

Abstract
Background
Our knowledge of natural genetic variation is increasing at an extremely rapid pace, affording an opportunity to come to a much richer understanding of how effects of specific genes are dependent on the genetic background. To achieve a systematic understanding of such GxG interactions, it is desirable to develop genome editing tools that can be rapidly deployed across many different genetic varieties.
Results
We present an efficient CRISPR/Cas9 toolbox of super module (SM) vectors. These vectors are based on a previously described fluorescence protein marker expressed in seeds allowing identification of transgene-free mutants. We have used this vector series to delete genomic regions ranging from 1.7 to 13 kb in different natural accessions of the wild plant Arabidopsis thaliana. Based on results from 53 pairs of sgRNAs targeting individual nucleotide binding site leucine-rich repeat (NLR) genes, we provide a comprehensive overview of obtaining heritable deletions.
Conclusions
The SM series of CRISPR/Cas9 vectors enables the rapid generation of transgene-free, genome edited plants for a diversity of functional studies.
Electronic supplementary material
The online version of this article (10.1186/s13007-018-0330-7) contains supplementary material, which is available to authorized users.
Keywords: Arabidopsis thaliana, Genome editing, CRISPR/Cas9, Natural variation, NLR genes
Background
A central question in biology is how genes direct organismal function and phenotype. Much of the mechanistic knowledge we have today about development and physiology has come from genetic analyses, but these have been usually restricted to a few standard genetic backgrounds. In the model plant Arabidopsis thaliana, this has typically been the reference accession Columbia-0 (Col-0) plus a few other popular accessions such as Landsberg erecta (Ler) or Wassilewskija-2 (Ws-2), chosen for convenience, rather than because they are the most typical representatives of their species.
An alternative approach to connecting genotype and phenotype is to exploit intraspecific variation, which has added the benefit of informing about genes and alleles that help organisms to adapt to their environment. This approach has in the past decade been fueled by the rapidly increasing knowledge of genetic variation in hundreds, if not thousands of individuals. In A. thaliana, this has culminated in the 1001 Genomes project [1], a major resource for genome wide association analyses (GWAS) [2–5]. However, while GWAS can provide a fast track to the preliminary identification of genetic variants that are likely to underlie phenotypic variation between different individuals, confirmation that the statistically identified variants have a causal role in a specific biological process requires genetic manipulation. Often, the fastest way for obtaining such supporting information is from knockout studies in one of the reference strains [6, 7]. However, that requires the reference strain to have a functional copy of the candidate gene in question. This may not be the case, either because the reference strain has an inactive allele, or because the reference strain lacks the gene all together. It is therefore desirable to carry out equivalent genetic tests in other accessions as well.
The Streptococcus pyogenes—derived CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas9 (CRISPR-associated protein 9) system has become the method of choice for targeted gene modifications in many organisms, including plants [8–10]. Cas9 can be programmed by a single guide RNA (sgRNA) to induce a DNA double strand break (DSB) at a specific genomic site that is complementary to a 20-base sequence in the sgRNA next to a short Protospacer Adjacent Motif (PAM sequence) [11]. If the DSB is not correctly repaired, short insertions/deletions (indels) or base substitutions can be caused by non-homologous end joining (NHEJ). If the DNA modification is introduced in the germline, the “trigger” transgene can be removed by segregation. That single sgRNAs can induce indels with high frequency is well known [12].
In addition to short indels, CRISPR/Cas9 technology can generate deletions of DNA fragments flanked by target sites for two different sgRNAs. Such larger deletions may be desirable when regulatory sequences need to be removed, or when several tandemly repeated genes need to be inactivated and when truncation of an open reading frame can lead to gain-of-function mutations, as is often the case for nucleotide binding domain leucine-rich repeat (NLR) protein genes [13, 14]. CRISPR/Cas9 induced larger deletions have been widely documented in vertebrates [15–18] and invertebrates [19, 20].
In plants, there have been reports not only of modest sized deletions (under 1 kb) in crops such as rice and tomato [21, 22], and in Arabidopsis [23, 24], but also much larger deletions, as large as 120 kb in Arabidopsis [24] or 245 kb in rice protoplasts [21]. Unfortunately, such events are rare, and a large number of plants needed to be screened to identify them. Whether they could be inherited is unclear.
We have developed an efficient and easy-to-use super module (SM) destination and sgRNA shuffle-in vector toolbox that facilitates the generation and identification of deletions in plants. We demonstrate its usefulness by targeting 53 different NLR genes, aiming for deletions ranging from 1.7 to 13 kb. At least half of the events were inherited independently of the Cas9-sgRNAs. Deletions were obtained in multiple accessions of A. thaliana, demonstrating the utility of these vectors for GxG studies.
Results and discussion
CRISPR/Cas9 SM-gRNA-shuffle-in vector toolbox
In plants, CRISPR/Cas9 components are typically introduced as transgenes. To confirm that a mutation has been established in the germline, the transgene must be removed by segregation [25]. Our super module (SM) sgRNA-shuffle-in vector (SM CRISPR vector) toolbox facilitates both the generation of transgenic constructs, and the identification of later-generation, transgene-free plants. The toolbox has two parts, the SM destination binary vector (Fig. 1a) and the sgRNA shuffle-in vectors (Fig. 1b).
Fig. 1.

