

Abstract
Critical aspects of maintaining glucose homeostasis in the face of chronic insulin resistance and type 2 diabetes (T2D) are increased insulin secretion and adaptive expansion of beta cell mass. Nutrient and hormone sensing G protein-coupled receptors are important mediators of these properties. A growing body of evidence now suggests that the G protein-coupled receptor, free fatty acid receptor 2 (FFA2), is capable of contributing to the maintenance of glucose homeostasis by acting at the pancreatic beta cell as well as at other metabolically active tissues. We have previously demonstrated that Gαq/11-biased agonism of FFA2 can potentiate glucose stimulated insulin secretion (GSIS) as well as promote beta cell proliferation. However, the currently available Gαq/11-biased agonists for FFA2 exhibit low potency, making them difficult to examine in vivo. This study sought to identify Gαq/11-biased FFA2-selective agonists with potent GSIS-stimulating effects. To do this, we generated an FFA2 homology model that was used to screen a library of 10 million drug-like compounds. Although FFA2 and the related short chain fatty acid receptor FFA3 share 52% sequence similarity, our virtual screen identified over 50 compounds with predicted selectivity and increased potency for FFA2 over FFA3. Subsequent in vitro calcium mobilization assays and GSIS assays resulted in the identification of a compound that can potentiate GSIS via activation of Gαq/11 with 100-fold increased potency compared with previously described Gαq/11-biased FFA2 agonists. These methods and findings provide a foundation for future discovery efforts to identify biased FFA2 agonists as potential T2D therapeutics.
INTRODUCTION
As type 2 diabetes (T2D) continues to grow as a global public health challenge, so too does the urgent need for new therapies. Nutrient sensing G-protein coupled receptors (GPCRs) in pancreatic beta (β) cells are particularly tractable targets, as these receptors are well known to contribute to the regulation of insulin secretion and β cell mass in response to the nutritional status of the host (reviewed in ref 1). A new and seemingly important group of nutrient sensing GPCRs, free fatty acid receptors (FFAs) have drawn a great deal of interest recently as potential therapeutic targets (reviewed in ref 2). For example, the long-chain FFA, FFA1 (GPR40), has been shown to regulate glucose stimulated insulin secretion (GSIS), and several FFA1 agonists have shown promise in early stage clinical trials, though concerns over potential toxicity have slowed the progression of some of these agonists through trials.2
Another member of the FFA family, FFA2 (GPR43), has recently gained attention as a potential metabolic regulator. This receptor is activated by the short-chain fatty acids acetate, propionate and butyrate.3,4 Expression of FFA2 in metabolically active tissues has led to multiple studies investigating its role in aspects of metabolism and energy balance.5–7 Among these, our group and others have reported increased expression of Ffar2 in mouse islets of Langerhans during the insulin-resistant phase of pregnancy,8,9 while data from others have reported a similar upregulation in diet-induced and genetic models of obesity.10 These data, along with recent reports from us and others that activation of FFA2 regulates insulin secretion,11–13 suggest that FFA2 may represent a novel T2D target through its involvement in the pancreatic β cell response to insulin resistance.
Our understanding of the role of FFA2 in β cell biology is complicated by several factors. Notably, another short-chain fatty acid receptor, FFA3, shares 52% sequence similarity with FFA2.4 As these receptors are both expressed in islets and share similar ligands, receptor-specific pharmacological tools are necessary to clearly define the role of each receptor in β cell biology. Furthermore, FFA2 has been demonstrated to couple to both Gαq/11 and Gαi/o.3 In the β cell, these pathways are predicted to exert opposing effects on insulin secretion and other aspects of β cell function and in fact, we have recently demonstrated that FFA2-selective agonists biased toward Gαq/11 or Gαi/o can either potentiate or inhibit insulin secretion, respectively.11 Thus, along with identifying receptor-specific agonists, the identification of compounds with specific Gαq/11 or Gαi/o bias will be crucial to the development of FFA2 as an effective therapeutic target.
To begin to address these issues, here we have identified a group of predicted FFA2-selective small molecule agonists using in silico homology modeling and high-throughput screening. To select for agonists with Gαq/11 signaling properties, the compounds were screened in calcium mobilization assays, and those compounds exhibiting the highest potency for calcium mobilization were tested for their ability to potentiate GSIS. These efforts led to the identification of a compound that potentiates GSIS at low micromolar concentrations, apparently via Gαq/11-biased signaling. The discovery and screening of these compounds provide insight into the molecular basis of biased FFA2 signaling and will serve as a foundation for future efforts to design Gαq/11-biased FFA2 agonists as potential T2D therapeutic candidates.
