

Abstract
Background:
Pulmonary Langerhans cell histiocytosis (PLCH) is an interstitial primary pulmonary disease, characterized by Langerhans cell proliferation. It is easily misdiagnosed in children. This study aimed to characterize the clinical manifestations and features of PLCH by retrospective analysis.
Methods:
A retrospective analysis was performed in 117 PLCH patients out of 338 LCH patients who were admitted in our center from November 2006 to October 2013. Variables between two groups were compared by Mann-Whitney U-test and Chi-square test. Kaplan-Meier curves were constructed to compare the survival rates and Cox regression to evaluate the effect of risk factors.
Results:
The median age of PLCH group was significantly lower than that of non-PLCH group (18.63 months vs. 43.4 months, P < 0.001). All PLCH children had other organ involvement and only 11 cases (9.4%) had respiratory symptoms. The most common radiologic finding was cystic lesions (29 cases, 24.8%). Pulmonary function abnormalities were dominated by obstructive ventilatory dysfunction (63 cases, 82.9%). The 5-year overall survival (OS) of PLCH children was 93.6% ± 2.3% and the event-free survival (EFS) was 55.7% ± 5.2%. Among the 38 cases with progressed or relapsed disease, five cases (13.2%) were due to progression or recurrence of lung damage. The 5-year OS of PLCH children with “risk organ” involvement was significantly lower than those without “risk organ” involvement (86.0% ± 4.9% vs. 100%, χ2 = 8.793, P = 0.003). The difference of EFS between two groups was also significant (43.7% ± 7.7% vs. 66.3% ± 6.5%, χ2 = 5.399, P = 0.020). The “risk organ” involvement had a significant impact on survival (hazard ratio = 1.9, P = 0.039).
Conclusions:
PLCH mainly occurs in young children, and only a small percentage of patients have respiratory symptoms. They generally have other organ involvement. Most of PLCH children have a good prognosis and most lung lesions could have improved or stabilized. Management of “risk organ” involvement is the key point to improving EFS.
Keywords: Children, Langerhans Cell Histiocytosis, Lung
摘要
背景:
肺朗格罕细胞组织细胞增生症(PLCH) 是一种原发于肺、以朗格罕细胞增生为特点的间质性疾病。它是一种特殊类型的朗格罕细胞组织细胞增生症(LCH),容易在儿童中误诊。本研究的目的是通过回顾性分析来描述PLCH的临床表现和特征。
方法:
回顾性分析2006年11月至2013年10月我院收治的338例LCH患者中的117例PLCH患者临床资料,采用Mann-Whitney U检验法比较连续变量,采用卡方检验或Fisher确切概率法比较分类变量。通过Kaplan-Meier曲线比较两组间生存率,并应用多因素Cox比例风险回归分析评估风险因素对生存率的影响。
结果:
PLCH患儿与非PLCH患儿相比无显著性别差异。PLCH患儿的中位年龄显著低于非PLCH患者(18.63个月vs.43.4个月,P<0.001)。在117例PLCH患儿中,仅有11例(9.4%)患儿有呼吸系统症状,全部患儿均伴有其他脏器受累。PLCH患儿肺部影像学表现主要包括囊泡(29例,24.8%)、磨玻璃(25例,21.4%)、结节(19例,16.2%)及其他特征。肺功能异常以小气道阻塞性通气功能障碍(63例,82.9%)为主。PLCH患儿的5年总生存率(OS)为93.6±2.3%,无事件生存率(EFS)为55.7±5.2%。在疾病进展或复发的患儿中,仅有13.2%是由于肺部病变的进展或复发导致的。有危险器官受累的PLCH患儿的5年总生存率显著低于无危险器官受累者(86.0±4.9% vs.100%, χ2=8.793, P = 0.003),无事件生存率也存在显著差异(43.7±7.7% vs. 66.3±6.5%, χ2=5.399, P=0.020)。在多因素分析中,危险器官受累对生存率有显著影响(风险比= 1.9,P = 0.039)。
结论:
PLCH主要发生于婴幼儿,仅有小部分患者伴有呼吸道症状。PLCH患儿通常伴有其他器官受累。大部分PLCH患儿预后良好,且肺部病变转归大多为好转或稳定。控制危险器官的受累是提高无事件生存率(EFS)的关键。
INTRODUCTION
Langerhans cell histiocytosis (LCH) is a specific type of histiocytic disorder characterized by infiltration of tissues or organs with a specific dendritic cell, the Langerhans cell.[1] The pathogenesis of LCH is still controversial. In recent years, LCH was called as “inflammatory myeloid tumors” in many studies because it has both inflammation and tumor characteristics.[2] Pulmonary LCH (PLCH) refers to a disease in which Langerhans cells are involved only in the lung or simultaneously with other organ involvement.[3] Narrow sense PLCH refers to the primary PLCH alone, in which LCH lesions are confined to the respiratory system, especially to the lungs. Generalized PLCH can be divided into two categories. In the first category, pulmonary involvement is only one part of multisystem involvement and it is more common in infants and children without significant pulmonary clinical manifestations. The other category, only involving lungs, with no other lesions, occurs mainly in adult smokers, predominantly affecting young smokers from the ages of 20–40 years, which is extremely rare in children.[4,5]
This study summarized the clinical data of 117 PLCH patients among 338 LCH patients admitted to our hospital between November 2006 and October 2013. Their clinical manifestations, imaging examinations, lung function, and treatment outcome were retrospectively analyzed.