SM (super module) Destination binary vectors and sgRNA shuffle-in vectors. a Schematic representation of SM Destination binary vectors. Plant codon optimized Cas9 (pcoCas9) is driven by the promoter of UBQ10 (proUBQ10). Transcriptional termination sequences from rbcS. Blue-white selection strategy with LacZ cassette. Seed coat expressed red fluorescence from At2S3:mcherry cassette as transgenic plant selection marker. Not drawn to scale. b Schematic representation of sgRNA shuffle-in vectors. sgRNA including 20-bp target sequences and shared sgRNA sequences transcribed by A. thaliana U6 promoter. c Overhang sequences used for Golden gate cloning in generating the final binary vectors. The numbers listed in left column are indicated in a and b
The SM destination vector was generated by Gibson cloning [26] using the pGreen binary vector pGGZ001 [27], which is Golden gate cloning compatible [28]. The bacterial counter selection ccdB/chloramphenicolR cassette was replaced with the lacZ gene driven by the lacZ promoter. This enables blue-white selection to monitor replacement of the lacZ cassette with sgRNA cassette. A plant codon optimized Cas9 open reading frame (pcoCas9) [29] was combined with a 3′ rbcS terminator in the modified pGGZ001 destination vector (Fig. 1a). NotI/BamHI double restriction sites were engineered in front of the pcoCas9 ORF sequence, for the insertion of different promoters that can induce pcoCas9 expression in stage- or tissue-specific manner. For example, we used the UBQ10 (AT4G05320) promoter to drive ubiquitous expression in all organs of A. thaliana [30, 31]. To select transgenic plants, we used mCherry expression under the control of the At2S3 (AT4G27160) promoter, which leads to red fluorescence in the seed coat [25]. This allows both positive (T1 generation) and negative selection (T2 generation) of the CRISPR/Cas9 transgene (Additional file 1: Figure S1).
sgRNA shuffle-in vectors were generated based on pGGD000 and pGGE000 vectors [27], which are also Golden Gate cloning compatible. A cassette consisting of the AtU6 promoter and sgRNA scaffold sequences was cloned into the above vectors. The proAtU6:target-sgRNA cassette can be tandemly shuffled into the SM destination binary vector by Golden Gate assembly with overhang sequences (Fig. 1c) in a simultaneous digestion/ ligation one tube reaction (Fig. 2).
Fig. 2.
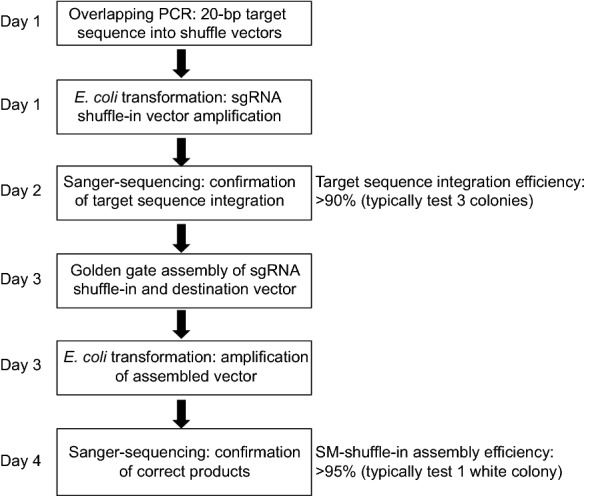
Workflow of using SM vectors
Application of the SM-gRNA-shuffle-in toolbox
The 20-bp portion of the sgRNA providing targeting specificity can be engineered into sgRNA shuffle-in vectors by mutagenesis PCR ("Methods" and Fig. 2), and in 90% of cases it was sufficient to test only three colonies to have at least one that was correct. PCR reaction conditions needed to be optimized, such as annealing temperatures and extension time for some primer pairs, depending on the GC content in the 20-bp-target sequence.
Swapping the proAtU6:target-sgRNA into the SM Destination vectors can be accomplished by Golden gate assembly [28]. White-blue selection is efficient, and over 95% of cases it was sufficient to test only one white colony (after swapping) to get the one harboring correctly assembled plasmids. Because the pcoCas9 sequence, especially at the 3’ end, was sometimes partially deleted in E. coli, it is recommended to confirm the assembled vectors by sequencing. This toolbox is efficient and straightforward to use, and the entire cloning process can be completed in 5 days (Fig. 2). We generated 40 constructs in 1 week.
Generation of genomic deletions in Arabidopsis thaliana natural accessions
Because premature stop codons or frame shifts can sometimes be associated with gain-of-function phenotypes, it may be advisable to partially or completely delete the open reading frame of a gene of interest. We therefore wanted to test whether there are limits to deletion size that can be induced by CRISPR/Cas9.
Fifty-three pairs of 20-bp target sequences were designed with CRISPR-P [32] based on the Col-0 reference genome (http://www.arabidopsis.org/), to target sequences flanking 53 individual NLR gene ORFs (Table 1 and Additional file 2: Table S1). Conservation of target sequences was ascertained with polymorphism information from 80 and 1135 natural A. thaliana accessions [1, 33] (Additional file 3: Figure S2), to ensure the sgRNAs can function to the same targets in other natural accessions. T-DNAs encoding proUBQ10:pcoCas9 and sgRNAs were introduced into the Col-0 reference accession (see "Methods") using Agrobacterium floral dip method [34]. Seeds carrying the T-DNA constructs were identified directly among the seeds harvested from the Agrobacterium treated plants based on red fluorescence from seed coat (Additional file 1: Figure S1a) under a dissecting microscope with dsRed filter (see "Methods"). Three oligonucleotide primers (Additional file 4: Table S2) were designed to genotype these T1 plants for deletions. Primers 1 and 2 were designed to flank the deletion (Fig. 3a, b), and control primers 1 and 3 were designed so as to span one of the two sgRNA target sites, with the binding site of primer 3 inside the expected deletion (Fig. 3a, e). Genotyping for a deletion event was first conducted in pooled T1 plants (pools of 6-25 plants for each target, Additional file 2: Table S1) with different transgene insertions. In the pools in which deletions were detected, individual T1 plants were analyzed.
