presented in (A) relative tumor volume and (B) relative body weight (mean ± SEM, n = 3, 4, *: p < 0.05).
CONCLUSION
In summary, we have demonstrated the formation of stereocomplex prodrugs using isotactic o(LLA)n and o(DLA)n as pro-moieties for stable incorporation in PEG-b-PLA micelles with greatly improved physical stability and a sustained drug release profile. In addition, we have shown that the presence of o(LLA)10-GEM and o(DLA)10-GEM in micelles can inhibit backbiting conversion and potentially evade esterase degradation. More importantly, the stability of o(L+DLA)10-GEM micelles can translate into enhanced antitumor efficacy in an A549 xenograft model compared to GEM and the saline control, providing a strong justification for a future preclinical study. The pharmacokinetic profiles and detailed understanding of drug−carrier interactions of o(L+DLA)10-GEM micelles await further characterization. The concept of PLA stereocomplexation as pro-moieties for prodrug micelle delivery systems could be widely applicable to other drug candidates with distinct physicochemical properties, potentially improving compatibility and stability within micelles for sustained drug release and improved efficacy.
METHODS
Materials.
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used as received. Analytical grade organic solvents and all other reagents were purchased from Fisher Scientific (Pittsburgh, PA, USA). Gemcitabine was purchased from LC Laboratories (Woburn, MA, USA). PEG-b-PLA was purchased from Advanced Polymer Materials Inc. (Montreal, Canada): the Mn of PEG and PLA was 4000 and 2200 g/mol, respectively; polydispersity index (PDI) 1.05. A549 human lung adenocarcinoma cells and PANC-1 pancreatic adenocarcinoma cells were purchased from ATCC (Manassas, VA, USA) and cultured in RPMI 1640 medium and Dulbecco’s modified Eagle’s medium (DMEM), respectively, supplemented with 10% fetal bovine serum, 100 IU/mL penicillin, 100 μg/mL streptomycin, and 2 mM L-glutamine in a 5% CO2 incubator at 37 °C. Heparinized Sprague−Dawley rat plasma was purchased from Innovative Research Inc. (Novi, MI, USA).
Instrumentation.
1H NMR data were recorded on a Varian Unity-Inova two-channel 500 MHz NMR spectrometer (Palo Alto, CA, USA) with a regulated temperature of 25 °C. Chemical shifts (δ) were reported in parts per million (ppm) relative to residual protonated solvent resonance at 7.26 ppm for CDCl3. Mass spectrometry data were obtained using a Waters LCT (electrospray ionization time-of-flight (ESI-TOF)) in the Chemical Instrumentation Center in the Department of Chemistry, University of Wisconsin−Madison. Samples were sprayed from a 10 mM NH4OAc/CH3CN solution. Reverse-phase high-performance liquid chromatography (RP-HPLC) analysis was carried out using a Shimadzu Prominence HPLC system (Shimadzu, Japan) equipped with an LC-20AT pump, a SIL-20AC HT autosampler, a CTO-20AC column oven, and a SPD-M20A diode array detector. Sample was separated by a Waters Symmetry Shield RP18 column (4.6 mm × 250 mm, 5 μm, 100 Å). In a typical experiment, 10 μL of sample was injected at a flow rate of 0.8 mL/min, column temperature at 25 °C, and UV detection wavelength at 270 nm for GEM and 300 nm for o(LA)n-GEM. The separation of o(LA)n-GEM conversion products was done in gradient mode with an organic phase containing 100% CH3CN as solvent A and an aqueous phase containing 100% Milli-Q water containing 0.1% formic acid as solvent B. Gradient elution was employed as follows: 0 min, 20% solvent A and 80% solvent B; 30 min, 68% solvent A and 32% solvent B; and 35 min for equilibration. Hydrodynamic diameters of PEG-b-PLA micelles were measured by dynamic light scattering using a Zetasizer Nano-ZS (Malvern Instruments Inc., UK) at 25 °C with a detection angle of 173° and a He−Ne ion laser as the light source (4 mW, 633 nm). Prior to measurements, PEG-b-PLA micelle solutions were diluted with Milli-Q water or phosphate-buffered saline (PBS) (10 mM, pH 7.4) to afford PEG-b-PLA at ~0.1 mg/mL, and 1 mL of each sample was placed into a disposable sizing cuvette (BrandTech Scientific Inc., Essex, CT, USA). The cumulant method was used to curve-fit the correlation function, and the z-average diameter and PDI of PEG-b-PLA micelles were calculated from the Stokes−Einstein equation and the slope of the correlation function, respectively. All measurements were performed in triplicate. The morphology of o(L+DLA)10-GEM-loaded PEG-b-PLA micelles was observed using AFM in ac mode after adsorption of the polymer at 50.0 mg/mL on mica. Micelles were imaged in Milli-Q water using an AC40 biolever on an Infinity Bioscope (Asylum Research, Santa Barbara, CA, USA).