MATERIALS AND METHODS
Homology Model Building of FFA2 and FFA3
In order to construct the homology models of FFA2 and FFA3, we applied the Prime module of the Schrodinger Suite.14 Prime 3.8 is a highly accurate protein structure prediction tool that integrates comparative modeling and fold recognition into a single interface. Considering the primary amino acid sequences of FFA2 and FFA3 as the queries and using the ‘blast’, ‘psi-blast’ and ‘fold’ recognition servers, we searched for template structures to build the comparative homology models for the two receptors. The search did not yield a single template with more than 30% sequence similarity to either receptor. In the absence of a good single template structure with reasonable sequence similarity to FFA2 and FFA3, we applied a multitemplate technique to build the homology models. Templates with sequence similarity greater than 25% were considered for constructing the models. For FFA2, four different templates were considered: human β-2 adrenergic receptor15 (2RH1.pdb), β-2 adrenergic receptor16 (2VT4. pdb), squid rhodopsin17 (2Z73. pdb) and substance-P receptor18 (2KSA.pdb). For FFA3, another set of three templates were chosen for the model building: turkey β1 adrenergic receptor19 (2Y00. pdb), bovine rhodopsin20 (1F88. pdb) and another form of bovine rhodopsin21 (1GZM. pdb). The templates were assigned to different regions of the FFA2 and FFA3 query sequences, and each part of the receptor was built using the most similar template structure. Using this multitemplate approach, we obtained homology models for each receptor that were then subjected to MolProbity analysis22 for validation. The initial validation tests did not generate acceptable data for both models. Hence, to obtain more accurate models, we subjected the models to prime minimization in optimized potential for liquid simulations (OPLS2005) force field implemented in the Schrodinger suite. After minimization of both models, we found <2% all atoms clash scores, <3% poor rotamers, <2% Ramachandran outliers, 98% Ramachandra favored residues, no residues with bad bonds and 0.3% residues with bad angles. Furthermore, analyzing the two models, we noticed that the cysteine residues present in the first and second extracellular loops form a disulfide bridge that contributes to the stability of the structures.
As both receptors traverse the lipid bilayer, we performed molecular dynamics simulation (MDS) studies of our FFA2 and FFA3 models embedded in 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), a ubiquitous cell membrane phospholipid bilayer, in a solvent box (TIP3P, water) to study the intrinsic behavior of the receptors. Using the Desmond MDS package,23,24 we built the system after setting up the membrane POPC at 300°K. A boundary condition with a cubic box with a volume of 20×20×20 Å3 was applied. The absolute box size calculation method was used to determine the size of the system. Keeping the number of particles constant as well as the pressure and temperature, an ensemble (NPT) was defined, and a 10 ns simulation was carried out for both models. The Desmond simulated three-dimensional (3D) models of both FFA2 and FFA3 are shown in figure 1. We used the site identification module (SITE-ID) of Tripos software25 in the Sybylx1.3 interface to determine the putative small molecule ligand binding pockets for both FFA2 and FFA3 as shown in figure 2. Our final model for FFA2 is deposited at https://bioinformatics.cineca.it/PMDB/insert_model.php?newtar=ckjg.
Figure 1.

Multicolor ribbon represents Desmond simulated FFA2 and FFA3 conformation in presence of POPC (gray sticks) and simulated water (red dots). FFA2, free fatty acid receptor 2; FFA3, free fatty acid receptor 3; POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine.
Figure 2.

Comparison of the Connolly surface of the ligand binding pockets of FFA2 and FFA3 with critical active site residues. FFA2, free fatty acid receptor 2; FFA3, free fatty acid receptor 3.
In Silico Filtering of the Small Molecule Database for Ligand Preparation
The ZINC database,26 which contains approximately 18 million commercially available compounds, was used for virtual high-throughput screening (vHTS). All compounds in the ZINC library were subjected to a panel of PAINS substructures filters with Smiles ARbitrary Target Specifications strings27 to eliminate promiscuous and non-drug-like molecules that interfere with functionality of the target proteins. Filtering generated a list of approximately 10 million commercially available compounds for further screening. Before screening this, 10 million compound data set with our earlier-defined small molecule ligand binding pocket, it was subjected to the LigPrep module of Schrodinger28 in OPLS2005 force field at pH 7.4±1 retaining the specific chirality. A low energetic 3D structure for each molecule was generated in this ligand preparation panel.