Table 1.
Summary of efficiency of inherited deletion events in Col-0 with UBQ10 promoter driving expression of pcoCas9
| Targeted genes | 53 |
| Genes with deletions in T1 pools | 34 |
| Size of deleted fragments | |
| < 3 kb (1.7–3 kb) | 3/6 (50%) |
| 3–7 kb | 26/42 (61.9%) |
| > 7 kb (7–13 kb) | 5/5 (100%) |
| Frequency of deletion events in T1 | 4.8–78.6% |
| Genes with inheritance of deletions in T1 | 17/23 |
| Deletion inheritance in T2 (≤100 T2 plants) | 1-90% |
Fig. 3.

An example of inherited deletions in different accessions. a Schematic representation of targeted gene At3g04220 (U17), with locations of sgRNA target sites and primer binding sites for genotyping. b Deletions of At3g04220 in Col-0 reference accession, as detected by PCR with primers designated in a. Nineteen individual T1 plants were tested. Expected size of PCR product for deletion is 404 bp. c Alignment of sequence of PCR product for deletion in b (red) with wild-type sequence, which is only partially shown. d Deletions of At3g04220 in T1 plants from natural accessions, as detected by PCR; number of pooled plants in square brackets. 1: Col-0 #1 sample in b; 2: TueSB30-3 [9]; 3: Nie1-2 [7]; 4: WalHaesB4 [13]; 5: Rue3.1-31 [6]; 6: TueWa1-2, TueV-13, and HKT2.4 [5] (transformation efficiency is low for these 3 accessions). e Inheritance of At3g04220 deletion in 28 transgene-free Col-0 T2 plants, progeny of #1 T1 plant in b. Several plants show only the mutant band (top), indicating that they are homozygous for the deletion
With UBQ10 promoter driving pcoCas9 (SM Destination vector pRW004, Additional file 5: Table S3), we found deletions in pooled T1 plants for 34 out of the 53 targeted genes. The sizes of deleted fragments ranged from 1.7 to 13 kb (Fig. 3a, Table 1 and Additional file 2: Table S1). The appearance of deletions appeared to be independent of size. Specifically, we found 50% (3/6) of expected deletions under 3 kb, 62% (26/42) of deletions between 3 and 7 kb, and 100% (5/5) of deletions over 7 kb.
Several factors could confound our estimates of successful deletion frequency. First, genotyping PCR might be a factor. The absence of a PCR product diagnostic of a deletion cannot be distinguished from a failed PCR reaction. Second, success and failure rates appear to depend to some extent on the targeted locus as well as the similarity in efficiency of the two sgRNAs. Local genome properties, such as methylation and chromatin compaction, may affect accessibility of sgRNA to target sequences, and DNA-bound transcription factors may block the loading of Cas9 endonuclease. Also, unequal targeting efficiency of the two sgRNAs might cause one of the sites to be consistently mutated before a deletion could occur by simultaneous cutting of both targets by Cas9. Changing one or both of the sgRNAs might be helpful in such cases (Additional file 6: Figure S3). Another potential contributing factor could be incomplete transfer of the proUBQ10:Cas9 cassette, which is immediately next to the left border (LB) of the T-DNA. The T-DNA LB is less stable than the right border (RB), often causing incomplete integration of adjacent transgene sequences [35]. We tested several loci and found that some individual transgenic plants had incomplete transgene sequences next to the LB, although we did not find a clear correlation between missing LB sequences and lack of target deletions (Additional file 7: Figure S4). To save on later genotyping efforts, it is recommended to ascertain completeness of Cas9 sequences adjacent to LB before testing for target deletions or mutations.
The next question we wanted to answer was how many T1 plants need to be screened for a deletion event. Based on the 34 genes for which we detected deletions, the frequency ranged from 5% (1 out of 21 T1 plants, U52/At4g08450) to 79% (11 out of 14 T1 plants, U7/At1g27170) (Table 1 and Additional file 2: Table S1), with frequency for most genes being between 20 and 40% (Fig. 4a). There was no correlation between the frequency of T1 plants with deletion and the size of deleted fragments.
Fig. 4.
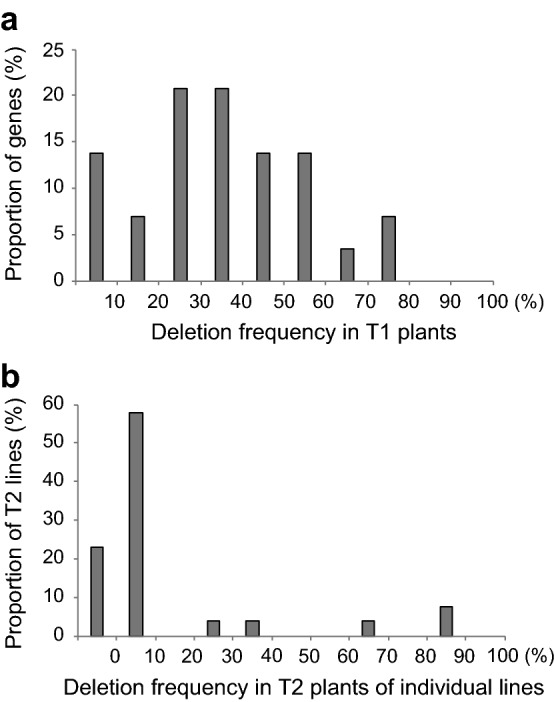
Frequencies of deletions and inheritance in T2 generation. a Deletion frequencies in T1 plants for the 34 out of 53 targeted genes where deletions were detected (Additional file 2: Table S1). b Frequencies of deletion inheritance in T2 for the 17 genes whose deletions were inherited (Additional file 2: Table S1)
We also wanted to test how well our toolbox works in other A. thaliana accessions. The UBQ10 promoter drives strong expression in A. thaliana accessions (http://jsp.weigelworld.org/AtGenExpress/resources/), and we introduced the construct targeting the At3g04220 gene (U17) into seven natural accessions. In Col-0, we detected in 6 out of 19 T1 plants the expected 3,536 bp deletion (Fig. 3b), which was confirmed by Sanger sequencing (Fig. 3c). The remaining 13 T1 plants were indistinguishable from our untransformed control. Because of lower transformation efficiency in the natural accessions, fewer T1 plants were obtained, but several carried deletions (Fig. 3d), with the frequency of 20 to 40% being similar to the one seen in Col-0 (Additional file 8: Table S4).
