

Abstract
BACKGROUND:
The dynamic role of autophagy in cancer development is a topic of considerable research and debate. Previously published studies have shown that anal cancer development can be promoted or prevented with the pharmacologic inhibition or induction, respectively, of autophagy in a human papillomavirus (HPV) mouse model. However, these results are confounded by the fact that the drugs utilized are known to affect other pathways besides autophagy. It has also been shown that autophagic inhibition occurs in the setting of HPV16 oncoprotein expression (E6 and E7) and correlates with increased susceptibility to anal carcinogenesis.
MATERIALS AND METHODS:
In this study, we employed a conditional, genetic, autophagic (Atg7) knockout mouse model to determine conclusively that autophagy has a role in anal cancer development, in the absence or presence of E6 and E7.
RESULTS:
In mice lacking both HPV16 oncogenes, knockout of autophagy followed by exposure to a carcinogen resulted in a tumor incidence of 40%, compared to 0% in mice treated with a carcinogen alone with an intact autophagic pathway (P = 0.007). In mice expressing either one or both HPV16 oncoproteins, the addition of genetic knockout of autophagy to carcinogen treatment did not lead to a significant difference in tumor incidence compared to carcinogen treatment alone, consistent with the ability of HPV oncogenes to inhibit autophagy in themselves.
CONCLUSIONS:
These results provide the first conclusive evidence for the distinct role of autophagy in anal carcinogenesis, and suggest that autophagy is a plausible target for therapies aimed at reducing anal dysplasia and anal cancer development.
Keywords: Anal cancer, autophagy, carcinogenesis, human papillomavirus
Introduction
Autophagy is a catabolic process where damaged intracellular components are removed.[1,2] Its specific role in anal cancer development is unknown. The primary risk factor for anal cancer is infection with the human papillomavirus (HPV). We have demonstrated that autophagy is inhibited by the expression of HPV oncoproteins, E6 and E7, and that cancer development is promoted or inhibited with pharmacologic inhibition or induction of autophagy, respectively.[3] These pharmacologic studies are hypothesis generating, but lack target specificity.[3] In this study, a genetic knockout mouse model of autophagy, (Atg7), is used to specify the role of autophagy in anal carcinogenesis.
Materials and Methods
Mice
K14E6/E7 mice were generated as previously described.[4] These mice constitutively express the HPV16 oncoproteins E6 and E7 in their epithelium, from an epithelial-specific keratin 14 (K14) transcriptional promoter. When treated with the carcinogen 7,12 dimethylbenz[a]anthracene (DMBA), these mice develop anal dysplasia that progresses to invasive squamous cell carcinoma at a high frequency, with pathologies and genetic changes similar to those seen in humans.[4] From these mice, we generated a conditional, genetic knockout mouse model for the essential autophagic protein Atg7. The generated mice carry both, one, or none of the HPV oncoproteins, E6 and E7, along with Atg7f/f and CreER. The inducible CreER transgene is expressed from the same epithelial-specific K14 transcriptional promotor as described above, and its activity is induced by topical exposure to 4-OH tamoxifen (4-OH Tam), resulting in the knockout of Atg7. K14E6+/-/K14E7+/-/K14CreERTM+/-/Atg7f/f, K14E6+/-/K14CreERTM+/-/Atg7f/f, K14E7+/-/K14CreERTM+/-/Atg7f/f and K14CreERTM+/-/Atg7f/f mice were generated from crosses between K14E6+/-/Atg7f/f mice (mixed strained of FVB/N and C57BL/6, respectively) and K14E7+/-/K14CreERTM+/-/Atg7f/f mice (FVB/N, CD-1 and C57BL/6, respectively). This resulted in the creation of a mixed mouse strain of FVB/N,CD-1, and C57BL/6. All mice in the study were littermates and thus have the same mixed genetic background. A minimum of 10 mice per group were studied to detect at least a 50% difference in tumor incidence (alpha < 0.05, and beta error of 80% between the treated and untreated groups). Mice were included based on genotype and randomized to treatment groups by cage before treatment initiation (with and without DMBA and/or with and without 4-OH Tam, as described below). Mice were assessed weekly for anal tumor appearance and growth. Animals were sacrificed at 20 weeks postinitiation of DMBA treatment or when they met euthanasia requirements. The 20 weeks time point was chosen based on previous work demonstrating that in mice with both HPV16 oncoproteins expressed, 100% will have an anal tumor at 20 weeks of DMBA treatment.