Protein preparation for small molecule screening and grid generation
The protein preparation (prot-prep) engine implemented in the Schrödinger software suite was utilized to prepare the protein. The FFA2 and FFA3 model structures were subjected to prime validation to correct for irrelevant side chains, missing atoms, and undesired orientations of Asn, Gln, or His residues, and to assign the OPLS charges. Next, the prot-prep module was used to prepare and refine the structure to generate the ‘receptor’, of FFA2 and FFA3 to be used for small molecule docking. The three-tier docking engine of the Schrödinger software program is built on a grid-based algorithm that requires grid generation in the active site of the target protein. Two separate 12 Å3 grids were generated considering the critical residues Glu166, Phe168, Arg180, His242, Lue183, Tyr238, and Arg255 for FFA2, and Phe173, Arg185, His 245, Met188, Tyr241, Arg258, and Lue171 for FFA3. The ligand binding pockets constituted by these residues were also previously reported.29
Virtual Screening Workflow
For vHTS, we began with the curated library of approximately 10 million drug-like compounds described above and the OPLS 2005 force field was set. The ligand van der Waals radii was scaled to 0.80 Å with partial atomic charges <0.15 electrostatic unit. A three-tier Glide docking algorithm30 was employed that incorporates vHTS followed by standard precision (SP) and extra precision (XP) docking protocols. The earlier-defined grid for FFA2 was used for the docking experiment. The output of this three-tier docking engine was analyzed using the XP visualization tools by considering the interactions of the compounds with the critical residues identified earlier by Schmidt et al.29 We selected 140 compounds having a Glide docking score <−6.0. The Glide score is a function of the binding energy.30 Then we carried out another three-tier docking of this hit set using the earlier generated grid for the FFA3 receptor. Though the ligand binding pockets of FFA2 and FFA3 were very different in shape, size, and other properties, we found 81 compounds with score <−6 in the FFA3 pocket. Based on our cross-docking results, we eliminated compounds with good interactions with FFA3 and obtained a set of 59 compounds predicted to be selective for FFA2 for testing in biological assays. From this set, 45 compounds were selected for screening in biological assays based on availability and synthetic tractability (structures of selected compounds shown in table 1). All compounds used in screening were purchased from ChemBridge (San Diego, California, USA).
Table 1.
Structures of acetate and selected FFA2-selective hits identified by in silico FFA2 modeling and virtual high-throughput screening of the ZINC library
| ID | Structure | ID | Structure |
|---|---|---|---|
| 1 |
|
7 |
|
| 2 |
|
8 |
|
| 3 |
|
9 |
|
| 4 |
|
10 |
|
| 5 |
|
11 |
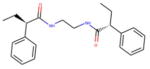
|
| 6 |

|
Acetate |

|
ID refers to the compound number.
Assessment of Ca2+ mobilization in the βTC pancreatic β cell line
Calcium mobilization was assessed in the mouse βTC3 β cell line (from Mauvais-Jarvis lab, Tulane University, see table 2) as previously described.11 Cells were loaded with 5 μM Fluo-8 (AAT Bioquest, Sunnyvale, California, USA) and incubated at 37°C for 60 min. Compounds were added at the indicated concentrations, and fluorescence was measured by fluorometric imaging plate reader. For each experiment, each compound was tested in quadruplicate at multiple concentrations. Raw fluorescence counts were normalized in each well by calculating delta F/f values (maximum fluorescent count obtained after stimulation – minimal fluorescent count obtained before stimulation/minimal fluorescent count obtained before stimulation). The Emax and pEC50 were determined using a non-linear regression algorithm (GraphPad PRISM).
Table 2.
Identification of cell lines used and expression pattern of receptors
In ref 11 and Kebede et al, 2009, data are presented on receptor expression in βTC3 and Min6 cells, respectively.
Preparation of poly-HEMA-coated plates
Poly(2-hydroxyethyl methacrylate) (Santa Cruz Biotech, Dallas, Texas, USA) was prepared at 20 mg/mL in 95% EtOH and stirred for 4–8 hours at 65°C. Five hundred microliters of prepared poly-HEMA was added to each well of a 6-well plate, and plates were covered and dried overnight at 37°C. Dried plates were washed twice with sterile water, UV treated, and stored at 4°C for future use.