CRISPR/Cas9-induced deletions are stably inherited
As discussed above, DNA fragment deletions have been reported [21–24], but there are few systematic studies of the inheritance of such deletions. We used the same pairs of primers to test for the presence of deletions in transgene-free T2 plants for 23 out of the 34 genes for which deletions had been found in the T1 generation (Table 1 and Additional file 2: Table S1). In a first round of screening, 30 to 50 T2 plants from each individual T1 line with a deletion event were pooled and genotyped by PCR, followed by screening of individual plants if a deletion was detected in the pool. If no inherited deletion could be detected, a second round of screening with 50 to 150 additional T2 plants was performed. In 17 of the 23 tested cases, deletions were recovered in transgene-free T2 plants, with frequencies of 1 to 90%, with most below 10% (Table 1 and Fig. 4b, Additional file 2: Table S1).
Several factors could explain the variation in stable inheritance of deletions. First, in the T1 generation, we tested the somatic tissue of leaves, and while the UBQ10 promoter drives strong expression of pcoCas9 in many tissues, it may be less effective in the germline. EC1.2en-EC1.1p [36], an egg-cell specific hybrid promoter, has been reported to increase the frequency of heritable CRISPR/Cas9-induced mutations compared to the more widely expressed UBQ10 promoter. Second, UBQ10 promoter activity in the germline may be particularly sensitive to the genomic insertion site of the transgene, explaining large variation in deletion inheritance, such as for At1g63750 (U13 in Additional file 2: Table S1). Some deletions were, however, observed at more consistent frequencies. For example, At3g04220 (U17) deletions were often inherited (35 to 83% in progenies from 6 T1 lines, Additional file 2: Table S1), while At5g45250 (U36) deletions were only rarely inherited (2% in 3 T1 lines Additional file 2: Table S1). In the U17 and U30 lines, we found homozygous and heterozygous mutants as well as wild-type progeny. In the other lines, most progeny was wild type, and only one or two plants were either homozygous or heterozygous for the deletions. For example, two out of 34 T2 plants for At4g33300 (U24, Additional file 2: Table S1) were both homozygous for the deletion, and the rest was wild type (Additional file 2: Table S1). Based on these analyses, we conclude that on the order of 30 T1 plants are in many cases sufficient to obtain heritable deletions, but a higher number of T2 transgene-free plants, at least 100, is advisable to obtain heterozygous or homozygous mutants.
Conclusions
In summary, we have set up an efficient toolbox that facilitates knocking out genes or deleting DNA fragments in A. thaliana. Based on 53 targeted NLR genes, we detected deletions of up to 13 kb in somatic tissue of transgene-carrying T1 plants in more than half of the cases. Although we cannot exclude that deletions may appear in the T2 generation even if none were detected in T1 plants, somatic deletions in T1 plants with UBQ10 promoter driving pcoCas9 are a reasonable predictor of heritable deletions in transgene-free T2 plants. With our tested constructs, about three quarter (17/23) of constructs with T1 deletions produced also T2 plants with deletions. Finally, we have shown that the success rate in natural A. thaliana accessions is similar to that in the Col-0 reference accession.
Methods
Plasmid propagation
Plasmids with pcoCas9 were propagated in E. coli strain DH5α at 30 °C. Bacteria containing binary vectors and sgRNA shuffle-in vectors were grown on LB plates with spectinomycin (50 µg/mL) plus X-gal (50 µg/mL) or ampicillin (50 µg/mL), respectively.
Construction of Cas9 destination binary vectors and sgRNA shuffle vectors
The pGGZ001 plasmid [27] was used as starting point for constructing SM Destination binary vectors. In pGGZ001, there are two BsaI sites flanking the ccdB/chloramphenicolR genes. The four-nucleotide overhangs next to each BsaI site were introduced by two rounds of overlap PCRs (Fig. 1c). A LacZ promoter:LacZ fragment was PCR amplified from pSE7 [37] and cloned between the HindIII and BcuI sites of pGGZ001 to replace the ccdB/ChloramphenicolR cassette. The At2S3:mCherry-terminator cassette was amplified from pRW003 [25] and inserted into an XbaI site next to the LacZα cassette. We modified a Cas9 (pcoCas9) construct with potato IV2 intron and nuclear localization signal that had been codon optimized for both monocotyledonous and dicotyledonous plants [29] (HBT-pcoCas9, Addgene plasmid #52254) by introducing synonymous mutations that altered 7 out of 9 BsaI recognition sites. A NotI/BamHI restriction site was added at the 5′ end of pcoCas9. pcoCas9 sequences were fused with rbcS 3′ transcriptional terminator sequences using Golden Gate cloning [28]. The pcoCas9-rbcS terminator cassette was added by Gibson assembly (NEB, Ipswich, USA) [26]. proUBQ10 promoter was inserted in front of pcoCas9 with NotI/BamHI.
