

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
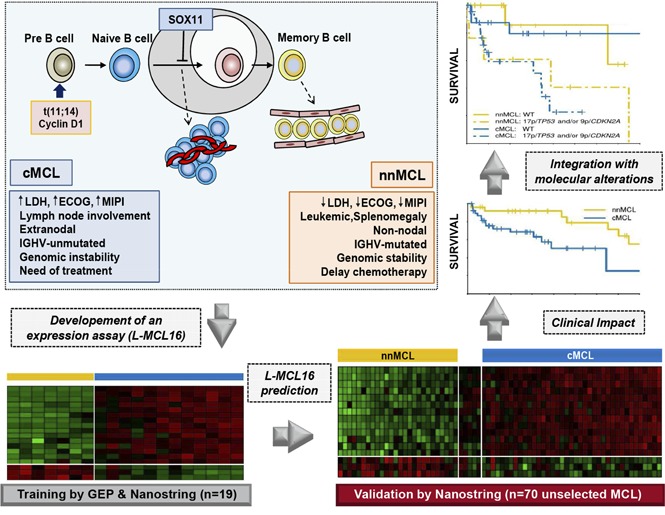
A new molecular assay identifies conventional and leukemic nonnodal MCL with differing clinicobiological features.
The integration of the novel assay with genetic alterations identifies subsets of MCL patients with different management and outcome.
Abstract
Mantle cell lymphoma (MCL) is an aggressive B-cell malignancy, but some patients have a very indolent evolution. This heterogeneous course is related, in part, to the different biological characteristics of conventional MCL (cMCL) and the distinct subgroup of leukemic nonnodal MCL (nnMCL). Robust criteria to distinguish these MCL subtypes and additional biological parameters that influence their evolution are not well defined. We describe a novel molecular assay that reliably distinguishes cMCL and nnMCL using blood samples. We trained a 16-gene assay (L-MCL16 assay) on the NanoString platform using 19 purified leukemic samples. The locked assay was applied to an independent cohort of 70 MCL patients with leukemic presentation. The assay assigned 37% of cases to nnMCL and 56% to cMCL. nnMCL and cMCL differed in nodal presentation, lactate dehydrogenase, immunoglobulin heavy chain gene mutational status, management options, genomic complexity, and CDKN2A/ATM deletions, but the proportion with 17p/TP53 aberrations was similar in both subgroups. Sequential samples showed that assay prediction was stable over time. nnMCL had a better overall survival (OS) than cMCL (3-year OS 92% vs 69%; P = .006) from the time of diagnosis and longer time to first treatment. Genomic complexity and TP53/CDKN2A aberrations predicted for shorter OS in the entire series and cMCL, whereas only genomic complexity was associated with shorter time to first treatment and OS in nnMCL. In conclusion, the newly developed assay robustly recognizes the 2 molecular subtypes of MCL in leukemic samples. Its combination with genetic alterations improves the prognostic evaluation and may provide useful biological information for management decisions.
Visual Abstract
Introduction
Mantle cell lymphoma (MCL) is considered one of the most aggressive lymphoid neoplasms with relatively short responses to therapy and frequent relapses in spite of early and intensive treatment strategies.1 However, different studies have recognized a subset of patients with indolent disease suitable for conservative management for long periods of time without negatively impacting their outcome.2-5 The majority of these latter cases seem to correspond to a distinct MCL subtype recently recognized in the updated World Health Organization classification as leukemic nonnodal MCL (nnMCL), which is clinically characterized by leukemic expression, splenomegaly, and no or minimal lymphadenopathy.2,6 These cases also have molecular, genomic, and epigenomic features that differ from those of conventional MCL (cMCL).2,3,7-12 Gene-expression profiling studies have identified a number of genes differentially expressed between these 2 MCL subtypes,2,3 but their implementation in clinical practice has been limited. One of the most discriminant genes is SOX11, whose expression is high in cMCL and very low in nnMCL. However, the use of this marker has been controversial as a result of different factors that include the identification of cases with borderline SOX11 expression levels, technical difficulties, and concomitant confounding factors, such as the presence of TP53 alterations that impair the outcome of cMCL and nnMCL.2,7,13-15 Given the clinical and biological differences between cMCL and nnMCL, a robust molecular classifier of these MCL subtypes, combined with additional genetic studies, may be useful for recognizing these subtypes in the clinic, assess prognosis, and support management decisions in patients with leukemic MCL.