Preparation of pseudoislets
MIN6 cells (from AddexBio, see table 2) were cultured as previously described,31 in Dulbecco’s modified Eagle medium (Invitrogen, Waltham, Massachusetts, USA), containing 25 mmol/L glucose and supplemented with 10% heat-inactivated fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin (Corning, Corning, New York), and 50 μM β mercaptoethanol (Sigma, St. Louis, Missouri, USA). MIN6 pseudoislets were formed by dispersing MIN6 monolayers to single cell suspension and seeding 1×106 cells per well in poly-HEMA coated 6-well plates. Cells were cultured for 5–7 days, and media changed after 2 days in culture.
In vitro insulin secretion assay
On the day of the experiment, following 30 min preincubation in Krebs Ringer Buffer (KRB; NaCl 130 mM, KCl 4.7 mM, NaH2PO40.5 mM, MgSO40.5 mM, CaCl21.5 mM, HEPES 10 mM, 0.1% BSA, pH 7.4), cells were transferred to KRB supplemented with 2.8 mM and incubated for 60 min at 37°C. Islets were then picked into groups of 10 and transferred to treatment conditions: 2.8 mM glucose alone, 16.7 mM glucose alone, or 16.7 mM glucose plus agonist at the indicated concentration for 60 min at 37°C. At the end of the incubation period, an aliquot of the supernatant was sampled for measurement of insulin secretion by ELISA (ALPCO, Salem, New Hampshire, USA). Pseudoislets were collected and sonicated in 1 mL acid ethanol for measurement of total insulin content.
RESULTS
Comparison of the ligand binding pockets of FFA2 and FFA3
While the overall structural similarity between FFA2 and FFA3 is 41%, a comparison of the small molecule ligand binding sites showed a 56% similarity. When we carried out the shape, lipophilic and electrostatic comparison, we observed that the shapes of the two pockets were very different and the electrostatic potential maps of the ligand binding pockets were also drastically different. These differences are mainly due to an increased presence of polar residues in the ligand binding site of FFA2 compared with FFA3. The Connolly surfaces of the ligand binding pockets are shown in figure 2.
Virtual docking of 10 million compounds derived from the ZINC database to FFA2 identified 140 compounds with predicted FFA2 activity. Comparing the surface area, volume, and the depth of FFA2 and FFA3 pockets, we observed that FFA2 has a larger surface area of 510 Å2 and volume 553 Å3 than FFA3. However, the depth of the FFA2 pocket was found to be 2 Å less than the FFA3 pocket. Using this differential analysis, the set of 140 compounds was cross-docked to the FFA3 binding pocket, and compounds that also demonstrated binding to FFA3 were eliminated, resulting in the identification of 59 compounds with predicted selectivity and enhanced potency for FFA2 over FFA3. Of these 59 compounds, based on availability and synthetic tractability, 45 compounds were used in subsequent biological assays.
Ca2+ mobilization screening of predicted agonists in vitro
Because Ca2+ mobilization is a primary mechanism by which a Gαq/11-coupled receptor potentiates insulin secretion, we screened the predicted agonists for activity at FFA2 by assessing the effect of these compounds on calcium mobilization in the βTC3 mouse β cell line. This cell line was selected due to its expression of Ffar2, but not the related receptor, Ffar3, as recently reported.11 Because FFA2 and FFA3 share some ligand specificity, this cell line allows us to specifically probe for FFA2 activity, without the confounding factor of FFA3 signaling. We have previously reported that this assay effectively detects FFA2 signaling induced by the endogenous ligand, acetate, as well as several previously described FFA2-selective agonists.11 Screening of these compounds in the calcium mobilization assay revealed that of the 49 compounds tested, 11 of the compounds activated FFA2 with increased potency and similar efficacy relative to the endogenous ligand, acetate (figure 3, table 3).
Figure 3.

Screening of 11 compounds for calcium mobilization in the βTC3 cell line. Serial dilutions of 11 compounds were tested for their ability to mobilize calcium. Representative data from a single experiment is presented. Each experiment was repeated in triplicate.