Starting material for sgRNA shuffle-in vectors were pGGD000 and pGGE000 [27]. A transcription unit containing the AtU6 promoter (with “G” at the end to enhance transcription), a 19-bp sgRNA sequence and a scaffold gRNA sequence [29] was generated using gene synthesis (GeneArt Gene Synthesis, Thermo Fisher Scientific Life Technologies, Regensburg, Germany). Two BsaI sites flank the cassette and overlapping PCR was used to place different four-nucleotide overhangs (Fig. 1c) next to the BsaI sites.
sgRNA sequence design and integration of sgRNA into shuffle vectors
Protospacer target sequences were designed with CRISPR-P [32]. In most cases, the suggestions with the highest scores were used. In cases where there was only a small number of automatic suggestions, sequences were chosen manually. Target sequences were inserted into the sgRNA shuffle-in vectors using overlapping PCR with primers that contained 20-bp vector sequences plus 20-bp guide RNA sequences. For example, forward primer with 5′ NV1NNNNV17NV18NV19NV20 + 20 bp-sgRNA, and reverse primer with 20 bp-sgRNA + NV21NV22NV23NV24NNNNV40 5′ were used for inserting a 20 bp-sgRNA between positions NV20 and NV21 of vector sequence NV1NNNNNNNV20NV21NNNNNNV40. The overlapping PCR reaction (10 μl) was set up as follows (final concentrations in brackets): Q5 Hi-Fidelity polymerase reaction buffer (1x, Thermo Scientific), dNTPs (200 μM), forward and reverse primers (0.2 μM each), ~60 ng vector as template, 0.1 μl Q5 polymerase, and ddH2O to 10 μl. PCR was according to the manual for Q5 polymerase (Thermo Scientific). We added 0.8 μl 10 x FD buffer and 0.2 μl DpnI (Thermo Scientific) to the final product, and the mix was directly transformed into E. coli strain DH5α. Three single colonies were cultured, and plasmids extracted using the GeneJET Plasmid Miniprep Kit (Thermo Scientific), followed by Sanger-sequencing to confirm sgRNA integration. sgRNA sequences and vectors are listed in Additional file 5: Table S3.
Assembly of SM-sgRNA shuffle-in vector
The sgRNA expression units were assembled into SM Destination vectors using Golden gate cloning [28]. Restriction/ligation reactions (15 μl) were set up as follows: 1× FastDigest buffer (Thermo Scientific, Waltham, MA, the USA), 1.0 mM ATP, 10 U of FD-Eco31I (BsaI, Thermo Scientific), 35 U of T4 DNA ligase (Thermo Scientific), 100 ng of SM Destination vector plasmid, 25 ng of each sgRNA shuffle-in vector. Reactions were incubated for 50 cycles (37 °C, 3 min; 16 °C, 5 min), followed by 50 °C for 5 min and 80 °C for 10 min. The reaction product was directly used for E. coli transformation, with antibiotics of Spectinomycin (Sigma) and X-Gal (X-galactopyranoside, Sigma). White colonies were grown in liquid culture (usually one) overnight and plasmids were extracted using the GeneJET Plasmid Miniprep Kit (Thermo Scientific). We confirmed integration of sgRNA units and the completeness of other components such as pcoCas9 sequences with Sanger sequencing.
Plant material and growth conditions
Non-reference A. thaliana accessions TueSB30-3, Nie1-2, WalHaesB4, Rue3.1-31, TueWa1-2, TueV-13, and HKT2.4 have been described [38]. Seeds were surface sterilized with 75% ethanol plus 0.005% TritonX-100, and germinated on soil. Plants were germinated and cultivated in growth rooms at a constant temperature of 23 °C (temperature variability about ± 0.1 °C), air humidity at 65% and long-day conditions (16 h day length), with light (125 to 175 μmol m−2 s−1) provided by a 1:1 mixture of Cool White and Gro-Lux Wide Spectrum fluorescent lights (Luxline plus F36 W/840, Sylvania, Germany).
Transgenic plants
The floral dip method [34] was used to transform plants, with Agrobacterium tumefaciens strain ASE [39] at an OD600 of 0.7 to 1. Presence and absence of transgenes in seeds was ascertained under a Leica MZFLIII Fluorescence Stereomicroscope (Leica, Wetzlar, Germany) with the setting of dsRed filter for the filter system FLUOIII. Luminescence light was provided by Light Engine Sola 365 SM II und Zubehör (Lumencor, Beaverton, OR, the USA).
Genotyping
Plant genomic DNA was extracted with a modified CTAB method [40]. Taq polymerase (NEB) or Q5 High-Fidelity DNA polymerase (Thermo Scientific) were used for PCR amplification to detect deletion events. Annealing temperatures were tested and optimized for each pair of genotyping primers, with extension time kept short enough to favor amplification of fragments carrying deletions under 1 kb, but not the original, non-deleted fragment. T7E1 (T7 Endonuclease I, NEB) was used for identification of mutations at individual sgRNA target sites [41]. DNA fragments were amplified from genomic DNA using a pair of primers spanning the targets with Q5 High-Fidelity DNA Polymerase (Thermo Scientific). The PCR product was purified using PCR Purification Column (Qiagen, Hilden, Germany) and concentration was determined with a Nanodrop 2000 Spectrophotometer (Thermo Scientific). 100 ng of purified PCR product was denatured-annealed under the following conditions: 95 °C for 2 min, ramp down to 85 °C, at 2 °C/s, followed by ramping down to 25 °C at 0.1 °C/s. Annealed PCR products were digested with 5 U of T7E1 for 20 min at 37 °C. The T7E1-digested products were separated on a 2% agarose gel. A similar amount of substrate DNA without T7E1 incubation was used as a negative control.