METHODS
Ethical approval
The study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of Beijing Children's Hospital (No. 2018-k-56). As a retrospective study and data analysis, this study was exempt from the informed consent from patients.
Study population
In total, 338 LCH cases were admitted to Beijing Children's Hospital from November 2006 to October 2013. Among them, 117 cases (34.6%) were found with pulmonary involvement (PLCH), while the other 221 cases (65.4%) were LCH patients without pulmonary involvement (non-PLCH).
Data collection
All patients received a complete physical examination, regular laboratory tests (complete blood counts, liver function, coagulation function, etc.), imaging examinations on bone, pituitary, lung, and so on, abdominal ultrasound scanning, bone marrow aspiration, and biopsy of the lesions. In this study, all PLCH patients were examined with computed tomography (CT) of the lungs, while the lung function tests were performed in 104 cases.
Definition of pulmonary involvement and “risk organ”
In this study, the term “pulmonary involvement” is defined as (1) disease proven by lung biopsy or (2) a positive biopsy of extrathoracic localization of the disease associated with characteristic high-resolution CT (HRCT) lung findings including the presence of nodular, cystic, patch, or cord shadows.
The definition of the “risk organ” involvement was based on the Histiocyte Society evaluation and treatment guidelines.[6]
Treatment and disease state assessment
All PLCH patients were classified as Group 1 and treated according to LCH-III-Group 1 chemotherapy (MTX group)[6] because the lung was taken as a “risk organ” in this study.
Disease state assessment after treatment was as follows:
Nonactive disease (NAD): The original lesions disappeared or improved, no new lesion
Stable disease: No change in the original lesion and no new lesion
Progressive disease: The original lesions progressed or new lesion(s) appeared.
Statistical analysis
The statistical analysis was performed with SPSS 17.0 (SPSS Inc., Chicago, IL, USA). Summary statistics of both groups were presented as frequencies, percentages, or median. For categorical variables, significant differences between groups were assessed with the Chi-square test or Fisher's exact test. Continuous variables were compared by Mann-Whitney U-test. Cumulative survival curves for end point events were constructed by Kaplan-Meier method and compared by log-rank test. Cox regression was used to evaluate the effect of risk factors on survival. A value of P < 0.05 was considered statistically significant.
RESULTS
General characteristics and clinical manifestation
The ratio of male (76 cases) to female (41 cases) was 1.85:1 in PLCH children, while it was 1.30:1 (125 cases vs. 96 cases) in non-PLCH group. However, there was no statistically significant difference in gender between two groups (χ2 = 2.237, P = 0.135).
The age of PLCH or non-PLCH patients ranged from 2 months to 162 months or 3 months to 165 months, respectively. The median age of PLCH children was significantly lower than that of non-PLCH patients (18.63 months vs. 43.4 months, P < 0.001). The number of children younger than 3 years in the PLCH group (95 cases) was significantly higher than that of non-PLCH group (98 cases) (81.2% vs. 44.4%, χ2 = 42.414, P < 0.001; Table 1).
Table 1.
