

Synopsis
Fanconi anemia (FA) is a DNA repair disorder associated with high risk of cancer and bone marrow failure. Patients with FA may present with certain dysmorphic features such as radial ray abnormalities, short stature, typical facies, bone marrow failure, or certain solid malignancies. Some patients may be recognized due to exquisite sensitivity after exposure to cancer therapy. FA is diagnosed by increased chromosomal breakage after exposure to clastogenic agents. It follows both autosomal recessive and X-linked inheritance depending on the underlying genomic alteration(s). Nineteen genes encode proteins comprising the Fanconi DNA damage response complex. To date, pathogenic variants in at least 15 FA pathway genes and at least 5 related genes have been identified as causative of FA. Recognizing patients with FA is important for therapeutic decisions, genetic counseling, and optimal clinical management.
Keywords: Bone Marrow Failure, Myelodysplastic Syndrome, Leukemia, Exquisite Therapeutic Sensitivity, DNA repair, BRCA1/2, Fanconi complex, Fanconi anemia, head and neck squamous cell cancer
INTRODUCTION TO FANCONI ANEMIA
Fanconi anemia (FA, MIM 607139) is a rare, cancer-prone inherited bone marrow failure syndrome with a wide range of clinical presentations including radial ray anomalies, short stature, microcephaly, café au lait spots, and other medical problems. The condition is eponymous for Dr. Guido Fanconi, who originally described the syndrome in 1927. At present there are more than 2000 patients reported in the literature.(1) Advances in supportive care including hematopoietic cell transplantation (HCT) have improved the lifespan for patients with FA but cancer, HCT-related complications, and other complex medical problems remain as significant causes of mortality. Herein we review the underlying biology and clinical manifestations of FA as well as the current recommendations for cancer surveillance and management.
The Biology of Fanconi Anemia
FA is a chromosomal instability disorder caused predominantly by autosomal recessive inheritance of pathogenic variants in key components of the DNA damage response(2, 3). There is one gene, FANCB, associated with X-linked recessive inheritance (4). Germline mutations (i.e., pathogenic variants) in at least 22 genes are associated with FA. There is consensus in the field regarding 18 genes, (FANCA, B, C, D1, D2, E, F, G, I, J, L, N, P, Q, T, U, V and W) (4–26) (Table 1) (27). An additional four genes (FANCM, FANCO, FANCR and FANCS) are considered FA-like, as they have not been described in patients with bone marrow failure (25, 28, 29). FANCM is not considered a bonafide FA gene, as only it has only occurred in a patient also reported to also harbor bi-allelic FANCA mutations. Biallelic pathogenic variants in FANCA are the most common cause of FA, with 65% of patients harboring mutations in this gene. This is followed by FANCC (14%) and FANCG (9%), with the remaining 14% covered by the other genes (30). (Table 1). The proteins of the FA pathway create a biochemical circuit that function in DNA repair, DNA damage response, and other cellular processes. As DNA is replicated, nucleotide incorporation and processing of the replication fork are prone to errors including wrong nucleotides, damaged bases within the DNA, abnormal DNA-protein complexes, creation of DNA-RNA hybrids (R-loops) and aberrant DNA structures, such as G quadraplexes (31). The specific role of proteins in the Fanconi pathway is removal of DNA interstrand cross-links (ICLs). Interstrand cross-links may arise from endogenous and exogenous compounds such as aldehydes and platinum drugs, respectively. Interstrand cross-links prevent DNA strand separation and can act to block the DNA replication process and/or transcription. These aberrant DNA strands, if left intact, promote cell death (32).
Table 1.