Additional files
Additional file 1: Figure S1. Fluorescence-based strategy for selection of transgenic plants. a. Fluorescent, transgenic T1 seed for At3g04220 deletion (white arrow). b. Non-fluorescent, transgene-free T2 seeds for At3g04220 gene deletion, line 5 (white arrows indicate a cluster of non-fluorescent seeds).
Additional file 2: Table S1. Frequency of deletion and inheritance for 53 targeted genes in Col-0 with UBQ10 promoter driving pcoCas9.
Additional file 3: Figure S2. Conservation of dual sgRNA target sequences and genotyping primers for At3g04220 gene deletion among natural accessions. “ref”, A. thaliana reference genome sequence. Sequence alignments were extracted from the genome matrix of 80 accessions [33, http://1001genomes.org/data/MPI/MPICao2010/releases/current/genome_matrix/TAIR10_genome_matrix_2012_03_13.txt.gz] using AWK, rotated by 90° and converted to HTML using Perl. Variants and uncalled sites were highlighted using CSS. a. Sequence alignment for the two target sites (Chr3:1108991..1109010 and Chr3:1115509..1115528) among 80 accessions [33]. b. Sequence alignment of locations of oligos for genotyping (Chr3:1108776..1108801 and Chr3:1112691..1112705) among accessions. “ref”, A. thaliana reference genome sequence.
Additional file 4: Table S2. sgRNA target sequences and oligonucleotides for genotyping.
Additional file 5: Table S3. Overview of vectors.
Additional file 6: Figure S3. Example showing simultaneous sgRNA efficiency in generating deletion in ACD6 gene. a. Schematic representation of ACD6 (At4g14400) gene structure and locations of sgRNA target sites and binding sites for genotyping primers (indicated by black numbers on top and the position of primer below). b. PCR and T7E1 assays showing mutations only at the target site of sgRNA1 but not sgRNA2. The left-most lane next to DNA ladder (Mix) is a wild-type control. c. PCR assay revealing deletion events with the combination of sgRNA1 and sgRNA3. The left-most lane next to DNA ladder (1 kb) is a wild-type control.
Additional file 7: Figure S4. PCR analyses of the T-DNA left border to determine completeness of integration of the proUBQ10:Cas9 expression cassette. a. Schematic representation of vectors integrated in the genome. Three fragments were PCR amplified. b. PCR products. The first lane (p) is the positive control with the SM destination vector as template. mCherry positive plants for four different targets were tested. Red stars indicate plants with target deletions. DNA ladder MIX was used for P1, and 1 kb ladder for P2 and P3 regions. A 404-bp genomic region from the actin gene AT2G37620 was used as the internal control for DNA quality (fourth panel labeled with CK). Samples for different gene loci are separated by lanes with DNA size markers (M). Arrows point to 1 kb for the first 3 panels and 500 bp for the fourth panel (CK).
Additional file 8: Table S4. Frequency of At3g04220 gene deletions in natural accessions.
Authors' contributions
RW and DW conceived and designed the experiments. RW, WSZ and WYX designed the sgRNAs for knocking out the genes. WSZ evaluated sgRNA efficiency for fragment deletions. WYX performed genotyping. ES and RS prepared starting plasmids and tested the initial system. CMW analyzed the conservation of sgRNAs target sequences and genotyping primers among 1135 natural accessions. JF generated the alignment of genomic regions of interest among 1135 Arabidopsis thaliana Pseudogenomes. RW, ML and YTJ conducted experiments, including generation of vectors, production of transgenic plants, and genotyping. RW and DW analyzed and interpreted the data, and wrote the paper with help from all authors. All authors read and approved the final manuscript.
Acknowledgements
We thank the laboratory of Jen Sheen for plasmid HBT-pcoCas9 (Addgene plasmid #52254), and the laboratory of Jan Lohmann for pZ001, pD000 and pE000. We thank Ulrich Lutz and Chang Liu for their valuable comments on the manuscript. Joffrey Fitz is an employee of Tropic IT Ltd.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Plasmids can be obtained from Addgene under accession #74812.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
The work, including study design, data collection, analysis and interpretation, and manuscript writing, was supported by Marie Curie Fellowship (European Commission) H2020-MSCA-IF-2014-655295 (RW), ERC AdG IMMUNEMESIS and the Max Planck Society (DW).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Rui Wu, Email: rui.wu@tuebingen.mpg.de.
Miriam Lucke, Email: miriam.lucke@student.uni-tuebingen.de.
Yun-ting Jang, Email: yun-ting.jang@student.uni-tuebingen.de.
Wangsheng Zhu, Email: wangsheng.zhu@tuebingen.mpg.de.
Efthymia Symeonidi, Email: efthymia.symeonidi@tuebingen.mpg.de.
Congmao Wang, Email: congmaowang@126.com.
Joffrey Fitz, Email: joffrey.fitz@tuebingen.mpg.de.
Wanyan Xi, Email: eukixi@gmail.com.
Rebecca Schwab, Email: rebecca.schwab@tuebingen.mpg.de.
Detlef Weigel, Email: weigel@weigelworld.org.