Table 3.
pEC50 and Emax values of 11 predicted FFA2 agonists in the calcium mobilization assays conducted in the βTC3 mouse β cell line are shown. Values are mean±SEM. N is the number of replicates
| pEC50 | Emax | N | |
|---|---|---|---|
| Acetate | 2.32±0.21 | 2.27±0.31 | 13 |
| Compound 1 | 6.37±1.40 | 2.00 | 3 |
| Compound 2 | 8.90±0.16 | 2.13±0.08 | 4 |
| Compound 3 | 6.05±0.37 | 2.08±0.08 | 3 |
| Compound 4 | 8.03±0.11 | 2.00 | 2 |
| Compound 5 | 4.84±0.35 | 3.02±0.88 | 2 |
| Compound 6 | 7.48 | 2.00 | 1 |
| Compound 7 | 5.32 | 2.00 | 1 |
| Compound 8 | 4.65±0.37 | 3.19±0.67 | 5 |
| Compound 9 | 6.08±2.06 | 2.37±0.37 | 2 |
| Compound 10 | 5.84±0.47 | 3.12±0.76 | 5 |
| Compound 11 | 8.97±0.62 | 2.00 | 2 |
pEC50 is the negative log of EC50 in Molar concentration and Emax is the fold increase in delta F/f values.
Identification of compounds that regulate GSIS via FFA2
Based on the results of the in vitro screening, we selected several compounds, based on availability, to test in GSIS assays in MIN6 cells in vitro. As previously described, MIN6 cells cultured as adherent monolayers may exhibit diminished glucose responsiveness and insulin secretion compared with rodent islets.32 However, when cultured in suspension, MIN6 cells self-aggregate into pseudoislets, which closely resemble freshly isolated mouse islets in size, shape, and capacity for insulin secretion.32,33 Using MIN6 pseudoislets, we conducted dose–response studies to determine the capacity for each compound to potentiate GSIS. As shown in figure 4a, compound 10 potentiated GSIS at 1 μM, demonstrating increased potency compared with acetate, which potentiates GSIS at 1 mM in pseudoislets (figure 4b) and isolated mouse islets.11 Additionally, the compound demonstrates 100-fold increased potency compared with other FFA2 agonists that potentiate GSIS, which require 100 μM concentrations to achieve potentiation of GSIS.11 As these compounds have previously been shown to potentiate GSIS via Gαq/11-biased signaling, and Gαq/11 activation is known to promote insulin secretion, we next examined whether compound 10 also potentiates GSIS by this mechanism. Pseudoislets were pretreated with the phospholipase C inhibitor, U73122. Inhibition of this key downstream effector of Gαq/11 abolished the GSIS-potentiating effect of compound 10, suggesting that the compound acts primarily by activating Gαq/11 (figure 4c). The docked pose of the compound ZINC03832747 in FFA2 model is shown in figure 5. It is interesting to note that one of the benzimidazole parts of the compound along with one hydroxyl group showed very strong hydrogen bond network with Arg180. Again, another hydroxyl group has also picked up a hydrogen bond with Arg255. Furthermore, the compound is also showing potential hydrogen bonding with Tyr90, Tyr 238 and Asn171. The docked pose of this compound reveals that it strongly interacts with the binding pocket of FFA2.
Figure 4.

Assessment of compound 10 in glucose stimulated insulin secretion assays. (A) Dose–response assessment of compound 10 in GSIS. Insulin secretion from cultured pseudoislets in response to treatment with 16.7 mM glucose and compound 10 at the indicated concentrations. (B) Dose–response assessment of acetate in GSIS. Insulin secretion from cultured pseudoislets in response to treatment with 16.7 mM glucose and acetate at the indicated concentrations. (C) Insulin secretion from cultured pseudoislets in response to treatment with 16.7 mM glucose and 1 μM compound 10, with or without pretreatment with the phospholipase C inhibitor, U73122. Dashed line represents insulin secretion in response to 16.7 mM glucose alone. All values are relative to insulin secretion from cultured pseudoislets in response to 16.7 mM glucose only. For A and B, comparison is between glucose-only and glucose plus agonist. For all experiments, n≥3; *p<0.05, **p<0.01 and ***p<0.001 as determined by Student’s t-test. GSIS, glucose stimulated insulin secretion.
Figure 5.

Docked pose of compound 10 in FFA2 homology model. The dotted yellow lines represent putative hydrogen bonds with Arg180, Arg255, Tyr238, Asn171 and Tyr90 residues in the orthosteric site.