The need for a robust assay that may identify MCL with different biological features is emphasized by the evolution of current treatment strategies from conventional chemotherapy, often with very intensive regimens, to new molecular targeted therapies.1,16-18 In addition, a “watch and wait” attitude is now considered reasonable for patients who do not have immediate need for therapy based on clinical criteria.5,19-21 A major factor determining the evolution of MCL is tumor cell proliferation.22 The evaluation of the Ki67-proliferation index has been incorporated with clinical features in the biological MCL International Prognostic Index.23,24 More recently, we have generated and validated a refined novel proliferation assay based on RNA extracted from formalin-fixed paraffin-embedded nodal biopsies that defines groups of patients with significantly different outcomes independently of conventional clinical prognostic parameters and improves upon the predictive value of the Ki67 index.25 However, a subset of MCL patients, particularly with nnMCL, have only leukemic disease without tissues accessible for biopsy. The value of the proliferation signature in these samples and in the nnMCL subtype is not known.
In this study, we have designed, analytically validated, and evaluated the clinical impact of a robust gene-expression assay based on the digital quantification of RNA from leukemic samples that reproducibly recognizes cMCL and nnMCL subtypes. We have also evaluated the prognostic impact of this assay combined with the genomic complexity, 17p/TP53, and 9p/CDKN2A alterations of the tumors.
Methods
Study design and samples
The design of the process for developing and validating the new gene expression–based assay for the recognition of nnMCL and cMCL in leukemic samples is summarized in supplemental Figure 1, available on the Blood Web site. The training cohort used to develop the assay included pretreatment peripheral blood (PB) samples from 12 cMCL and 7 nnMCL cases. The tumor cells were purified (>95% as determined by flow cytometry), as previously described.2 The selected cases were considered representative of these 2 MCL subgroups based on previously defined criteria (supplemental Table 1).2,6-8 All cases tested (n = 15) had the t(11;14)(q13;q32) translocation, and all cases had cyclin D1 overexpression. Compared with a set of 54 chronic lymphocytic leukemia (CLL) samples described by Navarro et al,26 the cMCL and nnMCL subgroups showed significantly higher CCND1 (MCL marker) and CD20, and lower LEF1, CD23, FMOD, and KSR2 (CLL markers) (supplemental Figure 2).26,27 All cases of cMCL were SOX11 positive, the patients required immediate treatment at diagnosis, and most of them had generalized lymphadenopathy in addition to the leukemic phase. The nnMCL patients had leukemic presentations with no or minimal lymphadenopathy, were SOX11 negative, and did not require treatment for ≥1 year after diagnosis.
Clinical validation of the assay was performed using the pretreatment PB samples from an independent cohort of 70 MCL patients. Tumor cells were purified in 39 of 70 (56%) samples, as previously described,2 and the final median tumor content was 96% (range, 60-100%). The study was approved by the Institutional Review Board of the Hospital Clinic of Barcelona.
Gene-expression analysis
Global gene-expression profiling of RNA extracted from the PB samples of the training cohort was performed using Affymetrix U133 plus 2.0 microarrays (Thermo Fisher Scientific, Waltham, MA), already deposited under Gene Expression Omnibus accession number GSE79196.26 The raw data were normalized using the frozen robust multiarray analysis method.28 Only the probe set with the highest interquartile range for each gene was used in all analyses. Gene expression of a subset of genes was subsequently evaluated using 200 ng of RNA on the NanoString platform (NanoString Technologies, Seattle, WA), using the “high sensitivity” setting on the nCounter Prep Station and 490 fields of view on the nCounter Analyzer. Normalization was done by dividing the digital counts of all genes of a sample by the geometric mean of the counts of the housekeeping genes of that sample and then multiplying by 1352 (which is the mean of the geometric means of the counts of the housekeeping genes of the training samples). The normalized data were log2 transformed.