References
- 1.1001 Genomes Consortium 1135 Genomes reveal the global pattern of polymorphism in Arabidopsis thaliana. Cell. 2016;166:481–491. doi: 10.1016/j.cell.2016.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atwell S, Huang YS, Vilhjálmsson BJ, Willems G, Horton M, Li Y, et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature. 2010;465:627–631. doi: 10.1038/nature08800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seren Ü, Vilhjálmsson BJ, Horton MW, Meng D, Forai P, Huang YS, et al. GWAPP: a web application for genome-wide association mapping in Arabidopsis. Plant Cell. 2012;24:4793–4805. doi: 10.1105/tpc.112.108068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seren Ü, Grimm D, Fitz J, Weigel D, Nordborg M, Borgwardt K, et al. AraPheno: a public database for Arabidopsis thaliana phenotypes. Nucleic Acids Res. 2017;45:D1054–D1059. doi: 10.1093/nar/gkw986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grimm DG, Roqueiro D, Salomé PA, Kleeberger S, Greshake B, Zhu W, et al. easyGWAS: a cloud-based platform for comparing the results of genome-wide association studies. Plant Cell. 2017;29:5–19. doi: 10.1105/tpc.16.00551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sessions A, Burke E, Presting G, Aux G, McElver J, Patton D, et al. A high-throughput Arabidopsis reverse genetics system. Plant Cell. 2002;14:2985–2994. doi: 10.1105/tpc.004630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alonso JM, Stepanova AN, Leisse TJ, Kim CJ, Chen H, Shinn P, et al. Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science. 2003;301:653–657. doi: 10.1126/science.1086391. [DOI] [PubMed] [Google Scholar]
- 8.Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 9.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puchta H. Applying CRISPR/Cas for genome engineering in plants: the best is yet to come. Curr Opin Plant Biol. 2017;36:1–8. doi: 10.1016/j.pbi.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 11.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bortesi L, Fischer R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol Adv. 2015;33:41–52. doi: 10.1016/j.biotechadv.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Michael Weaver L, Swiderski MR, Li Y, Jones JDG. The Arabidopsis thaliana TIR-NB-LRR R-protein, RPP1A; protein localization and constitutive activation of defence by truncated alleles in tobacco and Arabidopsis. Plant J. 2006;47:829–840. doi: 10.1111/j.1365-313X.2006.02834.x. [DOI] [PubMed] [Google Scholar]
- 14.DeYoung BJ, Innes RW. Plant NBS-LRR proteins in pathogen sensing and host defense. Nat Immunol. 2006;7:1243–1249. doi: 10.1038/ni1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canver MC, Bauer DE, Dass A, Yien YY, Chung J, Masuda T, et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem. 2014;289:21312–21324. doi: 10.1074/jbc.M114.564625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song Y, Yuan L, Wang Y, Chen M, Deng J, Lv Q, et al. Efficient dual sgRNA-directed large gene deletion in rabbit with CRISPR/Cas9 system. Cell Mol Life Sci. 2016;73:2959–2968. doi: 10.1007/s00018-016-2143-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao A, Wang Z, Hu Y, Wu Y, Luo Z, Yang Z, et al. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res. 2013;41:e141. doi: 10.1093/nar/gkt464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kraft K, Geuer S, Will AJ, Chan WL, Paliou C, Borschiwer M, et al. Deletions, inversions, duplications: engineering of structural variants using CRISPR/Cas in mice. Cell Rep. 2015;10:833–839. doi: 10.1016/j.celrep.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 19.Kistler KE, Vosshall LB, Matthews BJ. Genome engineering with CRISPR-Cas9 in the mosquito Aedes aegypti. Cell Rep. 2015;11:51–60. doi: 10.1016/j.celrep.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedland AE, Tzur YB, Esvelt KM, Colaiácovo MP, Church GM, Calarco JA. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods. 2013;10:741–743. doi: 10.1038/nmeth.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou H, Liu B, Weeks DP, Spalding MH, Yang B. Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res. 2014;42:10903–10914. doi: 10.1093/nar/gku806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nekrasov V, Wang C, Win J, Lanz C, Weigel D, Kamoun S. Rapid generation of a transgene-free powdery mildew resistant tomato by genome deletion. Sci Rep. 2017;7:482. doi: 10.1038/s41598-017-00578-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Y, Zhang C, Liu W, Gao W, Liu C, Song G, et al. An alternative strategy for targeted gene replacement in plants using a dual-sgRNA/Cas9 design. Sci Rep. 2016;6:23890. doi: 10.1038/srep23890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ordon J, Gantner J, Kemna J, Schwalgun L, Reschke M, Streubel J, et al. Generation of chromosomal deletions in dicotyledonous plants employing a user-friendly genome editing toolkit. Plant J. 2017;89:155–168. doi: 10.1111/tpj.13319. [DOI] [PubMed] [Google Scholar]
- 25.Gao X, Chen J, Dai X, Zhang D, Zhao Y. An effective strategy for reliably isolating heritable and cas9-free arabidopsis mutants generated by CRISPR/Cas9-mediated genome editing. Plant Physiol. 2016;171:1794–1800. doi: 10.1104/pp.16.00663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 27.Lampropoulos A, Sutikovic Z, Wenzl C, Maegele I, Lohmann JU, Forner J. GreenGate—a novel, versatile, and efficient cloning system for plant transgenesis. PLoS ONE. 2013;8:e83043. doi: 10.1371/journal.pone.0083043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engler C, Gruetzner R, Kandzia R, Marillonnet S. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS ONE. 2009;4:e5553. doi: 10.1371/journal.pone.0005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J-F, Norville JE, Aach J, McCormack M, Zhang D, Bush J, et al. Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotechnol. 2013;31:688–691. doi: 10.1038/nbt.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Callis J, Carpenter T, Sun CW, Vierstra RD. Structure and evolution of genes encoding polyubiquitin and ubiquitin-like proteins in Arabidopsis thaliana ecotype Columbia. Genetics. 1995;139:921–939. doi: 10.1093/genetics/139.2.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, et al. A gene expression map of Arabidopsis thaliana development. Nat Genet. 2005;37:501–506. doi: 10.1038/ng1543. [DOI] [PubMed] [Google Scholar]