The Biology of Fanconi Anemia
| Fanconi Genes | Inheritance Patterns | Classic Triad (bone marrow failure, chromosomal fragility, malformations) | Number of patients with specific mutation and cancer reported by NCI (without history of transplant) n=163 patients | Adult Onset Cancers for Carriers | Key References |
|---|---|---|---|---|---|
| FANCA | AR | Yes | 11 / 70 | Apostolu et al., Nature Genetics 1996 | |
| FANCB | X-Linked Recessive | Yes | 1 | Meetei et al., Nature Genetics, 1996 | |
| FANCC | AR | Yes | 3 / 19 | Strathdee et al., Nature, 1992 | |
| FANCD1/BRCA2 | AR / AD | Yes | 2 / 3 | Breast, Ovarian, Prostate, Pancreatic, Skin | Timmers et al., Molecular Cell, 2001; Kauff et al. NEJM, 2002, Pritchard et al. NEJM, 2016, |
| FANCD2 | AR | Yes | 7 | Timmers et al., Molecular Cell, 2001 | |
| FANCE | AR | Yes | – | de Winter et al., AJHG, 2000 | |
| FANCF | AR | Yes | 2 | de Winter et al., AJHG, 2000 | |
| FANCG | AR | Yes | 1 / 7 | Garcia-Higuera et al., MCB, 1999 | |
| FANCI | AR | Yes | 2 / 3 | Dorsman et al., Cell, 2007; Sims et al., Nature Structural Molecular Biology, Smogorzewska et al., Cell, 2007 | |
| FANCJ/BRIP1 | AR / AD | Yes | – | Ovarian | Cantor et al., Cell, 2001;Tung et al., Nature Reviews, 2016 |
| FANCL | AR | Yes | – | Meetei et al., Nature Genetics, 2003 | |
| FANCN/PALB2 | AR / AD | Yes | – | Breast, Pancreatic, Metastatic Prostate | Xia et al., Molecular Cell, 2006; Tung et al., Nature Reviews, 2016 |
| FANCP/SLX4 | AR | Yes | – | Svendsen et al., Cell, 2009 | |
| FANCQ/ERCC4 | AR | Yes | – | Bogliolo et al., AJHG, 2013 | |
| FANCT/UBE2T | AR | Yes | – | Zhang et al., Genome Research 2000; Hira et al., AJHG 2015 | |
| FANCU/XRCC2 | AR | Yes | – | Jones et al., Genomics, 1995; Shamselden et al. JMG, 2012 | |
| FANCV/REV7 | AR | Yes | – | Li and Benezra et al. Science 1996; Bluteau et al. JCI, 2016 | |
| FANCW/RFWD3 | AR | Yes | – | Knies et al. JCI, 2017 | |
| FANCM | AR | No | – | Meetei et al., Nature Genetics,2005 | |
| FACNO/RAD51C | AR / AD | No | – | Ovarian / Metastatic Prostate | Vaz et al. Nature Genetics, 2010 ; Tung et al., Nature Reviews, 2016 |
| FANCR/RAD51 | AR / AD | No | 1 | Ovarian / Metastatic Prostate | Ameziane et al. Nature Communications, 2015 ; Tung et al., Nature Reviews, 2016 |
AR= autosomal recessive, AD=autosomal dominant
Clinical Manifestations
Clinical characteristics of FA may vary significantly from patient to patient. Typical FA findings include radial ray abnormalities with missing or unusual thumbs, café au lait macules, and typical “Fanconi” facies. Abnormal thumbs are an important differentiating feature of FA and differentiate this syndrome from thrombocytopenia absent radius syndrome (TAR) in which thumbs are present(33). Other significant FA-associated anomalies include: kidney and urinary tract malformations, vertebral anomalies, esophageal atresia, hydrocephalus, short stature, and small eyes. Almost all organ systems may be involved with the highest number of congenital malformations associated with the FANCD1/BRCA2 genotype(34). Clinical features of FA may significantly overlap with those of the VACTERL-H association: VACTERL-H is the acronym for vertebral anomalies, anal atresia, cardiac anomalies, trachea-esophageal fistula, esophageal or duodenal atresia, renal structural anomalies, limb deformities and hydrocephalus(35). Additional manifestations of FA may include metabolic abnormalities, endocrinopathies, and hearing impairments. While patients with FA may exhibit the VACTERL-H phenotype, it is not specific to FA. This led Alter and Giri to propose PHENOS (Pigmentation, small Head, small Eyes, central Nervous system [not hydrocephalus], Otology, and Short stature) as an acronym that includes the major dysmorphic features of FA as an aid to identify FA patients within the VACTERL-H phenotype (36).
Patients with FA are at very high risk of bone marrow failure (BMF), myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), head and neck squamous cell carcinoma (HNSCC), and other malignancies. In some patients, BMF or cancer may be the first presenting sign of FA. It should be suspected in patients who have higher than expected toxicities related to cancer treatment.
Diagnosing Fanconi Anemia
FA is diagnosed by chromosome breakage studies on patient-derived cells. In this assay, the patient’s peripheral blood T lymphocytes are exposed to diepoxybutane or mitomycin C and the number of resultant abnormal chromosomes are counted. False negatives may occur in samples obtained from individuals with hematopoietic somatic mosaicism and in such instances testing of skin fibroblasts is required to diagnose FA (37).
Genetic testing for FA now includes multiplex (i.e., gene panel) testing, as well as whole exome or whole genome sequencing. Identification of the causative pathogenic variants is helpful in confirming the diagnosis of FA and essential for genetic counseling of the patient and their family members. The availability of multiplex genetic testing has led to widespread testing of patients in whom FA may not have been considered. In such instances, chromosomal breakage studies have been particularly important in confirming whether the genetic variants are contributory to disease.