Comparison of general characteristics in PLCH and non-PLCH groups
| Parameters | Number of cases, n (%) | χ2 | P | |
|---|---|---|---|---|
| PLCH | Non-PLCH | |||
| Gender | ||||
| Male | 76 (64.9) | 125 (56.6) | 2.237 | 0.135 |
| Female | 41 (35.1) | 96 (43.4) | ||
| Age (years) | ||||
| <3 | 95 (81.2) | 98 (44.4) | 42.414 | <0.001 |
| ≥3 | 22 (18.8) | 123 (55.6) | ||
The median age of PLCH children was significantly lower than that of non-PLCH patients (18.63 months vs. 43.4 months, P<0.001). PLCH: Pulmonary Langerhans cell histiocytosis.
Among the 117 PLCH children in this study, 106 cases (90.6%) had no obvious respiratory symptom, while only 11 cases (9.4%) had cough, wheezing, or shortness of breath.
Organ involvement
All PLCH children had other organ involvement and there were 56 cases (47.9%) with other “risk organ” involvement. There were 91 cases (77.8%) with bone destruction, 75 cases (64.1%) with rash, 51 cases (43.6%) with liver involvement, 33 cases (28.2%) with spleen involvement, 13 cases (11.1%) with hematopoietic system involvement, and 12 cases (10.3%) with pituitary involvement. Moreover, hearing damage and thyroid, lymph node, or other organs' involvement were not common.
Imaging findings
HRCT scanning was carried out in all PLCH patients in this study. The most common manifestation was cystic lesions, which was found in 29 cases (24.8%). Moreover, there were some other radiologic findings such as ground glass opacity in 25 cases (21.4%), micronodular pattern in 19 cases (16.2%), and emphysema in 16 cases (13.7%) [Figure 1]. In this study, pulmonary imaging findings could be limited to a certain type of lesion or include the coexistence of several types of lesions.
Figure 1.
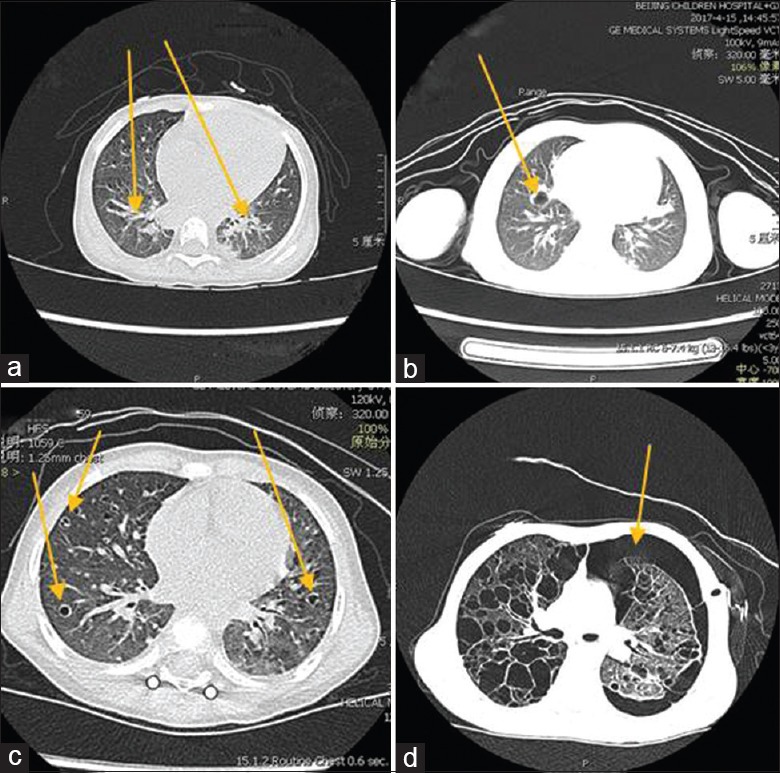
Common pulmonary imaging findings in children with pulmonary Langerhans cell histiocytosis. (a) Patch and cords' shadows. (b) Mass and nodules. (c) Interstitial lesions and multiple cysts. (d) Cysts rupture to form pneumothorax.