- 32.Lei Y, Lu L, Liu H-Y, Li S, Xing F, Chen L-L. CRISPR-P: a web tool for synthetic single-guide RNA design of CRISPR-system in plants. Mol Plant. 2014;7:1494–1496. doi: 10.1093/mp/ssu044. [DOI] [PubMed] [Google Scholar]
- 33.Cao J, Schneeberger K, Ossowski S, Günther T, Bender S, Fitz J, et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet. 2011;43:956–963. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- 34.Clough SJ, Bent AF. Floral dip: a simplified method forAgrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998;16:735–743. doi: 10.1046/j.1365-313x.1998.00343.x. [DOI] [PubMed] [Google Scholar]
- 35.Forsbach A, Schubert D, Lechtenberg B, Gils M, Schmidt R. A comprehensive characterization of single-copy T-DNA insertions in the Arabidopsis thaliana genome. Plant Mol Biol. 2003;52:161–176. doi: 10.1023/A:1023929630687. [DOI] [PubMed] [Google Scholar]
- 36.Wang Z-P, Xing H-L, Dong L, Zhang H-Y, Han C-Y, Wang X-C, et al. Egg cell-specific promoter-controlled CRISPR/Cas9 efficiently generates homozygous mutants for multiple target genes in Arabidopsis in a single generation. Genome Biol. 2015;16:144. doi: 10.1186/s13059-015-0715-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emami S, Yee M-C, Dinneny JR. A robust family of Golden Gate Agrobacterium vectors for plant synthetic biology. Front Plant Sci. 2013;4:339. doi: 10.3389/fpls.2013.00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bomblies K, Yant L, Laitinen RA, Kim S-T, Hollister JD, Warthmann N, et al. Local-scale patterns of genetic variability, outcrossing, and spatial structure in natural stands of Arabidopsis thaliana. PLoS Genet. 2010;6:e1000890. doi: 10.1371/journal.pgen.1000890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hood EE, Helmer GL, Fraley RT, Chilton MD. The hypervirulence of Agrobacterium tumefaciens A281 is encoded in a region of pTiBo542 outside of T-DNA. J Bacteriol. 1986;168:1291–1301. doi: 10.1128/jb.168.3.1291-1301.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murray MG, Thompson WF. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980;8:4321–4325. doi: 10.1093/nar/8.19.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim HJ, Lee HJ, Kim H, Cho SW, Kim J-S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res. 2009;19:1279–1288. doi: 10.1101/gr.089417.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Fluorescence-based strategy for selection of transgenic plants. a. Fluorescent, transgenic T1 seed for At3g04220 deletion (white arrow). b. Non-fluorescent, transgene-free T2 seeds for At3g04220 gene deletion, line 5 (white arrows indicate a cluster of non-fluorescent seeds).
Additional file 2: Table S1. Frequency of deletion and inheritance for 53 targeted genes in Col-0 with UBQ10 promoter driving pcoCas9.
Additional file 3: Figure S2. Conservation of dual sgRNA target sequences and genotyping primers for At3g04220 gene deletion among natural accessions. “ref”, A. thaliana reference genome sequence. Sequence alignments were extracted from the genome matrix of 80 accessions [33, http://1001genomes.org/data/MPI/MPICao2010/releases/current/genome_matrix/TAIR10_genome_matrix_2012_03_13.txt.gz] using AWK, rotated by 90° and converted to HTML using Perl. Variants and uncalled sites were highlighted using CSS. a. Sequence alignment for the two target sites (Chr3:1108991..1109010 and Chr3:1115509..1115528) among 80 accessions [33]. b. Sequence alignment of locations of oligos for genotyping (Chr3:1108776..1108801 and Chr3:1112691..1112705) among accessions. “ref”, A. thaliana reference genome sequence.
Additional file 4: Table S2. sgRNA target sequences and oligonucleotides for genotyping.
Additional file 5: Table S3. Overview of vectors.
Additional file 6: Figure S3. Example showing simultaneous sgRNA efficiency in generating deletion in ACD6 gene. a. Schematic representation of ACD6 (At4g14400) gene structure and locations of sgRNA target sites and binding sites for genotyping primers (indicated by black numbers on top and the position of primer below). b. PCR and T7E1 assays showing mutations only at the target site of sgRNA1 but not sgRNA2. The left-most lane next to DNA ladder (Mix) is a wild-type control. c. PCR assay revealing deletion events with the combination of sgRNA1 and sgRNA3. The left-most lane next to DNA ladder (1 kb) is a wild-type control.
Additional file 7: Figure S4. PCR analyses of the T-DNA left border to determine completeness of integration of the proUBQ10:Cas9 expression cassette. a. Schematic representation of vectors integrated in the genome. Three fragments were PCR amplified. b. PCR products. The first lane (p) is the positive control with the SM destination vector as template. mCherry positive plants for four different targets were tested. Red stars indicate plants with target deletions. DNA ladder MIX was used for P1, and 1 kb ladder for P2 and P3 regions. A 404-bp genomic region from the actin gene AT2G37620 was used as the internal control for DNA quality (fourth panel labeled with CK). Samples for different gene loci are separated by lanes with DNA size markers (M). Arrows point to 1 kb for the first 3 panels and 500 bp for the fourth panel (CK).
Additional file 8: Table S4. Frequency of At3g04220 gene deletions in natural accessions.
Data Availability Statement
Plasmids can be obtained from Addgene under accession #74812.