CONSEQUENSES AND MANAGEMENT OF HEMATOPOIETIC STEM CELL ATTRITION
Bone marrow failure and leukemia
BMF in FA is attributed to attrition of the hematopoietic stem and progenitor cell compartment due to elevated DNA damage response and apoptosis.(38) The bone marrow of patients with FA has been shown to be proinflammatory; specifically, TNF-α may contribute to apoptosis of FA hematopoietic stem cells (HSCs) through activation of the extrinsic apoptotic pathway.(38) Moreover, functional studies have illustrated that aberrantly activated oligoclonal T-cell populations suppress hematopoiesis by releasing cytokines (including INF-γ and TNF-α). (39, 40) This cascade of events is cytotoxic to HSCs and subsequently to lymphoid populations. (41) This is thought to result in a stem cell compartment favoring the evolution of somatically mutated cells with the capacity to resist the attack of T-cells and the evolution of covert leukemic clones (Figure 1) (42–45).
Figure 1.

Fanconi Triple Assessment: Patients being evaluated for Fanconi anemia require a triple assessment including: (A) history/physical, (B) clastogenic testing or chromosomal breakage testing with diepoxybutane +/− mitomycin C, and (C) genetic testing.
In addition to adverse effects of INF-γ, TNF-α and other cytokines in FA patients, there is evidence that BMF may be exacerbated by endogenous aldehyde-induced toxicity and /or DNA damage-induced p53 activation, both of which result in HSC attrition.(46, 47) One report found that FA patients with ALDH2 mutations have accelerated progression of bone marrow failure and murine models harboring ALDH2 mutations independent of FA genetics have exhibited bone marrow failure. (48)
In the absence of typical congenital anomalies, cytopenias may be a presenting sign of FA. Additionally, a clue to an IBMFS may also be elevated hemoglobin F and high mean corpuscular volume for age without another cause. Most patients with FA manifest clinically significant cytopenias during their lifetimes. The cumulative incidence of severe BMF of 70% by age 50 and a peak hazard rate of 4% at 12 to 15 years of age (49). The age of BMF onset varies amongst patients with FA, even among siblings. (50)
Patients with FA and BMF should be monitored for the development of MDS and progression to AML by following both cytogenetic and morphologic abnormalities. Long term studies of FA have shown that the risk of developing MDS or AML within three years of observing an abnormal clone was roughly 1 in 3 (35%) compared to 1 in 30 (3.3%) for those without an identifiable clone.(49, 51) Cytogenetic findings in the bone marrow samples of FA patients may reveal recurring chromosomal changes of 1, 3 and 7 and these findings need to be followed closely given the risk of AML in FA. A notable finding in bone marrow of FA patients is the presence of aberrations of chromosome 3 (52, 53). In these patients with identifiable chromosome 3 abnormalities, the 3-year risk of MDS/AML was 90% and 17% respectively. In general, the classification scheme for AML is evolving and becoming more molecularly centric.(54) How applicable this evolving schema will be to FA patients is still to be determined and needs to be further analyzed in larger numbers of FA patients.
MDS and AML occur in a significant percentage of patients with Fanconi anemia. (51) In a recent report from Alter et al., interrogating the National Cancer Institute’s registry between 1945-2014, one hundred and thirty families have been reported to date with FA, with approximately one hundred and sixty individuals who are presently living. Leukemia had a cumulative incidence of 5% in Fanconi anemia by the age of 30. The cumulative incidence of MDS in FA patients by the age of 50 was 50% (CI 35-65%).(49)
Of note, AML is significantly more common in FA than acute lymphoblastic leukemia and lymphomas. The latter hematopoietic malignancies have been reported in patients with FA harboring FANCD1/BRCA2 gene mutations (51, 52, 55–57).
Clinical management
In general, patients with BMF require treatment for when BMF becomes severe, which is generally defined as hemoglobin <8 g/dL, platelets <30,000/mm3, absolute neutrophil count <500/mm3.(2) The decision to undergo hematopoietic cell transplant (HCT) is a serious one, which requires a multidisciplinary approach and assessment. The complexities of decision making in the context of FA-associated co-morbidities led to the development of an interesting mathematical decision model to estimate event free-survival (EES) conditional on age and per-year cause specific hazard rates, based on the assumption that bone marrow transplant eliminates the risk of bone marrow failure and acute myeloid leukemia but not the risk of solid tumors (2, 49, 58). This approach provides perspective in deciding when or if a patient with FA should undergo HCT and suggests that transplantation at a younger age offers a benefit that is not as clear in older patients.
Non-myeloablative HCT conditioning regimens are recommended for patients with FA undergoing HCT for BMF without MDS or AML due to their exquisite sensitivity to chemotherapy and ionizing radiation(56, 59–64). Matched sibling donors are recommended, when available, and mutation identification and testing is essential in this context. It is important for patients and their families to understand that HCT can cure BMF, but it does not cure non-hematopoietic complications of FA (49).
All patients with FA in whom HCT is considered should be evaluated and treated at a center specializing in FA(2). This is important to ensure the optimal supportive care and clinical expertise in the management of the complex FA co-morbidities. Patients with FA who evolved to MDS or AML require a highly specialized approach due to their sensitivity to standard cancer therapeutics (2, 65).