DISCUSSION
At the beginning of the 21st century, over 170 million individuals worldwide were estimated to have developed T2D, and this number is projected to increase to 366 million by the year 2030.34 As the diabetic population continues to grow, so too does the need for improved therapeutics and alternative therapeutic targets. Many currently marketed T2D drugs are associated with undesirable side effects, most commonly hypoglycemia and weight gain, and occasionally, complications such as increased risk of cardiovascular disease, liver damage, and pancreatitis may occur. Thus, there is a need for new therapies that can selectively enhance insulin secretion in a glucose-dependent manner.
Increasing evidence suggests that FFA2 may be a viable target to promote GSIS. Recent studies have described the role of FFA2 in maintaining gestational glucose homeostasis by contributing to insulin secretion as well as adaptive beta cell mass expansion during the insulin resistant phase of pregnancy.35 Additionally, reports from our lab and another group have suggested that Gαq/11-biased signaling can potentiate GSIS11,13 and promote beta cell proliferation,13 another important factor in promoting beta cell function in T2D. Despite the potential of these compounds to potentiate beta cell function, the study of FFA2 to date has been complicated by multiple conflicting studies relating to the characterization of FFA2 signaling. Specifically, while the above studies have found that FFA2 may potentiate insulin secretion, a separate study has reported that FFA2 may inhibit insulin secretion and suggested that antagonism of FFA2 may be most beneficial to the treatment of T2D.12 While some of these differences may relate to the use of different mouse models, another important factor that has recently come to light is the ability of FFA2 agonists to exhibit biased Gαq/11 or Gαi/o signaling. Thus, the identification of new Gαq/11-biased agonists will be especially useful in identifying potential therapeutic compounds.
We have previously assessed the signaling bias of several FFA2 selective compounds identified by Schmidt et al.29 These compounds were identified from the screen of a small carboxylic acid library and exhibited potency in the range of 100 μM, only a 10-fold increase in potency over the endogenous ligand, acetate. Of the compounds that we found to potentiate GSIS, all did so by activating the Gαq/11 signaling pathway. In the present study, we conducted homology modeling of FFA2 and FFA3 in order to screen the 35 million compound ZINC database for FFA2-selective compounds. We filtered these compounds to select for drug-like properties, and conducted homology modeling and virtual docking to identify compounds with selectivity for FFA2 over FFA3. From these compounds, we assessed the potency and efficacy of selected compounds to mobilize calcium in vitro, in order to identify compounds that activate the Gαq/11 pathway. These studies have led to the identification of a FFA2-selective agonist that potentiates GSIS from cultured beta cells with increased potency relative to previously studied agonists by signaling specifically through Gαq/11.
Some limitations of our study need to be mentioned. First, we cannot rule out that our compounds are influencing insulin secretion through non-FFA2 mechanisms. Additionally, comprehensive profiling of whether these molecules are modulating other GPCRs will also be needed. It is also possible, but not expected, that these compounds could act as FFA3 antagonists, which would lead to enhanced GSIS. Thus, more extensive structure activity profiling of these molecules as agonists, antagonists, and allosteric properties is needed, along with greater in vitro profiling of their effects on GSIS.
Given the multiple conflicting findings relating to the role of FFA2 in regulating glucose homeostasis, and recent characterization of the dual, opposing effects on insulin secretion that may be elicited by FFA2 agonists, additional well-controlled studies with potent biased agonists will be necessary to shed light on the role of this receptor in regulating glucose homeostasis and its utility as a therapeutic target. In addition to its utility as a preclinical research tool, strongly biased Gαq/11 agonists such as the compound we have described here may hold particular therapeutic potential compared with unbiased FFA2 agonists. Specifically, Gαq/11-biased FFA2 agonism may benefit glucose homeostasis by acting at tissues other than the islet. For example, it has been proposed that Gαq/11 signaling by FFA2 in intestinal L cells can promote GLP-1 secretion, thereby indirectly further potentiating GSIS and promoting beta cell proliferation.6 Furthermore, activation of FFA2 may have the potential to regulate inflammation3,7,36,37 and inhibit lipolysis,7,38,39 both of which may benefit insulin sensitivity and glucose homeostasis, but more studies are necessary to fully understand how biased FFA2 agonism can be exploited to achieve the desired effects. The present study represents the targeted efforts to identify FFA2 agonists that can potentiate GSIS and to characterize the receptor-ligand interactions that mediate this activity. Further efforts to identify similarly Gαi/o-biased agonists that inhibit GSIS and to improve the potency and pharmacological properties of these agonists will significantly aid in the study of FFA2 biology and development of FFA2 as a therapeutic target.
Significance of this study.