All mice were maintained in an American Association for Accreditation of Laboratory Animal Care-approved Wisconsin Institute for Medical Research Animal Care Facility. The experiments were performed in accordance with approved the Institutional Animal Care and Use Committee Protocol M005946.
4-OH Tam administration
The Cre-recombinase is on a K14 promoter and is induced selectively in the setting of 4-OH Tam (70% Z isoform, remainder primarily E isoform; Sigma H6278, St Louis, MO). Mice in the 4-OH Tam treatment group received a topical application of 4-OH Tam (0.1 mg) dissolved in ethanol/corn oil, applied to the anus for 5 days consecutively, beginning at 5 weeks of age. The following week, mice in the DMBA treatment group were started on DMBA treatment, as described below. Age-matched mice that were not treated with 4-OH Tam served as controls.
7,12 Dimethylbenz[a]anthracene induced anal carcinogenesis
Once weekly, 0.12 μmole of DMBA (Sigma D3524-1G; in 60% acetone/40% dimethylsulfoxide) was applied topically to the anus of mice, as previously described, in mice in the DMBA treatment group.[4] Age-matched mice that were not treated with DMBA served as controls.
Histopathological analysis
Anal tissue was collected at the time of euthanasia and fixed in 4% paraformaldehyde (J. T. Baker S898-07) for 24 h, and then placed in 70% ethanol. After fixation, the tissues were embedded in paraffin and underwent serial 5 μm sectioning. Every fourth slide underwent hematoxylin and eosin (H and E) staining and was evaluated by a gastrointestinal fellowship-trained, board-certified surgical pathologist for evidence of dysplasia (low-grade vs. high-grade), carcinoma in situ, or invasive squamous cell carcinoma. Histology was scored in the following manner: 0 for normal, 1 for low-grade dysplasia, 2 for high-grade dysplasia, 3 for carcinoma in situ, and 4 for invasive carcinoma.
Immunofluorescence for autophagic proteins
Paraffin sections were de-paraffinized, rehydrated, subjected to antigen retrieval with 10 mM sodium citrate buffer pH 6.0 and heat, permeabilized with 0.2% Triton X-100, and blocked with 10% donkey serum in phosphate buffered saline (1X PBS).[5] To examine autophagy, the sections were stained overnight at 4°C with monoclonal rabbit antibody against Atg7 (diluted 1:100 in 10% donkey serum in PBS; Santa Cruz Biotechnology, Dallas, TX, USA) and rabbit antibody against light chain 3 (LC3) β (diluted 1:50 in 10% donkey serum in PBS; Santa Cruz Biotechnology, Dallas, TX, USA). Sections were then washed and stained for 1 h in the dark at room temperature, with donkey anti-rabbit Fluor 488 (diluted 1:500 in 10% donkey serum in PBS; Life Technologies, Carlsbad, CA, USA). Slides were then counterstained with a 4’,6-Diamidino-2-Phenylindole, dihydrochloride (DAPI) nuclear stain. Slides were imaged using the Zeiss Axio Imager M2 imaging system. Images at ×200 magnification were obtained for each sample, focusing on the anorectal transition zone (ATZ). Each image was imported into ImageJ version 2.0.0 (National Institutes of Health, Bethesda, Maryland, USA) (Fiji distribution) and underwent processing as follows: images were split into two channels (488 and DAPI). All images were thresholded using the default dark background. The areas of interest were manually selected to focus on the ATZ and then RawIntDen was used to measure the intensity of the fluorescent signal in regions of interest. The RawIntDen was then normalized for the area of the region selected (RawIntDen/Area).
Statistical analysis
Fisher's two-sided exact test was used to determine differences in tumor incidence between treatment groups at 20 weeks. A Kaplan–Meier analysis was used to determine differences in time to tumor onset. Comparisons in levels of expression of Atg7 and LC3 β between treatment groups were made using an independent samples t-test. SPSS version 24 (IBM, Armonk, North Castle, NY, USA) and SAS version 9.4 (SAS Institute Inc., Cary, NC, USA) were utilized to perform these analyses. Statistical significance was defined as P < 0.05.