Androgens are an option for BMF in patients with FA who cannot undergo HCT (66, 67). There are multiple well recognized side effects of androgens including but not limited to: virilization, premature closure of growth plates, behavioral changes, elevated liver enzymes, hepatic adenomas, hepatocellular carcinoma, peliosis hepatis, acne and hypertension. Half to three-quarters of patients respond to androgens within three months. Androgens do not prevent progression to AML, which once developed, may increase the risks associated with transplant and when patients are older additional viral infections may have been acquired which may impact transplant outcome.(68)
SOLID MALIGNANCIES
FA patients are at very high risk of solid tumors. In the NCI’s prospective longitudinal cohort study of 130 families including 163 patients, 21/163 (12%) patients were diagnosed with solid tumors: HNSCC n=10, vulva n=3, esophagus n=2, brain n=2, anus n=1, lung n=1, cervix n=1 and breast n=1 and an additional 35/163 (21%) were diagnosed including squamous cell cancer (SCC) n=23 and basal cell cancer (BCC) n=12.(49) The observed to expected ratio, for any malignancy in non-transplanted patients for FA was nineteen. FA patients having undergone hematopoietic stem cell transplant, n=63/163, the frequency of solid tumors was higher at 33 (51%): HNSCC n=5, larynx n=1, vulva n=2, brain n=1, thyroid n=2, BCC n=9 and SCC n=12. The effect of HSCT on increased cancer rates is reflected in the higher observed to expected ratios for transplanted vs. the non-transplanted FA patients 55 versus 19. (49) According to Alter et al. the cumulative incidence of solid tumors reported in FA patients is roughly 20% by the age of 65. As FA patients exhibit extreme sensitivity to alkylating and radiation therapies, there continue to be significant challenges in treating patients and avoiding second malignancies.
CANCER SCREENING AND RISK REDUCTION
The guidelines for diagnosis management of FA are described in detail at http://fanconi.org/index.php/publications/guidelines. Fanconi anemia patient require multidisciplinary care and self-directed screening. Proper health maintenance includes coordination of care with ear, nose and throat (ENT) physicians, hematologists, oncologists, gastroenterologists, orthopedic surgeons, gynecologists (females), endocrinologists, dermatologists, dentists and pediatric developmental physicians.
HNSCC is the most common solid tumor in FA patients. Approximately 1 in 7 (14%) of patients with FA who survive to the age 40 will be diagnosed with HNSCC during their lifetime.(49, 51) Surveillance should begin at age 10 years, which is based on literature reports of the earliest age of diagnosis with head and neck cancer. The sites at risk for the development of HNSCC include all areas of the upper aerodigestive tract. Therefore, all mucosal surfaces of the head and neck should be examined. This exam should also include the proximal and distal oropharynx and should include appropriate equipment such as a transoral mirror and flexible fiberoptic laryngoscope. This exam should occur every six months and suspicious lesions should be biopsied. In instances where a premalignant or malignant lesion is identified it should be appropriately treated and screening should increase to every 2-3 months Patients need to be diligent and perform self-exam bringing any suspicious findings to their physicians’ attention. (http://fanconi.org/index.php/publications/guidelines_for_diagnosis_and_management).
Fanconi patients are at increased risk of skin cancers, thus individuals should perform routine skin checks for suspicious lesions following the ABCD acronym for nevus inspection (asymmetry, bleeding/border changes, color changes and difference in diameter). Patient should also protect their skin with hats, clothing and sunscreen. FA patients require application of sunscreen with an SPF of at least 30 or higher. Individuals should see their dermatologist once a year and strive to maintain adequate vitamin D levels through supplements if necessary.
In regard to hematologic monitoring, most physicians agree that a complete blood count (CBC) and bone marrow aspirate and biopsy are recommended at diagnosis. The bone marrow evaluation should be repeated annually. The CBC should be monitored more frequently for proactive management of cytopenias and myelodysplastic syndrome (Figure 2) (65)
Figure 2.
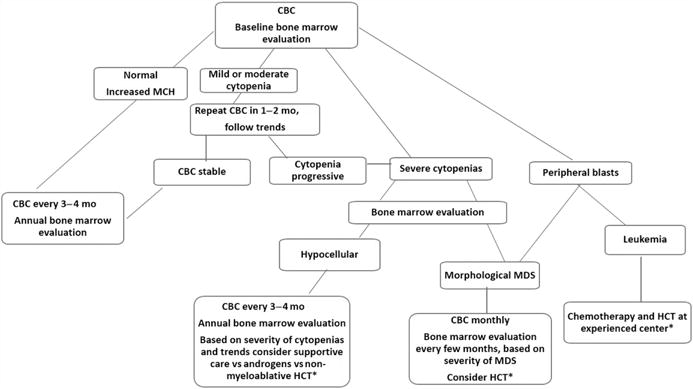
Screening for hematologic abnormalities in Fanconi anemia.