Synthesis of Monodisperse Benzyl-oligo(L-lactic acid)n (Bno(LLA)n) or Benzyl-oligo(D-lactic acid)n (Bn-o(DLA)n).
Synthesis of polydisperse Bn-o(LLA)n or Bn-o(DLA)n was achieved with tin(II) ethylhexanoate (Sn(Oct)2) according to the previously reported procedure with modifications.17 For example, at an average degree of polymerization of 8, cyclic L-lactide or cyclic D-lactide was mixed with benzyl alcohol in a molar ratio of 4 to 1. The mixture was stirred at 130 °C until molten. Subsequently, 0.01 equiv of Sn(Oct)2 in toluene (100 mg/mL) was added. The mixture was stirred at 130 °C for 4 h and allowed to cool to room temperature, to obtain polydisperse Bno(LLA)n or Bn-o(DLA)n. Monodisperse Bn-o(LLA)6, Bn-o(LLA)10, Bn-o(DLA)6, or Bn-o(DLA)10 was fractionated via a CombiFlash Rf 4x system using C18 reverse-phase column chromatography. Gradient elution of acetonitrile in 0.1% formic acid and water in 0.1% formic acid was applied. The purified product was concentrated under reduced pressure to provide a colorless liquid (yield: 8.3% for Bno(LLA)6, 8.0% for Bn-o(DLA)6, 6.7% for Bn-o(LLA)10, and 7.9% for Bn-o(DLA)10). ESI-TOF of Bn-o(LLA)6: m/z calcd C25H32O13 [M + Na]+ 558.2181, found 558.2179. ESI-TOF of Bn-o(DLA)6: m/z calcd C25H32O13 [M + Na]+ 558.2181, found 558.2178. ESI-TOF of Bno(LLA)10: m/z calcd C37H48O21 [M + Na]+ 846.3026, found 846.3027. ESI-TOF of Bn-o(DLA)10: m/z calcd C37H48O21 [M + Na]+ 846.3026, found 846.3023.
Synthesis of Oligo(L-lactic acid)n-gemcitabine (o(LLA)n-GEM) or Oligo(D-lactic acid)n-gemcitabine (o(DLA)n-GEM).
The synthesis of o(LLA)n-GEM or o(DLA)n-GEM was prepared through the amidation of a carboxylic acid group on o(LLA)n or o(DLA)n and an amine group on a gemcitabine molecule. In general, direct hydrogenation of Bn-o(LLA)n or Bn-o(DLA)n was achieved over palladium on activated carbon (10 wt %) to afford o(LLA)n or o(DLA)n according to a previously published report (yield: ~90− 99%).18 Subsequently, 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (1.5 equiv) and N-hydroxysuccinimide (1.5 equiv) were added to a solution of o(LLA)n or o(DLA)n (1 equiv) in dry dimethylformamide (DMF) and stirred at room temperature under argon for 2 h. GEM (1 equiv) was dissolved in dry DMF and was added dropwise into the above mixture. The reaction mixture was stirred for 24 h at room temperature under argon. The crude products were diluted with ethyl acetate (20-fold) and washed with 0.5% w/v hydrochloric acid, saturated NaHCO3, and brine solution. The organic layer was then collected, dried over MgSO4, and filtered. Solvent was removed under reduced pressure, and the resulting concentrate was purified via a CombiFlash Rf 4x system using gradient elution of hexane and ethyl acetate. The purified product was concentrated under reduced pressure to provide a white solid (yield: 80% for o(LLA)6-GEM, 63% for o(DLA)6-GEM, 87% for o(LLA)10-GEM, and 74% for o(DLA)10-GEM). 