Results
Autophagic and LC3 β immunofluorescence
Using immunofluorescence (IF), we confirmed the knockdown of Atg7 in K14CreERTM+/-/Atg7f/f mice [Figure 1A] when treated with 4-OH Tam (mean ± standard error of the mean [SEM]; 0.068 ± 0.009 vs. 0.048 ± 0.002; P < 0.05). Figure 1B shows representative IF images of Atg7 staining of anal tissue from K14CreERTM+/-/Atg7f/f mice, following treatment with DMBA alone. Panel B i is Atg7, Panel B ii is the DAPI nuclear stain, while Panel B iii shows the fused image with Atg7 in green and DAPI in blue. Figure 1C shows a representative IF image of a mouse treated with with 4-OH Tam treatment followed by DMBA. Panel C i shows Atg7, Panel C ii is the DAPI nuclear stain, and Panel C iii. shows the fused image with Atg7 in green and DAPI in blue. All anal tissue samples were isolated at 20 weeks following 4-OH Tam administration, thus indicating the durable and long-term knockout of Atg7 when mice were treated with 4-OH Tam.
Figure 1.

Atg7 Immunofluorescence. In panel A K14CreERTM+/-/Atg7f/f mice receiving DMBA alone or with 4-OH Tam were analyzed for Atg7. Panels B (DMBA alone) and C (4-OH Tam followed by DMBA) show Atg7 (i.), nuclear staining (ii), and Atg7 (green) and nuclear staining (blue) (iii.)
Anal tissues from the same mice were also assessed for LC3 β, a marker of Atg7 induction, via IF [Figure 2]. As expected, mice treated with both 4-OH Tam and DMBA had lower LC3 β expression, although the mean difference in comparison with those mice treated with DMBA alone did not quite reach statistical significance [Figure 2a; mean ± SEM; 0.68 ± 0.14 vs. 0.38 ± 0.08; P = 0.078]. Figures 2b and c show representative IF images of LC3 β protein expression (green) and nuclear staining with DAPI (blue) from K14CreERTM+/-/Atg7f/f mice without and with 4-OH Tam treatment, respectively, along with DMBA treatment.
Figure 2.
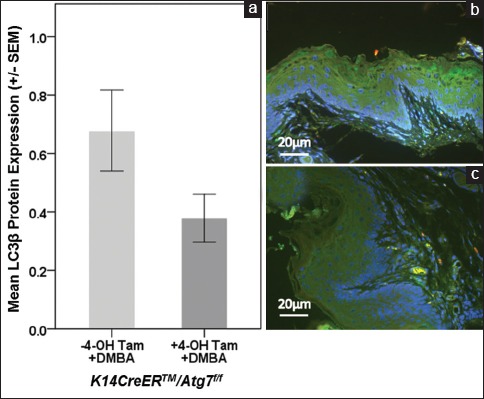
LC3β protein expression in K14CreERTM+/-\/Atg7f/f mice. Panel A shows that in K14CreERTM+/-/Atg7f/f mice there was a trend toward lower LC3β (green) in 4-OH TAM and DMBA compared to DMBA alone. Panels B (DMBA alone) and C (4-OH TAM followed by DMBA) are representative images
Tumor free survival
As shown in Figure 3, and consistent with previous publications using the K14E6/E7 mouse model,[4,6] treatment with DMBA alone (without tamoxifen) in the presence of both oncogenes (K14E6+/-/K14E7+/-/K14CreERTM+/-/Atg7f/f) leads to the highest rate of tumor development (13 of 14 mice, 92.8%). E7 expression alone, again consistent with previous data, leads to a much higher rate of tumor incidence than E6 expression alone (75.0% [9 of 12] vs. 27.3% [3 of 11]). Mice not expressing either oncogene (K14CreERTM+/-/Atg7f/f) did not develop tumors over 20 weeks of DMBA treatment without 4-OH Tam treatment (0 of 16), indicating no significant perturbations in risk of anal carcinogenesis in this mixed genetic background (FVB,CD-1 and C57BL/6) compared to prior studies on the pure FVB/N background.[6] Fisher's exact tests comparing all four groups yielded a significant difference in tumor incidence (P = 0.0014).