ˆ Persistent drop or rise in blood counts without apparent cause warrants bone marrow evaluation
ˆ ˆ Specific clonal abnormalities may warrant immediate treatment intervention or closer monitoring
Adapted from Alter BP, Hanenberg H, Kinsey S, et al. Hematologic Abnormalities in Patients with Fanconi Anemia. In: Hays L, Frohnmayer D, Frohnmayer L, et al., editors. Fanconi Anemia: Guidelines for Diagnosis and Management, 4th edition. Eugene: Fanconi Anemia Research Fund, Inc.: 2014; with permission.
SUMMARY AND DISCUSSION
Fanconi anemia is a DNA damage syndrome. FA in inherited in an autosomal recessive inheritance disease except for patients with FANCB mutations, in which case the disease is X-linked. Multidisciplinary care for Fanconi anemia patients is required.(65) The field’s understanding of FA continues to evolve with greater than 20 genes described to play a role in Fanconi biology, a description of the environmental milieu of FA patient’s bone marrow, tailored non-myeloablative bone marrow transplant regimens and candidate therapeutics.
At present HCT continues to be the only curative treatment for hematopoietic disease in FA patients.
Chromosomal breakage studies are the gold standard testing assay and there is a continued need for genotypic/phenotypic description and studies. Triple assessment including clinical characteristics, chromosomal breakage studies and molecular testing provide the greatest context for advancing our understanding of FA and better understanding atypical findings. Studies need to be powered well in order to make valid conclusions from integrated genotype/phenotype analyses. Moreover, uniform annotation of variant calling following rigorous classification guidelines will allow for meaningful clinical translation. At present, identification of FANC complex carriers has broad clinical relevance depending on the specific gene and variant detected. In some instances, such as FANCD1, PALB2, RAD51C, RAD51, and BRIP1 heterozygous carriers are at risk for adult onset cancers but the extent to which heterozygous carriers of pathogenic variants in other FA genes are at risk is not known and is an area of active study. Carriers should be counseled for reproductive risks as well.(69–71) The continued international effort focused on comprehensive care for Fanconi anemia patients is crucial given the rarity of this disease and for further advancements in caring for patients to be made.
Key Points (3–5).
Fanconi anemia is a DNA damage repair syndrome caused by pathogenic variants in key components of the Fanconi DNA repair complex.
Patients with Fanconi anemia are at very high risk of bone marrow failure, myelodysplastic syndrome, leukemia, head and neck squamous cell carcinoma, and other malignancies.
Treatment for patients with Fanconi anemia and cancer must be carefully tailored due to exquisite sensitivity to ionizing radiation and alkylating drugs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alter BP. Fanconi anemia and the development of leukemia. Best Pract Res Clin Haematol. 2014;27(3-4):214–21. doi: 10.1016/j.beha.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fanconi Anemia Reserach Fund. Guidelines for Diagnosis and Management, 2008. Eugene OR: Fanconi Anemia Reserach Fund, Inc; 2008. Fanconi Anemia. [Google Scholar]
- 3.Perona G, Cetto GL, Bernardi F, Todeschini G, D’Andrea F. Fanconi’s anaemia in adults: study of three families. Haematologica. 1977;62(6):615–28. [PubMed] [Google Scholar]
- 4.Meetei AR, Levitus M, Xue Y, Medhurst AL, Zwaan M, Ling C, et al. X-linked inheritance of Fanconi anemia complementation group B. Nat Genet. 2004;36(11):1219–24. doi: 10.1038/ng1458. [DOI] [PubMed] [Google Scholar]
- 5.Fanconi anaemia/Breast cancer c. Positional cloning of the Fanconi anaemia group A gene. Nat Genet. 1996;14(3):324–8. doi: 10.1038/ng1196-324. [DOI] [PubMed] [Google Scholar]
- 6.Timmers C, Taniguchi T, Hejna J, Reifsteck C, Lucas L, Bruun D, et al. Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol Cell. 2001;7(2):241–8. doi: 10.1016/s1097-2765(01)00172-1. [DOI] [PubMed] [Google Scholar]
- 7.Strathdee CA, Gavish H, Shannon WR, Buchwald M. Cloning of cDNAs for Fanconi’s anaemia by functional complementation. Nature. 1992;358(6385):434. doi: 10.1038/358434a0. [DOI] [PubMed] [Google Scholar]