1H NMR of o(LLA)6-GEM (CDCl3, 500 MHz): δ 8.09 (d, J = 7.6 Hz, 1 H), 7.36 (br s, 1 H), 6.20 (br s, 1 H), 5.30 (s, 1 H), 5.14−5.29 (m, 5 H), 4.45−4.57 (m, 1 H), 4.37 (q, J = 7.1 Hz, 1 H), 4.00−4.09 (m, 2 H), 3.92 (d, J = 10.5 Hz, 1 H), 1.46−1.62 ppm (m, 18 H). ESI-TOF: m/z calcd C27H35F2N3O16 [M + H]+ 696.2058, found 696.2061. 1H NMR of o(DLA)6-GEM (CDCl3, 500 MHz): δ 8.10 (d, J = 7.3 Hz, 1 H), 7.40 (br s, 1 H), 6.17 (br s, 1 H), 5.34 (d, J = 7.1 Hz, 1 H), 5.10−5.31 (m, 5 H), 4.58 (br s, 1 H), 4.37 (q, J = 6.8 Hz, 1 H), 3.99−4.09 (m, 2 H), 3.92 (d, J = 10.7 Hz, 1 H), 1.47−1.61 ppm (m, 18 H). ESI-TOF: m/z calcd C27H35F2N3O16 [M + H]+ 696.2058, found 696.2058. 1H NMR of o(LLA)10-GEM (CDCl3, 500 MHz): δ 8.08 (d, J = 7.3 Hz, 1 H), 7.39 (d, J = 5.9 Hz, 1 H), 6.19 (br s, 1 H), 5.13−5.33 (m, 10 H), 4.54 (br s, 1 H), 4.37 (q, J = 6.8 Hz, 1 H), 4.05 (d, J = 10.3 Hz, 2 H), 3.93 (d, J = 10.0 Hz, 1 H), 1.46−1.63 ppm (m, 30 H). ESI-TOF: m/z calcd C39H51F2N3O24 [M + H]+ 984.2903, found 984.2907. 1H NMR of o(DLA)10-GEM (CDCl3, 500 MHz): δ 8.04 (d, J = 8.1 Hz, 1 H),7.32−7.49 (m, 1 H), 6.18 (br s, 1 H), 5.11−5.41 (m, 10 H), 4.56−4.70 (m, 1 H), 4.36 (d, J = 6.3 Hz, 1 H), 4.02−4.09 (m, 2 H), 3.93 (d, J = 10.0 Hz, 1 H), 1.48−1.65 ppm (m, 30 H). ESI-TOF: m/z calcd C39H51F2N3O24 [M + H]+ 984.2903, found 984.2905.
Formation and Characterization of Stereocomplex of o(LLA)10-GEM and o(DLA)10-GEM.
DSC measurements were performed in crimped aluminum pans with approximately 5−10 mg of materials, using a TA Q2000 differential scanning calorimeter. The samples were cooled and heated at 10 K/min under a 50 mL/min N2 purge. PXRD patterns were collected at room temperature from 2° to 40° 2θ with a step size of 0.02° and an integration time of 1 s, using a Bruker D8 Advance diffractometer. The morphology of stereo-complex crystals was recorded using an Olympus BH2-UMA polarized light microscope equipped with a digital camera. To prepare the stereocomplex of o(LLA)10-GEM and o(DLA)10-GEM, equal amounts of o(LLA)10-GEM (4 mg) and o(DLA)10-GEM (4 mg) were first dissolved in 100 μL of acetonitrile in a glass vial and swirled for 30 s. The solution was then placed on the DSC substrates or the silicon wafer and dried under vacuum at 70 °C for 12 h to yield the solid.
Preparation and Characterization of PEG-b-PLA Micelles Containing o(L+DLA)10-GEM, o(LLA)10-GEM, or o(LLA)6-GEM.