Figure 3.
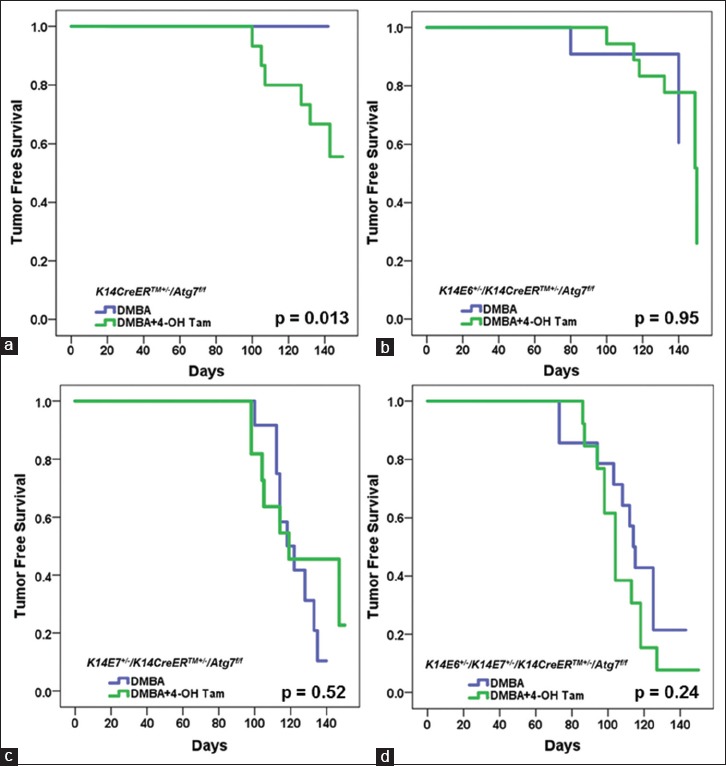
Variation in tumor free survival by genotype and treatment group. Within genotype comparisons showed that only K14CreERTM+/-/Atg7f/f mice treated with 4-OH Tam and DMBA showed a significant difference compared to DMBA alone (Panel a; Log-Rank P = 0.013). Panel a K14CreERTM+/-/Atg7f/f, Panel b K14E6+/-/K14CreERTM+/-/Atg7f/f, Panel c K14E7+/-/K14CreERTM+/-/Atg7f/f, and Panel d K14E6+/-/K14E7+/-/K14CreERTM+/-/Atg7f/f
Figure 3 also shows K14E6+/-/K14E7+/-/K14CreERTM+/-/Atg7f/f, K14E7+/-/K14CreERTM+/-/Atg7f/f, K14E6+/-/K14CreERTM+/-/Atg7f/f and K14CreERTM+/-/Atg7f/f mice, exposed to both 4-OH Tam and DMBA. In mice lacking both HPV oncogenes (K14CreERTM+/-/Atg7f/f), a significant number of mice exposed to both 4-OH Tam and DMBA developed tumors (40% [6 of 15]), whereas none of the mice treated with DMBA alone developed tumors (0 of 16; Log-Rank P = 0.013) [Figure 3a]. This finding clearly demonstrates that the inhibition of autophagy increases the susceptibility to anal cancer. As expected, this effect of Atg7 knockout was not observed in the setting of HPV16 oncogene expression, which we have shown previously to inhibit autophagy independently.[6,7] Thirteen of 14 K14E6+/-/K14E7+/-/K14CreERTM+/-/Atg7f/f mice (85.7%) treated with DMBA alone developed tumors, whereas 12 of 13 mice (92.3%) treated with both 4-OH Tam and DMBA developed tumors [Figure 3d; Log-Rank, P = 0.24]. The lack of effect of the genetic inhibition of autophagy in the presence of HPV oncogenes was also seen in mice expressing just E6 (27.3% [3 of 11] vs. 33.3% [6 of 18]; Log-Rank P = 0.95) [Figure 3b] or just E7 (63.6% [7 of 11] vs. 75% [9 of 12]; Log-Rank P = 0.52) [Figure 3c]. These results indicate that in the absence of HPV oncoprotein expression, conditional knockout of Atg7 promotes anal carcinogenesis. In the presence of HPV oncoproteins, this effect is obscured, reflecting the ability of HPV oncogenes to inhibit autophagy on their own.