What is already known about this subject?
New drugs are needed for type 2 diabetes.
Signaling through free fatty acid receptor 2 (FFA2) mediates insulin secretion.
Biased FFA2 agonists will enhance insulin secretion.
What are the new findings?
We show that biased FFA2 agonists can be developed.
These biased agonists can mediate insulin secretion.
A new chemical scaffold for FFA2 is revealed.
How might these results change the focus of research or clinical practice?
These data may serve the foundation for new drug discovery.
Acknowledgments
Funding
BTL is supported by the National Institutes of Health under award number, R01DK104927-01A1, The University of Chicago DR&TC (P30DK020595) and Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Career Development (grant no. 1IK2BX001587-01). SRV is supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health (F31DK102371), and the Northwestern University Program in Endocrinology, Diabetes and Hormone Action (NIH T32 DK007169). A part of this work was supported by the Northwestern University Medicinal and Synthetic Chemistry Cgpcsore (ChemCgpcsore the Center for Molecular Innovation and Drug Discovery (CMIDD), which is funded by the Chicago Biomedical Consortium with support from The Searle Funds aSearleChicago Community Trust and Cancer Center Support Grant P30 CA060553 from the National Cancer Institute awarded to the Robert H. Lurie Comprehe Lurie Cancer Center.
Footnotes
Contributors
Each author contributed to this article.
Competing interests
None declared.
Provenance and peer review
Not commissioned; externally peer reviewed.
References
- 1.Ahrén B. Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov. 2009;8:369–85. doi: 10.1038/nrd2782. [DOI] [PubMed] [Google Scholar]
- 2.Watterson KR, Hudson BD, Ulven T, et al. Treatment of type 2 diabetes by free Fatty acid receptor agonists. Front Endocrinol. 2014;5:137. doi: 10.3389/fendo.2014.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le Poul E, Loison C, Struyf S, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem. 2003;278:25481–9. doi: 10.1074/jbc.M301403200. [DOI] [PubMed] [Google Scholar]
- 4.Brown AJ, Goldsworthy SM, Barnes AA, et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem. 2003;278:11312–9. doi: 10.1074/jbc.M211609200. [DOI] [PubMed] [Google Scholar]
- 5.Bjursell M, Admyre T, Göransson M, et al. Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. Am J Physiol Endocrinol Metab. 2011;300:E211–E220. doi: 10.1152/ajpendo.00229.2010. [DOI] [PubMed] [Google Scholar]
- 6.Tolhurst G, Heffron H, Lam YS, et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes. 2012;61:364–71. doi: 10.2337/db11-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kimura I, Ozawa K, Inoue D, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun. 2013;4:1829. doi: 10.1038/ncomms2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Layden BT, Durai V, Newman MV, et al. Regulation of pancreatic islet gene expression in mouse islets by pregnancy. J Endocrinol. 2010;207:265–79. doi: 10.1677/JOE-10-0298. [DOI] [PubMed] [Google Scholar]
- 9.Rieck S, White P, Schug J, et al. The transcriptional response of the islet to pregnancy in mice. Mol Endocrinol. 2009;23:1702–12. doi: 10.1210/me.2009-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keller MP, Choi Y, Wang P, et al. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res. 2008;18:706–16. doi: 10.1101/gr.074914.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Priyadarshini M, Villa SR, Fuller M, et al. An Acetate-Specific GPCR, FFAR2, regulates insulin secretion. Mol Endocrinol. 2015;29:1055–66. doi: 10.1210/me.2015-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang C, Ahmed K, Gille A, et al. Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat Med. 2015;21:173–7. doi: 10.1038/nm.3779. [DOI] [PubMed] [Google Scholar]
- 13.McNelis JC, Lee YS, Mayoral R, et al. GPR43 potentiates β-Cell function in obesity. Diabetes. 2015;64:3203–17. doi: 10.2337/db14-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schrodinger P. Schrödinger Release 2014-4: prime, version 3.8. New York, NY: Schrödinger, LLC; 2014. [Google Scholar]
- 15.Cherezov V, Rosenbaum DM, Hanson MA, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–65. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warne T, Serrano-Vega MJ, Baker JG, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–91. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murakami M, Kouyama T. Crystal structure of squid rhodopsin. Nature. 2008;453:363–7. doi: 10.1038/nature06925. [DOI] [PubMed] [Google Scholar]