- 8.de Winter JP, Leveille F, van Berkel CG, Rooimans MA, van Der Weel L, Steltenpool J, et al. Isolation of a cDNA representing the Fanconi anemia complementation group E gene. Am J Hum Genet. 2000;67(5):1306–8. doi: 10.1016/s0002-9297(07)62959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Winter JP, Rooimans MA, van Der Weel L, van Berkel CG, Alon N, Bosnoyan-Collins L, et al. The Fanconi anaemia gene FANCF encodes a novel protein with homology to ROM. Nat Genet. 2000;24(1):15–6. doi: 10.1038/71626. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Higuera I, Kuang Y, Naf D, Wasik J, D’Andrea AD. Fanconi anemia proteins FANCA, FANCC, and FANCG/XRCC9 interact in a functional nuclear complex. Mol Cell Biol. 1999;19(7):4866–73. doi: 10.1128/mcb.19.7.4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorsman JC, Levitus M, Rockx D, Rooimans MA, Oostra AB, Haitjema A, et al. Identification of the Fanconi anemia complementation group I gene, FANCI. Cell Oncol. 2007;29(3):211–8. doi: 10.1155/2007/151968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sims AE, Spiteri E, Sims RJ, 3rd, Arita AG, Lach FP, Landers T, et al. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat Struct Mol Biol. 2007;14(6):564–7. doi: 10.1038/nsmb1252. [DOI] [PubMed] [Google Scholar]
- 13.Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129(2):289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105(1):149–60. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- 15.Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet. 2003;35(2):165–70. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- 16.Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22(6):719–29. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 17.Svendsen JM, Smogorzewska A, Sowa ME, O’Connell BC, Gygi SP, Elledge SJ, et al. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell. 2009;138(1):63–77. doi: 10.1016/j.cell.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet. 2013;92(5):800–6. doi: 10.1016/j.ajhg.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang QH, Ye M, Wu XY, Ren SX, Zhao M, Zhao CJ, et al. Cloning and functional analysis of cDNAs with open reading frames for 300 previously undefined genes expressed in CD34+ hematopoietic stem/progenitor cells. Genome Res. 2000;10(10):1546–60. doi: 10.1101/gr.140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hira A, Yoshida K, Sato K, Okuno Y, Shiraishi Y, Chiba K, et al. Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. Am J Hum Genet. 2015;96(6):1001–7. doi: 10.1016/j.ajhg.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones NJ, Zhao Y, Siciliano MJ, Thompson LH. Assignment of the XRCC2 human DNA repair gene to chromosome 7q36 by complementation analysis. Genomics. 1995;26(3):619–22. doi: 10.1016/0888-7543(95)80187-q. [DOI] [PubMed] [Google Scholar]
- 22.Shamseldin HE, Elfaki M, Alkuraya FS. Exome sequencing reveals a novel Fanconi group defined by XRCC2 mutation. J Med Genet. 2012;49(3):184–6. doi: 10.1136/jmedgenet-2011-100585. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Benezra R. Identification of a human mitotic checkpoint gene: hsMAD2. Science. 1996;274(5285):246–8. doi: 10.1126/science.274.5285.246. [DOI] [PubMed] [Google Scholar]
- 24.Bluteau D, Masliah-Planchon J, Clairmont C, Rousseau A, Ceccaldi R, Dubois d’Enghien C, et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest. 2016;126(9):3580–4. doi: 10.1172/JCI88010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet. 2005;37(9):958–63. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knies K, Inano S, Ramirez MJ, Ishiai M, Surralles J, Takata M, et al. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J Clin Invest. 2017;127(8):3013–27. doi: 10.1172/JCI92069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bogliolo M, Surralles J. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev. 2015;33:32–40. doi: 10.1016/j.gde.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 28.Vaz F, Hanenberg H, Schuster B, Barker K, Wiek C, Erven V, et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet. 2010;42(5):406–9. doi: 10.1038/ng.570. [DOI] [PubMed] [Google Scholar]
- 29.Ameziane N, May P, Haitjema A, van de Vrugt HJ, van Rossum-Fikkert SE, Ristic D, et al. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat Commun. 2015;6:8829. doi: 10.1038/ncomms9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong H, Nebert DW, Bruford EA, Thompson DC, Joenje H, Vasiliou V. Update of the human and mouse Fanconi anemia genes. Hum Genomics. 2015;9:32. doi: 10.1186/s40246-015-0054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodriguez A, D’Andrea A. Fanconi anemia pathway. Curr Biol. 2017;27(18):R986–R8. doi: 10.1016/j.cub.2017.07.043. [DOI] [PubMed] [Google Scholar]