Histopathology
Similar trends were observed when tissue was histopathologically scored for the presence or absence of squamous cell carcinoma of the anus (SCCA) in the different cohorts of mice. K14CreERTM+/-/Atg7f/f, K14E6+/-/K14CreERTM+/-/Atg7f/f, K14E7+/-/K14CreERTM+/-/Atg7f/f, and K14E6+/-/K14E7+/-/K14CreERTM+/-/Atg7f/f mice given weekly DMBA alone, or in combination with 4-OH Tam, underwent histological assessment [Table 1]. Of the K14CreERTM+/-/Atg7f/f mice treated with DMBA and 4-OH Tam, 40% (six out of 15) developed SCCA compared to 0% (0 out of 16) of K14CreERTM+/-/Atg7f/f mice treated with DMBA only, trending towards significance (Fisher's exact test P = 0.0068). In the HPV16 transgenic backgrounds, there were no differences in the incidence of SCCA [Table 1: P =0.65 for E6, P = 0.67 for E7, P = 1.0 for E6/E7 mice]. Figure 4a and b show representative H and E images (200X) of tissues from K14CreERTM+/-/Atg7f/f mice not treated or treated with 4-OH Tam in the presence of DMBA treatment, respectively. Panel A illustrates a representative, low-grade dysplasia in a K14CreERTM+/-/Atg7f/f mouse treated with DMBA alone, while Panel B illustrates a representative, grade 1 squamous cell carcinoma in the K14CreERTM+/-/Atg7f/f mouse treated with 4-OH Tam and DMBA. Panels C and D show representative microscopic images (200X) of tissues from K14E6+/-/K14E7+/-/K14CreERTM+/-/Atg7f/f mice not treated or treated with 4-OH Tam, respectively, in the presence of DMBA treatment. Panel C illustrates a representative grade 1 squamous cell carcinoma in a K14E6+/-/K14E7+/-/K14CreERTM+/-/Atg7f/f mouse treated with DMBA alone, while Panel D illustrates a representative grade 1 squamous cell carcinoma in a K14E6+/-/K14E7+/-/K14CreERTM+/-/Atg7f/f mouse treated with 4-OH Tam and DMBA.
Table 1.
Distribution of cancer incidence by genotype and treatment group. K14CreERTM+/-/Atg7f/f mice treated with 4-OH Tam and 7,12 dimethylbenz[a]anthracene showed increased cancer compared to 7,12 dimethylbenz[a] anthracene alone (P=0.0068). The addition of 4-OH Tam did not impact cancer incidence in the remaining genotypes
Figure 4.
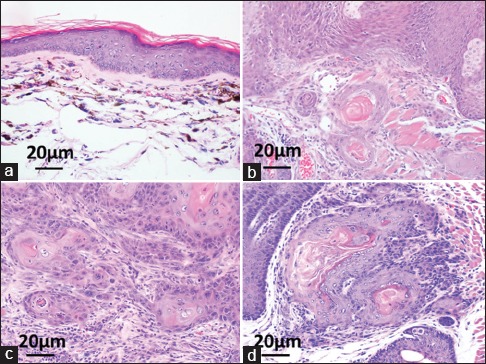
Tissue Histology. (Panel a) shows low-grade dysplasia in K14CreERTM+/-/ Atg7f/f mice with 7,12 dimethylbenz[a]anthracene alone, whereas (Panel b) shows SCCA when adding 4-OH Tam. (Panels c and d) show SCCA in K14E6+/-/K14E7+/-/ K14CreERTM+/-/Atg7f/f mice without and with 4-OH Tam, respectively
Discussion
The role of autophagy in cancer development is controversial. This manuscript presents a conditional, genetic Atg7 knockout mouse model wherein the localized knockout of Atg7 allowed testing of the hypothesis that autophagy is important in preventing in anal carcinogenesis. We chose Atg7 for our target protein because it is essential for the Atg7 process, as described in the introduction. Since anal carcinogenesis is a localized disease, we chose to knock out autophagy at the anus only, instead of using a global mouse knockout model. IF of Atg7 in this mouse model demonstrated that 4-OH Tam-induced knockout of Atg7 expression at the anus was both robust and sustained over the 20-week DMBA treatment period [Figure 1]. In Figure 2, there was a trend to a decrease in LC3β with Atg7 knockout, however it did not reach statistical significance. This may be related to one or both of the following: not enough mice to power the analysis of LC3β or the fact that our measurement of LC3β protein expression occurred 20 weeks following Atg7 knockout in area of the body with frequent epithelial turnover. From our results, we cannot conclude what impact this may have had on median tumor-free survival. Regardless, in the absence of HPV16 oncogene expression, the knockout of autophagy, as measured by essential Atg7 knockout, led to a significant increase in tumor development in mice treated with DMBA compared to mice where autophagy was left intact [Figure 3a]. This finding clearly demonstrates the importance of autophagy in anal cancer development. In the HPV transgenic mice, the genetic disruption of autophagy did not lead to substantial increases in tumor development, consistent with our previous data and with the data of other groups, demonstrating a strong inhibitory effect of E6 and E7 on autophagy.