O(L+DLA)10-GEM, o(LLA)10-GEM, or o(LLA)6-GEM loaded PEG-b-PLA micelles were prepared by freeze-drying from a tert-butanol/water mixture.26 In a typical experiment, 1.0 mg of o(LLA)10-GEM,1.0 mg of o(DLA)10-GEM, and 10.0 or 20.0 mg of PEG-b-PLA were dissolved in 1.0 mL of tert-butanol at 60 °C, followed by addition of 1.0 mL of prewarmed double-distilled water at 60 °C with vigorous mixing. The mixture was allowed to freeze in a dry ice/ethanol cooling bath at −70 °C for 2 h. Lyophilization was then performed on a VirTis AdVantage Pro freeze-dryer (SP Scientific, Gardiner, NY, USA) at −20 °C shelf inlet temperature for 72 h at 100 μbar throughout the experiment. The lyophilized cake was then rehydrated with 1.0 mL of Milli-Q water or 0.9% saline solution at 60 °C, centrifuged, and filtered using a 0.2 μm filter. The drug content in the supernatant was characterized by RP-HPLC. A similar method was employed to prepare o(LLA)6-GEM or o(LLA)10-GEM loaded PEG-b-PLA micelles.
Conversion of o(L+DLA)10-GEM Prodrugs in a CH3CN/PBS Mixture at pH 7.4.
A mixture of o(LLA)10-GEM (1.0 mg/mL) and o(DLA)10-GEM (1.0 mg/mL) was first dissolved in a 1:1 (v/v) mixture of CH3CN and PBS (10 mM, pH 7.4), placed in a 1.5 mL Eppendorf tube, and incubated at 37 °C in a temperature-adjusted water-bath (GCA Corporation, IL, USA).7 A 10 μL amount of solution was withdrawn at predetermined time points and diluted with 90 μL of CH3CN prior to RP-HPLC analysis. Similarly, conversion of o(LLA)6-GEM or o(LLA)10-GEM in a 1:1 (v/v) mixture of CH3CN and PBS (10 mM, pH 7.4) was measured by RPHPLC. First-order constants were calculated for the degradation kinetics of a mixture of o(LLA)10-GEM and o(DLA)10-GEM, o(LLA)10-GEM, or o(LLA)6-GEM. Each experiment was evaluated three times and reported with mean and standard deviation.
Stability of o(L+DLA)10-GEM-Loaded PEG-b-PLA Micelles in Water.
O(L+DLA)10-GEM-loaded PEG-b-PLA micelles were prepared (2.0 mg/mL) via lyophilization and rehydrated with Milli-Q water. The stability of o(L+DLA)10-GEM micelle in water was monitored at room temperature. A 10 μL amount of solution was withdrawn at predetermined time points and diluted with 90 μL of CH3CN prior to RP-HPLC analysis.
Stability of o(L+DLA)10-GEM-Loaded PEG-b-PLA Micelles, o(L+DLA)10-GEM, and GEM in Rat Plasma.
The stability of o(L+DLA)10-GEM-loaded PEG-b-PLA micelles, a mixture of o(L+DLA)10-GEM, or GEM in rat plasma was determined using heparinized Sprague−Dawley rat plasma (Innovative Research Inc., Novi, MI, USA). Frozen plasma samples were incubated at 37 °C for 5 min before use. A stock solution of o(L+DLA)10-GEM micelles or GEM was prepared in water at 2.0 mg/mL. A 100 μL amount of o(L+DLA)10-GEM micelles or GEM in water was added to 900 μL plasma samples to reach a final concentration of 0.2 mg/mL. For o(L+DLA)10-GEM, a stock solution was prepared in DMSO at 4.0 mg/mL. A 10 μL amount of the mixture in DMSO was added to 990 μL plasma samples to reach a final concentration of 0.04 mg/mL. Samples were incubated at 37 °C in a temperature-adjusted water-bath (GCA Corporation, IL, USA). At predetermined time intervals (0, 0.5, 1, 2, 4, and 24 h), 50 μL of plasma samples was withdrawn and diluted with 100 μL of acetonitrile containing 0.1% formic acid. Precipitated samples were centrifuged at 13 000 rpm for 10 min, and the resultant supernatants were analyzed by RP-HPLC.
In Vitro Release Studies.