- 32.Noll DM, Mason TM, Miller PS. Formation and repair of interstrand cross-links in DNA. Chem Rev. 2006;106(2):277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaw S, Oliver RA. Primary heamorrhagic thrombocythaemia. Proc R Soc Med. 1958;51(9):768–72. [PMC free article] [PubMed] [Google Scholar]
- 34.Savage SA, Dufour C. Classical inherited bone marrow failure syndromes with high risk for myelodysplastic syndrome and acute myelogenous leukemia. Semin Hematol. 2017;54(2):105–14. doi: 10.1053/j.seminhematol.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 35.Bluteau D, Masliah-Planchon J, Clairmont C, Rousseau A, Ceccaldi R, d’Enghien CD, et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest. 2017;127(3):1117. doi: 10.1172/JCI92946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alter BP, Giri N. Thinking of VACTERL-H? Rule out Fanconi Anemia according to PHENOS. Am J Med Genet A. 2016;170(6):1520–4. doi: 10.1002/ajmg.a.37637. [DOI] [PubMed] [Google Scholar]
- 37.Alter BP, Joenje H, Oostra AB, Pals G. Fanconi anemia: adult head and neck cancer and hematopoietic mosaicism. Arch Otolaryngol Head Neck Surg. 2005;131(7):635–9. doi: 10.1001/archotol.131.7.635. [DOI] [PubMed] [Google Scholar]
- 38.Ceccaldi R, Sarangi P, D’Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016;17(6):337–49. doi: 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- 39.Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108(8):2509–19. doi: 10.1182/blood-2006-03-010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Risitano AM, Maciejewski JP, Green S, Plasilova M, Zeng W, Young NS. In-vivo dominant immune responses in aplastic anaemia: molecular tracking of putatively pathogenetic T-cell clones by TCR beta-CDR3 sequencing. Lancet. 2004;364(9431):355–64. doi: 10.1016/S0140-6736(04)16724-X. [DOI] [PubMed] [Google Scholar]
- 41.Sloand E, Kim S, Maciejewski JP, Tisdale J, Follmann D, Young NS. Intracellular interferon-gamma in circulating and marrow T cells detected by flow cytometry and the response to immunosuppressive therapy in patients with aplastic anemia. Blood. 2002;100(4):1185–91. doi: 10.1182/blood-2002-01-0035. [DOI] [PubMed] [Google Scholar]
- 42.Dufour C, Corcione A, Svahn J, Haupt R, Battilana N, Pistoia V. Interferon gamma and tumour necrosis factor alpha are overexpressed in bone marrow T lymphocytes from paediatric patients with aplastic anaemia. Br J Haematol. 2001;115(4):1023–31. doi: 10.1046/j.1365-2141.2001.03212.x. [DOI] [PubMed] [Google Scholar]
- 43.Dufour C, Corcione A, Svahn J, Haupt R, Poggi V, Beka’ssy AN, et al. TNF-alpha and IFN-gamma are overexpressed in the bone marrow of Fanconi anemia patients and TNF-alpha suppresses erythropoiesis in vitro. Blood. 2003;102(6):2053–9. doi: 10.1182/blood-2003-01-0114. [DOI] [PubMed] [Google Scholar]
- 44.Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C, et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A. 2002;99(12):8242–7. doi: 10.1073/pnas.112218799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Whitney MA, Royle G, Low MJ, Kelly MA, Axthelm MK, Reifsteck C, et al. Germ cell defects and hematopoietic hypersensitivity to gamma-interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood. 1996;88(1):49–58. [PubMed] [Google Scholar]
- 46.Ceccaldi R, Parmar K, Mouly E, Delord M, Kim JM, Regairaz M, et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell. 2012;11(1):36–49. doi: 10.1016/j.stem.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475(7354):53–8. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- 48.Hira A, Yabe H, Yoshida K, Okuno Y, Shiraishi Y, Chiba K, et al. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood. 2013;122(18):3206–9. doi: 10.1182/blood-2013-06-507962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after 15 years of followup. Haematologica. 2017 doi: 10.3324/haematol.2017.178111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Faivre L, Guardiola P, Lewis C, Dokal I, Ebell W, Zatterale A, et al. Association of complementation group and mutation type with clinical outcome in fanconi anemia. European Fanconi Anemia Research Group. Blood. 2000;96(13):4064–70. [PubMed] [Google Scholar]
- 51.Kutler DI, Singh B, Satagopan J, Batish SD, Berwick M, Giampietro PF, et al. A 20-year perspective on the International Fanconi Anemia Registry (IFAR) Blood. 2003;101(4):1249–56. doi: 10.1182/blood-2002-07-2170. [DOI] [PubMed] [Google Scholar]
- 52.Tonnies H, Huber S, Kuhl JS, Gerlach A, Ebell W, Neitzel H. Clonal chromosomal aberrations in bone marrow cells of Fanconi anemia patients: gains of the chromosomal segment 3q26q29 as an adverse risk factor. Blood. 2003;101(10):3872–4. doi: 10.1182/blood-2002-10-3243. [DOI] [PubMed] [Google Scholar]