Pulmonary function
In this study, 104 cases of children with PLCH accepted the lung function tests. The results showed that 76 patients (73.1%) had abnormal pulmonary function, among whom, there were 63 cases (82.9%) with obstructive ventilation dysfunction, three cases (3.9%) with restrictive ventilation dysfunction, and ten cases (13.2%) with mixed ventilation dysfunction. Fifteen of 28 patients with normal pulmonary function had improved or stabilized, while 49 of 73 cases with abnormal pulmonary function had improved or stabilized. No significant difference in the prognosis was found between the two groups (53.6% vs. 67.1%, χ2 = 1.601, P = 0.206).
Treatment outcome
Among the 117 children with PLCH, four patients gave up treatment and lost to follow-up in the early stage of chemotherapy due to economic reasons, while the remaining 113 patients were treated according to Histiocyte Society LCH-III protocol. As a whole, 5-year event-free survival (EFS) and overall survival (OS) were 55.7% ± 5.2% and 93.6% ± 2.3%, respectively. Sixty-eight cases (60.2%) were always in NAD or stable disease state. Thirty-eight cases (33.6%) had progressed or relapsed disease. Among them, progression or recurrence was more common in other organs than that in lung, there is 33 (86.8%) and five cases (13.2%), respectively. Seven cases gave up treatment or died in hospital due to multiple organ failure, which occurred before chemotherapy or in the early stage of chemotherapy. Notably, lesions such as patch, cords, or ground glass opacity could be restored, while micronodular, cysts, or pneumothorax often left residual lesions.
We also compared 5-year OS and EFS between PLCH children with (RO+ group) or without (RO− group) “risk organ” involvement. The results showed that the prognosis of RO− group was significantly better than RO+ group [Figure 2].
Figure 2.

Treatment outcome of PLCH patients grouped by “risk organ” involvement. (a) EFS: There was a significant difference in the EFS between children with and without “risk organ” involvement (43.7% ± 7.7% vs. 66.3% ± 6.5%, χ2 = 5.399, P = 0.020). (b) OS: The 5-year OS of PLCH children with “risk organ” involvement was significantly lower than those without “risk organ” involvement (86.0% ± 4.9% vs. 100%, χ2 = 8.793, P = 0.003). PLCH: Pulmonary langerhans cell histiocytosis; RO−: Without “risk organ” involvement; RO+: With “risk organ” involvement; EFS: Event-free survival; OS: Overall survival.
Moreover, multifactor Cox regression analysis showed that the “risk organ” involvement was an independent risk factor affecting prognosis (hazard ratio [HR] = 1.9, P = 0.039), while age was not an independent risk factor (HR = 1.3, P = 0.550).
DISCUSSION
PLCH is an interstitial primary pulmonary disease, characterized by Langerhans cell proliferation. PLCH in adults typically involves lung alone and predominantly affects young smokers from the ages of 20 to 40 years, while it generally involves multiple organ systems in children.[4,5] The etiology of children suffering from isolated PLCH is still not clear. Some studies[7,8] have shown that passive smoking may play a role. Increased use of lung biopsy in children with lung lesions may increase the number of pediatric patients diagnosed with isolated PLCH. Noninvasive techniques such as bronchoalveolar lavage cytology based on morphologic features and immunocytochemistry can also help in diagnosis.[9] In this study, there were 117 PLCH cases, accounting for 34.6% of the total LCH children. The proportion was higher than the results of recent studies.[10,11,12] This might be related to regional differences such as passive smoking, air pollution, or genetic difference, but more research is still needed to prove these. All the PLCH children in the study had other organ involvement, in which bone destruction (77.8%), rash (64.1%), and liver damage (43.6%) were very common. In addition, some children also had other system involvement such as pituitary, spleen, hematopoietic system, hearing system, thyroid, and lymph nodes. Of the 117 children with PLCH, 56 cases (47.9%) had “risk organ” involvement, similar to the results of Ronceray et al.[11]
Early diagnosis and treatment are essential to prognosis of PLCH. However, in this study, only 11 cases of the 117 children with PLCH had respiratory symptoms included cough, wheezing, and shortness of breath, none of which were initial symptoms. This suggested that the respiratory symptoms in PLCH children were difficult to recognize at an early stage. Early diagnosis of PLCH was difficult. Studies have shown that the proportion of PLCH in multiple cystic lung disease (MCLD) is not low. Gang et al.[13] conducted a retrospective study of 25 children with MCLD, in which three cases were confirmed with PLCH, and four cases were suspected LCH, altogether accounting for up to 28% of total cases. The treatment of PLCH is widely different from other MCLDs such as bronchopulmonary dysplasia and idiopathic pulmonary fibrosis. Lesions such as patch, cords, or ground glass opacity are reversible if timely treatment is given. However, when lesions develop to micronodular, cysts, or pneumothorax, the patients might suffer from serious sequelae. To avoid misdiagnosis and treatment delay, early identification and diagnosis of PLCH is essential. PLCH should be considered in the differential diagnosis of extensive interstitial lung disease in children, and systemic examination and lung biopsy should be performed if necessary.