[6] E6 has been shown to lead to sustained Akt/mammalian target of rapamycin activity (upstream inhibitor of autophagy), which enhances cell growth and survival[8] and inhibits p53, leading to genomic instability and carcinogenesis.[9] E7 has been demonstrated to inhibit the retinoblastoma tumor suppressor, leading to sustained Akt activity (again an upstream inhibitor of autophagy).[10,11]
This study provides, for the first time, clear genetic proof that autophagy plays a critical role in anal carcinogenesis. Given the strong effect of autophagic inhibition on anal carcinogenesis, our data provide solid evidence to support further research on modulating autophagy to prevent the ever-growing health problem of anal cancer.
Financial support and sponsorship
This work is supported by the NIH/NCI (E. H. C., K. A. M., grant number - P30 CA14520), the Society for Surgery of the Alimentary Tract (E. H. C., grant number - PRJ96MZ) and the National Cancer Institute of the National Institutes of Health (B. L. R., grant number - T32CA090217).
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We are grateful to Dr. Paul Lambert for providing the mouse models used in the above studies and for his critical reading of this manuscript. The authors would like to thank the University of Wisconsin Carbone Cancer Center for the use of its Experimental Pathology Laboratory to conduct paraffin embedding and serial sectioning of pathological specimens.
References
- 1.Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis. 2011;32:955–63. doi: 10.1093/carcin/bgr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glick D, Barth S, Macleod KF. Autophagy: Cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rademacher BL, Matkowskyj KA, Meske LM, Romero A, Sleiman H, Carchman EH, et al. The role of pharmacologic modulation of autophagy on anal cancer development in an HPV mouse model of carcinogenesis. Virology. 2017;507:82–8. doi: 10.1016/j.virol.2017.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stelzer MK, Pitot HC, Liem A, Schweizer J, Mahoney C, Lambert PF, et al. A mouse model for human anal cancer. Cancer Prev Res (Phila) 2010;3:1534–41. doi: 10.1158/1940-6207.CAPR-10-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barth S, Glick D, Macleod KF. Autophagy: Assays and artifacts. J Pathol. 2010;221:117–24. doi: 10.1002/path.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carchman EH, Matkowskyj KA, Meske L, Lambert PF. Dysregulation of autophagy contributes to anal carcinogenesis. PLoS One. 2016;11:1–21. doi: 10.1371/journal.pone.0164273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carchman EH, Matkowskyj KA, Meske L, Lambert PF. Dysregulation of autophagy contributes to anal carcinogenesis. PLoS One. 2016;11:e0164273. doi: 10.1371/journal.pone.0164273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L, Wu J, Ling MT, Zhao L, Zhao KN. The role of the PI3K/Akt/mTOR signalling pathway in human cancers induced by infection with human papillomaviruses. Mol Cancer. 2015;14:87. doi: 10.1186/s12943-015-0361-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moody CA, Laimins LA. Human papillomavirus oncoproteins: Pathways to transformation. Nat Rev Cancer. 2010;10:550–60. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 10.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–64. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 11.Menges CW, Baglia LA, Lapoint R, McCance DJ. Human papillomavirus type 16 E7 up-regulates AKT activity through the retinoblastoma protein. Cancer Res. 2006;66:5555–9. doi: 10.1158/0008-5472.CAN-06-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]