O(L+DLA)10-GEM, o(LLA)10-GEM, or o(LLA)6-GEM loaded PEG-b-PLA micelles were diluted to 0.5 mg/mL in 10 mM PBS solution at pH 7.4. A Slide-A-Lyzer dialysis cassette with MWCO 20K (ThermoFisher, Waltham, MA, USA) was used to load 2.5 mL of diluted micelle solution. Three dialysis cassettes were placed in a 4 L PBS solution (10 mM, pH 7.4) on a Corning hot plate stirrer (Corning, NY, USA) at 37 °C. At 0, 1, 2, 3, 6, 9, 24, 48, 72, 120, 168, 216, 312, and 504 h, 100 μL of sample was withdrawn from the dialysis cassette, and the cassette was replenished with 100 μL of fresh PBS solution (10 mM, pH 7.4). The external medium was replaced with 4 L of fresh buffer at 2, 6, 24, 72, and 168 h to approximate sink conditions. Drug quantification of o(L+DLA)10-GEM, o(LLA)10-GEM, or o(LLA)6-GEM in PEG-b-PLA micelles was determined by RP-HPLC. Cumulative percent drug release was calculated, and drug release t1/2 was calculated according to a first-order rate equation.
In Vitro Cytotoxicity Studies.
The cytotoxicity of GEM or o(L+DLA)10-GEM micelles at 15% loading against human A549 non-small-cell lung cancer cells and PANC-1 pancreatic cancer cells was investigated by the CellTiter-Blue Cell Viability Assay (Promega, WI, USA). A549 cells were seeded into a 96-well plate at a seeding density of 1500 cells/100 μL/well and cultured in RPMI 1640 medium, or PANC-1 cells were seeded into a 96-well plate at a seeding density of 5000 cells/100 μL/well and cultured in DMEM, at 37 °C in a 5% CO2 incubator for 24 h. GEM or o(L+DLA)10-GEM micelles at 15% loading in PBS solution (10 mM, pH 7.4) were added into the wells to attain a final concentration of 1−10 000 nM. Cells cultured with diluted PBS in medium were used as controls. Drug-treated cells were placed in an incubator at 5% CO2 at 37 °C for 72 h. The medium in each well was aspirated, and 100 μL of 20% (v/v) CellTiter-Blue reagent in serum-free RPMI 1640 or DMEM was added, followed by incubation at 37 °C in a 5% CO2 atmosphere for 1.5 h. Fluorescence intensity was measured by a SpectraMax M2 plate reader (Molecular Devices, CA, USA) with excitation and emission at 560 and 590 nm, respectively. The half-maximal inhibitory drug concentration (IC50) was determined by using GraphPad Prism version 5.00 for Windows (GraphPad Software, CA, USA).
In vivo Antitumor Efficacy Studies.
All animal experiments were conducted under the protocol approved by Institutional Animal Care and Use Committee in University of Wisconsin−Madison in accordance with institutional and NIH guidance for the Care and Use of Laboratory Animals. Female athymic nude mice (6−8-week-old, 20−25 g each) were acquired from laboratory animal resources at School of Medicine and Public Health, University of Wisconsin− Madison. Mice were housed in ventilated cages with free access to water and food and acclimated for 1 week prior to tumor cell injection. A549 cells (2 × 106 cells in 100 μL of serum-free RPMI 1640 medium) were harvested from subconfluent cultures after trypsinization and were injected subcutaneously into the right flank of each mouse. When tumor volume had reached approximately 200−400 mm3, mice were randomly divided into 3 treatment groups (n = 3, 4/group): GEM at 10 mg/kg, o(L+DLA)10-GEM micelles at 5% loading at 10 mg/kg GEM equivalents, and saline control. Drugs were administered via tail vein for weekly injections for 3 weeks. Body weight and tumor volume were monitored over the course of study. Tumor volume was calculated using the formula V = (a × b2)/2, where V is tumor volume, a is tumor length, and b is tumor width.
Statistical Analysis.
Student’s t-test at the 5% significance level was used for statistical analysis. All data analyses were performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, CA, USA).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the NIH (R01AI01157). The purchase of the LCT was partially funded by NSF Award #CHE-9974839. We thank Prof. L. Yu (Department of Pharmaceutical Sciences and Chemistry, UW−Madison) for instrumentation (DSC and PXRD) and Prof. S. Hong (Department of Pharmaceutical Sciences, UW−Madison) for instrumentation (AFM).
The authors declare no competing financial interest
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.8b04205.