In this study, the age distribution of children in PLCH group was significantly younger than that in non-PLCH group, which was consistent with the results of Ronceray et al.[11] This indicates that PLCH is more frequent in infants and young children, which may be because of younger children's relatively narrow respiratory track compared with adults, immature immune system, and Th2-dominant immune response. These may be related to low age preference of PLCH. Considering that the boys had a higher risk of airway obstruction than girls for their smaller airway capacity and higher airway resistance,[14] we compared the gender ratio differences between the PLCH group and non-PLCH group. The results showed that the ratio in PLCH group (1.85:1) was higher than that in non-PLCH group (1.30:1), but the difference was not significant (P = 0.135). The anatomical differences in the respiratory tract between male and female children may not be sufficient to cause a difference in the incidence of PLCH. Further studies with larger sample sizes may be needed.
PLCH mainly involved the distal respiratory bronchioles. The lung function of most early-stage PLCH patients was normal or only showed dysfunction of alveolar dispersion. However, there was obstruction, restriction, or mixed ventilation dysfunction in the late stage. Thus, in the differential diagnosis of diffuse interstitial lung disease with obstructive ventilatory dysfunction, PLCH needs to be taken into consideration. In this study, 73.1% of PLCH children had abnormal pulmonary function, mainly with obstructive ventilatory dysfunction (82.9%). However, we did not find significant correlation between the pulmonary function and the disease prognosis. There are many factors affecting pulmonary function including the development of the lung itself, infection, and other lung diseases, so its diagnostic value for PLCH is limited. Pulmonary function, therefore, cannot serve as an independent diagnostic criterion for PLCH, but it can be used as an auxiliary means for diagnosis and judgment of disease stage.
Other organ systems are typically involved in most children diagnosed as PLCH. The treatment of these patients was basically in accordance with the histiocyte society LCH-III protocol with the treatment of isolated PLCH cases based on chemotherapy.[15] Furthermore, for some refractory patients, cladribine was found to improve respiratory function and reduce the size of cysts, even in patients with advanced disease.[16,17]
In recent years, many studies[10,11,12,18] showed that pulmonary involvement did not play an important role on the prognosis of children with LCH, and the Histiocyte Society has gradually not classified lung as a “risk organ.” The results of this study showed that the 5-year OS rate of PLCH patients without “risk organ” involvement was significantly higher than that of children with “risk organ” involvement (100% vs. 86%), and both groups had better outcomes compared to the results of Ronceray et al.'s study.[11] Moreover, among the children with progressed or relapsed disease, only 13.2% were mainly due to progression or recurrence of lung damage. The lung lesions in the vast majority of PLCH children could be restored (or maintained stable) after treatment, which also showed that pulmonary involvement might not be a prognostic risk factor. Management of “risk organ” involvement was essential to improve progression-free survival.
As we all know, pulmonary involvement is no longer an independent prognostic risk factor, but there will be serious progression or complication in the absence of timely and effective treatment. Nagy et al.[19] reported a 2-year-old girl with isolated PLCH. Since LCH was not medically treated for 3 years due to parental refusal, the disease could be regarded as having followed its natural course. In the beginning, the X-ray revealed significant bilateral and multifocal reticulonodular infiltrates in all lung lobes. After 3 years of follow-up, HRCT revealed widespread, severe cystic fibrotic destruction characteristic of the honeycomb lungs. In this study, a 4-year-old boy with recurrent rash, cough, wheezing, and other symptoms for up to 1 year had not received regular treatment before admission to our hospital. With the lung, skin, and liver involvement, he was treated with LCH-III-Group 1 protocol. Pneumothorax occurred repeatedly during chemotherapy [Figure 3], and finally, the boy died of severe tension pneumothorax at 22 weeks of chemotherapy.