- 53.Cioc AM, Wagner JE, MacMillan ML, DeFor T, Hirsch B. Diagnosis of myelodysplastic syndrome among a cohort of 119 patients with fanconi anemia: morphologic and cytogenetic characteristics. Am J Clin Pathol. 2010;133(1):92–100. doi: 10.1309/AJCP7W9VMJENZOVG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374(23):2209–21. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alter BP, Rosenberg PS, Brody LC. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet. 2007;44(1):1–9. doi: 10.1136/jmg.2006.043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Myers K, Davies SM, Harris RE, Spunt SL, Smolarek T, Zimmerman S, et al. The clinical phenotype of children with Fanconi anemia caused by biallelic FANCD1/BRCA2 mutations. Pediatr Blood Cancer. 2012;58(3):462–5. doi: 10.1002/pbc.23168. [DOI] [PubMed] [Google Scholar]
- 57.Wagner JE, Tolar J, Levran O, Scholl T, Deffenbaugh A, Satagopan J, et al. Germline mutations in BRCA2: shared genetic susceptibility to breast cancer, early onset leukemia, and Fanconi anemia. Blood. 2004;103(8):3226–9. doi: 10.1182/blood-2003-09-3138. [DOI] [PubMed] [Google Scholar]
- 58.Khan NE, Rosenberg PS, Alter BP. Preemptive Bone Marrow Transplantation and Event-Free Survival in Fanconi Anemia. Biol Blood Marrow Transplant. 2016;22(10):1888–92. doi: 10.1016/j.bbmt.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ayas M, Saber W, Davies SM, Harris RE, Hale GA, Socie G, et al. Allogeneic hematopoietic cell transplantation for fanconi anemia in patients with pretransplantation cytogenetic abnormalities, myelodysplastic syndrome, or acute leukemia. J Clin Oncol. 2013;31(13):1669–76. doi: 10.1200/JCO.2012.45.9719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chaudhury S, Auerbach AD, Kernan NA, Small TN, Prockop SE, Scaradavou A, et al. Fludarabine-based cytoreductive regimen and T-cell-depleted grafts from alternative donors for the treatment of high-risk patients with Fanconi anaemia. Br J Haematol. 2008;140(6):644–55. doi: 10.1111/j.1365-2141.2007.06975.x. [DOI] [PubMed] [Google Scholar]
- 61.MacMillan ML, DeFor TE, Young JA, Dusenbery KE, Blazar BR, Slungaard A, et al. Alternative donor hematopoietic cell transplantation for Fanconi anemia. Blood. 2015;125(24):3798–804. doi: 10.1182/blood-2015-02-626002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.MacMillan ML, Hughes MR, Agarwal S, Daley GQ. Cellular therapy for fanconi anemia: the past, present, and future. Biol Blood Marrow Transplant. 2011;17(1 Suppl):S109–14. doi: 10.1016/j.bbmt.2010.11.027. [DOI] [PubMed] [Google Scholar]
- 63.Stepensky P, Shapira MY, Balashov D, Trakhtman P, Skorobogatova E, Rheingold L, et al. Bone marrow transplantation for Fanconi anemia using fludarabine-based conditioning. Biol Blood Marrow Transplant. 2011;17(9):1282–8. doi: 10.1016/j.bbmt.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 64.Mehta PA, Davies SM, Leemhuis T, Myers K, Kernan NA, Prockop SE, et al. Radiation-free, alternative-donor HCT for Fanconi anemia patients: results from a prospective multi-institutional study. Blood. 2017;129(16):2308–15. doi: 10.1182/blood-2016-09-743112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walsh MF, Chang VY, Kohlmann WK, Scott HS, Cunniff C, Bourdeaut F, et al. Recommendations for Childhood Cancer Screening and Surveillance in DNA Repair Disorders. Clin Cancer Res. 2017;23(11):e23–e31. doi: 10.1158/1078-0432.CCR-17-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Diamond LK, Shahidi NT. Treatment of aplastic anemia in children. Semin Hematol. 1967;4(3):278–88. [PubMed] [Google Scholar]
- 67.Shahidi NT, Diamond LK. Testosterone-induced remission in aplastic anemia of both acquired and congenital types. Further observations in 24 cases. N Engl J Med. 1961;264:953–67. doi: 10.1056/NEJM196105112641901. [DOI] [PubMed] [Google Scholar]
- 68.Guidelines FA. Chapter 3 Hematolgoic Abnormatilites in Patients with Fanconi Anemia. 2014 [Google Scholar]
- 69.Kauff ND, Satagopan JM, Robson ME, Scheuer L, Hensley M, Hudis CA, et al. Risk-reducing salpingo-oophorectomy in women with a BRCA1 or BRCA2 mutation. N Engl J Med. 2002;346(21):1609–15. doi: 10.1056/NEJMoa020119. [DOI] [PubMed] [Google Scholar]
- 70.Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N Engl J Med. 2016;375(5):443–53. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tung N, Domchek SM, Stadler Z, Nathanson KL, Couch F, Garber JE, et al. Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat Rev Clin Oncol. 2016;13(9):581–8. doi: 10.1038/nrclinonc.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]