PXRD profiles and DSC thermograms of o(LLA)6-GEM, o(DLA)6-GEM, and a mixture of both prodrugs; 1H NMR and MS spectra of o(LLA)n-GEM and o(DLA)n-GEM prodrugs; HPLC chromatogram of polydisperse Bn-o(LA)n; and backbiting conversion of o(LLA)n-GEM prodrug (PDF)
REFERENCES
- (1).Slager J; Domb AJ Biopolymer Stereocomplexes. Adv. Drug Delivery Rev. 2003, 55, 549–583. [DOI] [PubMed] [Google Scholar]
- (2).Ikada Y; Jamshidi K; Tsuji H; Hyon SH Stereocomplex Formation between Enantiomeric Poly(lactides). Macromolecules 1987, 20, 904–906. [Google Scholar]
- (3).Soleymani Abyaneh H; Vakili MR; Shafaati A; Lavasanifar A Block Copolymer Stereoregularity and Its Impact on Polymeric Micellar Nanodrug Delivery. Mol. Pharmaceutics 2017, 14, 2487–2502. [DOI] [PubMed] [Google Scholar]
- (4).Hennink WE; De Jong SJ; Bos GW; Veldhuis TFJ; Van Nostrum CF Biodegradable Dextran Hydrogels Crosslinked by Stereocomplex Formation for the Controlled Release of Pharmaceutical Proteins. Int. J. Pharm 2004, 277, 99–104. [DOI] [PubMed] [Google Scholar]
- (5).Kang N; Perron M È Prud’Homme, R. E.; Zhang, Y.; Gaucher, G.; Leroux, J. C. Stereocomplex Block Copolymer Micelles: Core-Shell Nanostructures with Enhanced Stability. Nano Lett. 2005, 5, 315–319. [DOI] [PubMed] [Google Scholar]
- 6.) Kersey FR; Zhang G; Palmer GM; Dewhirst MW; Fraser CL Stereocomplexed Poly (lactic acid)-Poly (ethylene glycol) Nanoparticles with Dual-Emissive Boron Dyes for Tumor Accumulation. ACS Nano 2010, 4, 4989–4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tam YT; Gao J; Kwon GS Oligo(lactic acid)n-Paclitaxel Prodrugs for Poly(ethylene glycol)-block-Poly(lactic acid) Micelles: Loading, Release, and Backbiting Conversion for Anticancer Activity. J. Am. Chem. Soc 2016, 138, 8674–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mini E; Nobili S; Caciagli B; Landini I; Mazzei T Cellular Pharmacology of Gemcitabine. Ann. Oncol. 2006, 17, v7–v12. [DOI] [PubMed] [Google Scholar]
- (9).Pili B; Bourgaux C; Meneau F; Couvreur P; Ollivon M Interaction of an Anticancer Drug, Gemcitabine, with PhosphoLipid Bilayers. J. Therm. Anal. Calorim 2009, 98, 19. [Google Scholar]
- (10).Moysan E; Bastiat G; Benoit JP Gemcitabine Versus Modified Gemcitabine: a Review of Several Promising Chemical Modifications. Mol. Pharmaceutics 2013, 10, 430–444. [DOI] [PubMed] [Google Scholar]
- (11).Abbruzzese JL; Grunewald R; Weeks EA; Gravel D; Adams T; Nowak B; Mineishi S; Tarassoff P; Satterlee W; Raber MN A Phase I Clinical, Plasma, and Cellular pharmacology Study of Gemcitabine. J. Clin. Oncol 1991, 9, 491–498. [DOI] [PubMed] [Google Scholar]
- (12).Immordino ML; Brusa P; Rocco F; Arpicco S; Ceruti M; Cattel L Preparation, Characterization, Cytotoxicity and Pharmacokinetics of Liposomes Containing Lipophilic Gemcitabine Prodrugs. J. Controlled Release 2004, 100, 331–346. [DOI] [PubMed] [Google Scholar]
- (13).Brusa P; Immordino ML; Rocco F; Cattel L Antitumor Activity and Pharmacokinetics of Liposomes Containing Lipophilic Gemcitabine Prodrugs. Anticancer Res. 2007, 27, 195–199. [PubMed] [Google Scholar]