Figure 3.

Repeated pneumothorax in the course of one patient with pulmonary Langerhans cell histiocytosis.
The present study is a retrospective observational study with inherent limitations. First, for lung biopsy was rarely performed, most of PLCH patients in this study were diagnosed by a positive biopsy of extrathoracic localization associated with characteristic HRCT lung findings. This may lead to misdiagnosis of some cases, especially the patients with isolated PLCH; moreover, some infants or young children had difficulty completing lung function tests which may be the reason for some imperfect data.
In summary, PLCH often occurred in infants and young children, with occult clinical manifestations. HRCT was important for diagnosis and assessing the disease. Children with PLCH were generally associated with other organ involvement. The involvement of “risk organs” was a poor prognostic factor for PLCH. Early diagnosis, standard treatment, and control of “risk organ” progression result in a good prognosis for PLCH; lung lesions were mostly reversible or stable, and relapse of lung disease was not common. Clinicians should improve the awareness of this disease to reduce misdiagnosis.
Financial support and sponsorship
This study was supported by grants from the National Science and Technology Key Projects (No. 2017ZX09304029004), Beijing Municipal Science and Technology Commission (No. Z171100001017050), National Natural Science Foundation of China (No. 81700186), Scientific Research Common Program of Beijing Municipal Commission of Education (No. KM201710025019), Pediatric Project of Ai You Foundation (No. AYEK201802), and Talent Training Project-Fostering Fund of National Natural Science Foundation of Beijing Children's Hospital, Capital Medical University (No. GPY201713).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Peng Lyu
REFERENCES
- 1.Favara BE, Feller AC, Pauli M, Jaffe ES, Weiss LM, Arico M, et al. Contemporary classification of histiocytic disorders. The WHO committee on histiocytic/Reticulum cell proliferations. Reclassification working group of the histiocyte society. Med Pediatr Oncol. 1997;29:157–66. doi: 10.1002/(sici)1096-911x(199709)29:3<157::aid-mpo1>3.0.co;2-c. doi: 10.1002/(SICI)1096-911X(199709) 29:3%3C157::AID-MPO1%3E3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 2.Berres ML, Allen CE, Merad M. Pathological consequence of misguided dendritic cell differentiation in histiocytic diseases. Adv Immunol. 2013;120:127–61. doi: 10.1016/B978-0-12-417028-5.00005-3. doi: 10.1016/B978-0-12-417028-5.00005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suri HS, Yi ES, Nowakowski GS, Vassallo R. Pulmonary Langerhans cell histiocytosis. Orphanet J Rare Dis. 2012;7:16. doi: 10.1186/1750-1172-7-16. doi: 10.1186/1750-1172-7- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim NR, Han J. Pathologic review of cystic and cavitary lung diseases. Korean J Pathol. 2012;46:407–14. doi: 10.4132/KoreanJPathol.2012.46.5.407. doi: 10.4132/KoreanJPathol.2012.46.5.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vassallo R, Ryu JH, Schroeder DR, Decker PA, Limper AH. Clinical outcomes of pulmonary Langerhans'-cell histiocytosis in adults. N Engl J Med. 2002;346:484–90. doi: 10.1056/NEJMoa012087. doi: 10.1056/NEJMoa012087. [DOI] [PubMed] [Google Scholar]
- 6.Minkov M, Grois N, Mcclain K, Nanduri V, Rodriguez-Galindo C, Simonitsch-Klupp I, et al. Langerhans Cell Histiocytosis: Histiocyte Society Evaluation and Treatment Guidelines –. [Last accessed on 2009 Apr]. Available from: https://www.histiocytesociety.org .