- 18.Gayen A, Goswami SK, Mukhopadhyay C. NMR evidence of GM1-induced conformational change of substance P using isotropic bicelles. Biochim Biophys Acta. 2011;1808:127–39. doi: 10.1016/j.bbamem.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 19.Warne T, Moukhametzianov R, Baker JG, et al. The structural basis for agonist and partial agonist action on a β(1)-adrenergic receptor. Nature. 2011;469:241–4. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palczewski K, Kumasaka T, Hori T, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–45. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Edwards PC, Burghammer M, et al. Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol. 2004;343:1409–38. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 22.Davis IW, Murray LW, Richardson JS, et al. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–19. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shan Y, Klepeis JL, Eastwood MP, et al. Gaussian split Ewald: A fast Ewald mesh method for molecular simulation. J Chem Phys. 2005;122:054101. doi: 10.1063/1.1839571. [DOI] [PubMed] [Google Scholar]
- 24.Dror RO, Mildorf TJ, Hilger D, et al. SIGNAL TRANSDUCTION. structural basis for nucleotide exchange in heterotrimeric G proteins. Science. 2015;348:1361–5. doi: 10.1126/science.aaa5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cramer RD. R-group template CoMFA combines benefits of “ad hoc” and topomer alignments using 3D-QSAR for lead optimization. J Comput Aided Mol Des. 2012;26:805–19. doi: 10.1007/s10822-012-9583-9. [DOI] [PubMed] [Google Scholar]
- 26.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–82. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baell JB, Holloway GA. New substructure filters for removal of Pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–40. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 28.Schrodinger LLC N, Release S. 2014-4: ligprep. version 3.2. Vol. 2014. Schrödinger, LLC; New York, NY: 2014. [Google Scholar]
- 29.Schmidt J, Smith NJ, Christiansen E, et al. Selective orthosteric free fatty acid receptor 2 (FFA2) agonists: identification of the structural and chemical requirements for selective activation of FFA2 versus FFA3. J Biol Chem. 2011;286:10628–40. doi: 10.1074/jbc.M110.210872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schrodinger LLC N. Small-Molecule drug Discovery Suite 2014-4: schrödinger Suite 2014-4 induced fit docking protocol; Glide version 6.5. New York, NY: Schrödinger, LLC; 2014. [Google Scholar]
- 31.Miyazaki Jun-i, Araki K, Yamato E, et al. Establishment of a pancreatic? cell line that retains Glucose-Inducible insulin secretion: special Reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–32. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- 32.Luther MJ, Hauge-Evans A, Souza KLA, et al. MIN6 ?-cell??-cell interactions influence insulin secretory responses to nutrients and non-nutrients. Biochem Biophys Res Commun. 2006;343:99–104. doi: 10.1016/j.bbrc.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 33.Kelly C, Guo H, McCluskey JT, et al. Comparison of insulin Release from MIN6 Pseudoislets and pancreatic islets of Langerhans reveals importance of homotypic cell interactions. Pancreas. 2010;39:1016–23. doi: 10.1097/MPA.0b013e3181dafaa2. [DOI] [PubMed] [Google Scholar]
- 34.Wild S, Roglic G, Green A, et al. Global prevalence of Diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–53. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 35.Fuller M, Priyadarshini M, Gibbons SM, et al. The short-chain fatty acid receptor, FFA2, contributes to gestational glucose homeostasis. Am J Physiol Endocrinol Metab. 2015;309 doi: 10.1152/ajpendo.00171.2015. ajpendo.00171.2015–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maslowski KM, Vieira AT, Ng A, et al. Regulation of inflammatory responses by gut Microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–6. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vinolo MAR, Ferguson GJ, Kulkarni S, et al. SCFAs induce mouse neutrophil chemotaxis through the GPR43 receptor. PLoS One. 2011;6:e21205. doi: 10.1371/journal.pone.0021205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong Y-H, Nishimura Y, Hishikawa D, et al. Acetate and Propionate Short Chain Fatty Acids stimulate adipogenesis via GPCR43. Endocrinology. 2005;146:5092–9. doi: 10.1210/en.2005-0545. [DOI] [PubMed] [Google Scholar]
- 39.Ge H, Li X, Weiszmann J, et al. Activation of G Protein-Coupled receptor 43 in adipocytes leads to inhibition of Lipolysis and suppression of plasma Free Fatty Acids. Endocrinology. 2008;149:4519–26. doi: 10.1210/en.2008-0059. [DOI] [PubMed] [Google Scholar]