- (14).Wang Y; Fan W; Dai X; Katragadda U; Mckinley D; Teng Q; Tan C Enhanced Tumor Delivery of Gemcitabine via PEG-DSPE/TPGS Mixed Micelles. Mol. Pharmaceutics 2014, 11, 1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Couvreur P; Stella B; Reddy LH; Hillaireau H; Dubernet C; Desmaele D; Lepetre-Mouelhi S; Rocco F; Dereuddre-Bosquet N; Clayette P; Rosilio V Squalenoyl Nanomedicines as Potential Therapeutics. Nano Lett. 2006, 6, 2544–2548. [DOI] [PubMed] [Google Scholar]
- (16).Réjiba S; Reddy LH; Bigand C; Parmentier C; Couvreur P; Hajri A Squalenoyl Gemcitabine Nanomedicine Overcomes the Low Efficacy of Gemcitabine Therapy in Pancreatic Cancer. Nanomedicine 2011, 7, 841–849. [DOI] [PubMed] [Google Scholar]
- (17).De Jong SJ; van Dijk-Wolthuis WNE; Kettenes-Van den Bosch JJ; Schuyl PJW; Hennink WE Monodisperse Enantiomeric Lactic Acid Oligomers: Preparation, Characterization, and Stereocomplex Formation. Macromolecules 1998, 31, 6397–6402. [Google Scholar]
- (18).Takizawa K; Nulwala H; Hu J; Yoshinaga K; Hawker CJ Molecularly Defined (L)-Lactic Acid Oligomers and Polymers: Synthesis and Characterization. J. Polym. Sci., Part A: Polym. Chem 2008, 46, 5977–5990. [Google Scholar]
- (19).Portinha D; Boué F; Bouteiller L; Carrot G; Chassenieux C; Pensec S; Reiter G Stable Dispersions of Highly Anisotropic Nanoparticles Formed by Cocrystallization of Enantiomeric Diblock Copolymers. Macromolecules 2007, 40, 4037–4042. [Google Scholar]
- (20).Sun L; Pitto-Barry A; Kirby N; Schiller TL; Sanchez AM; Dyson MA; Sloan J; Wilson NR; O’Reilly RK; Dove AP Structural Reorganization of Cylindrical Nanoparticles Triggered by Polylactide Stereocomplexation. Nat. Commun 2014, 5, 5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Xiao L; Xiong X; Sun X; Zhu Y; Yang H; Chen H; Gan L; Xu H; Yang X Role of Cellular Uptake in the Reversal of Multidrug Resistance by PEG-b-PLA Polymeric Micelles. Biomaterials 2011, 32, 5148–5157. [DOI] [PubMed] [Google Scholar]
- (22).Reck M; Von Pawel J; Zatloukal PV; Ramlau R; Gorbounova V; Hirsh V; Leighl N; Mezger J; Archer V; Moore N; Manegold C Overall Survival with Cisplatin−Gemcitabine and Bevacizumab or Placebo as First-Line Therapy for Nonsquamous Non-Small-Cell Lung Cancer: Results from a Randomised Phase III Trial (AVAiL). Ann. Oncol 2010, 21, 1804–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Burris HA 3rd; Moore MJ; Andersen J; Green MR; Rothenberg ML; Modiano MR; Cripps MC; Portenoy RK; Storniolo AM; Tarassoff P; Nelson R Improvements in Survival and Clinical Benefit with Gemcitabine as First-Line Therapy for Patients with Advanced Pancreas Cancer: a Randomized Trial. J. Clin. Oncol 1997, 15, 2403–2413. [DOI] [PubMed] [Google Scholar]
- (24).Pili B; Reddy LH; Bourgaux C; Lepetre-Mouelhi S; Desmaele D; Couvreur P Liposomal Squalenoyl-Gemcitabine: Formulation, Characterization and Anticancer Activity Evaluation. Nanoscale 2010, 2, 1521–1526. [DOI] [PubMed] [Google Scholar]
- (25).Reddy LH; Marque PE; Dubernet C; Mouelhi SL; Desmaele D; Couvreur P Preclinical Toxicology (Subacute and Acute) and Efficacy of a New Squalenoyl Gemcitabine Anticancer Nanomedicine. J. Pharmacol. Exp. Ther 2008, 325, 484–490. [DOI] [PubMed] [Google Scholar]
- (26).Fournier E; Dufresne MH; Smith DC; Ranger M; Leroux JC A Novel One-Step Drug-Loading Procedure for Water-Soluble Amphiphilic Nanocarriers. Pharm. Res 2004, 21, 962–968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.