- 7.Aydin GB, Kibar E, Han U, Kale Y, Aslan A, Kose G, et al. Pulmonary Langerhans cell histiocytosis in an infant: Can passive smoking accelerate the disease progress? Pediatr Pulmonol. 2007;42:565–7. doi: 10.1002/ppul.20617. doi: 10.1002/ppul.20617. [DOI] [PubMed] [Google Scholar]
- 8.Vassallo R, Ryu JH, Colby TV, Hartman T, Limper AH. Pulmonary Langerhans'-cell histiocytosis. N Engl J Med. 2000;342:1969–78. doi: 10.1056/NEJM200006293422607. doi: 10.1056/NEJM200006293422607. [DOI] [PubMed] [Google Scholar]
- 9.Sharma S, Dey P. Childhood pulmonary Langerhans cell histiocytosis in bronchoalveolar lavage: A case report along with review of literature. Diagn Cytopathol. 2016;44:1102–6. doi: 10.1002/dc.23561. doi: 10.1002/dc.23561. [DOI] [PubMed] [Google Scholar]
- 10.Braier J, Latella A, Balancini B, Castaños C, Rosso D, Chantada G, et al. Outcome in children with pulmonary Langerhans cell histiocytosis. Pediatr Blood Cancer. 2004;43:765–9. doi: 10.1002/pbc.20112. doi: 10.1002/pbc.20112. [DOI] [PubMed] [Google Scholar]
- 11.Ronceray L, Pötschger U, Janka G, Gadner H, Minkov M, German Society for Pediatric Hematology and Oncology, Langerhans Cell Histiocytosis Study Group Pulmonary involvement in pediatric-onset multisystem Langerhans cell histiocytosis: Effect on course and outcome. J Pediatr. 2012;161:129–330. doi: 10.1016/j.jpeds.2011.12.035. doi: 10.1016/j.jpeds.2011.12.035. [DOI] [PubMed] [Google Scholar]
- 12.Odame I, Li P, Lau L, Doda W, Noseworthy M, Babyn P, et al. Pulmonary Langerhans cell histiocytosis: A variable disease in childhood. Pediatr Blood Cancer. 2006;47:889–93. doi: 10.1002/pbc.20676. doi: 10.1002/pbc.20676. [DOI] [PubMed] [Google Scholar]
- 13.Gang G, Guiju L, Jian L, Zhengxiu L, Liu E, Fu Z, et al. Clinical analysis of etiology of 25 cases of multiple cystic lung disease in children. J Chongqing Med Univ. 2016;41:945–9. doi: 10.13406/j.cnki.cyxb.000897. [Google Scholar]
- 14.Becklake MR, Kauffmann F. Gender differences in airway behaviour over the human life span. Thorax. 1999;54:1119–38. doi: 10.1136/thx.54.12.1119. doi: 10.1136/thx.54.12.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varkki S, Tergestina M, Bhonsle VS, Moses PD, Mathai J, Korula S, et al. Isolated pulmonary Langerhans cell histiocytosis. Indian J Pediatr. 2013;80:700–3. doi: 10.1007/s12098-012-0866-x. doi: 10.1007/s12098-012-0866-x. [DOI] [PubMed] [Google Scholar]
- 16.Grobost V, Khouatra C, Lazor R, Cordier JF, Cottin V. Effectiveness of cladribine therapy in patients with pulmonary Langerhans cell histiocytosis. Orphanet J Rare Dis. 2014;9:191. doi: 10.1186/s13023-014-0191-8. doi: 10.1186/s13023-014-0191-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lorillon G, Bergeron A, Detourmignies L, Jouneau S, Wallaert B, Frija J, et al. Cladribine is effective against cystic pulmonary Langerhans cell histiocytosis. Am J Respir Crit Care Med. 2012;186:930–2. doi: 10.1164/ajrccm.186.9.930. doi: 10.1164/ajrccm.186.9.930. [DOI] [PubMed] [Google Scholar]
- 18.Smets A, Mortelé K, de Praeter G, François O, Benoit Y, Kunnen M, et al. Pulmonary and mediastinal lesions in children with Langerhans cell histiocytosis. Pediatr Radiol. 1997;27:873–6. doi: 10.1007/s002470050260. doi: 10.1007/s002470050260. [DOI] [PubMed] [Google Scholar]
- 19.Nagy B, Soós G, Nagy K, Dezso B. Natural course of isolated pulmonary Langerhans' cell histiocytosis in a toddler 3-year follow-up. Respiration. 2008;75:215–20. doi: 10.1159/000090159. doi: 10.1159/000090159. [DOI] [PubMed] [Google Scholar]