Molecular characterization
TP53 mutations in exons 4-11 were investigated by Sanger sequencing7 or whole-exome sequencing,29 and copy number alterations (CNA) were studied using Affymetrix SNP6.0 microarrays (Thermo Fisher Scientific) and analyzed with Nexus BioDiscovery version 9 software (BioDiscovery, Hawthorne, CA).29 TP53 was considered altered when it was mutated and/or deleted. The CDKN2A locus was considered altered when it was deleted (homozygous or heterozygous), and 11q was considered altered when there was a deletion including the ATM gene. The total number of CNAs for each case was computed as a measure of genomic complexity. Immunoglobulin heavy chain genes (IGHVs) were considered mutated when the identity with the germ line was <97%, as previously described.8
Statistical analysis
Limma30 was used to identify differentially expressed genes in the microarray data. A modification of the Bayesian Compound Covariate method, named BCCm,31 was used to build the predictor. The intra- and interlaboratory variability was estimated using mixed models, as implemented in the R package lme4.32 Paired and nonpaired Student t tests were used to compare the gene expression or the predictor scores between groups, as appropriate. Spearman correlation was used to measure the association between continuous variables. The Fisher exact and Mann–Whitney U tests were used to examine the significance of differences in categorical and continuous variables between groups, respectively. The median follow-up was estimated using the reverse-censoring method.33 The end point of the study was the overall survival (OS), which was calculated from the date of diagnosis or the date of sampling to the date of death from any cause. OS and time to first treatment (TTT) were estimated using the Kaplan-Meier (KM) method. The log-rank test and Cox regression were used to assess the relationship between OS (or TTT) and categorical and continuous variables, respectively.
Results
Development of the leukemic MCL assay
To develop a highly reproducible assay that could identify cMCL and nnMCL in leukemic samples, we first analyzed the global gene-expression profiling of the 19 training samples (12 cMCL and 7 nnMCL). Eighteen differentially expressed genes with high fold-change, including 15 upregulated in cMCL and 3 upregulated in nnMCL (supplemental Figure 3A; supplemental Table 2), were selected to include in a pilot NanoString code set, alongside 30 housekeeping genes that were previously described.25 The digital gene expression of these 48 genes was analyzed in the same 19 samples. These data were used to refine the initial 18-gene signature. First, 1 gene (PLEKHG4B) with low expression levels on the NanoString platform was removed. Then, a leave-one-out cross-validation strategy was used to identify the optimal number of genes with minimum classification error, which was minimized when the number of genes included in the model was ≥16 (supplemental Figure 3B). These results suggested that we could remove 1 more gene without losing predictive power. We decided to remove GLUL, given that this gene had the lowest t statistic in this pilot NanoString series and, moreover, had the poorest correlation with the microarray data (supplemental Figure 3C).
Thus, the refined signature consisted of 16 genes: 13 genes upregulated in cMCL (including the previously identified SOX11, HDGFRP3, and DBN1)2 and 3 novel genes upregulated in nnMCL (CD200, BTLA, and SLAMF1). This signature, together with 18 housekeeping genes, was remeasured on the same 19 samples using a smaller NanoString code set. After data normalization, the BCCm was used to build the final model.31 The output of this model is a score that can be used to obtain an estimate of the probability that a given sample belongs to the nnMCL or cMCL category. The cases were classified as cMCL or nnMCL if they had a ≥90% probability of belonging to either subgroup, or they were considered “Undetermined” when the probability was below this cutoff. The final model, named the leukemic MCL (L-MCL16) assay, was locked and used to assign an MCL subgroup to each patient in the validation cohort (Figure 1; supplemental Figure 1).
Figure 1.

Gene-expression–based L-MCL16 scores in the validation cohort. L-MCL16 score for each validation sample in ascending order (top panel) and associated probability of membership in the nnMCL subgroup (middle panel). L-MCL16 signature heat map (13 upregulated genes in cMCL and 3 upregulated genes in nnMCL) (bottom panel); genes are shown in rows, and cases are shown in columns (red indicates high expression). Two clear gene expression patterns can be observed, with high scores corresponding to cMCL samples (blue) and low scores corresponding to nnMCL samples (red). Only 5 samples have an intermediate gene-expression pattern and are considered Undetermined (gray).
Reproducibility and dilution analysis of the L-MCL16 assay
To determine the intra- and interlaboratory reproducibility of the L-MCL16 assay, we selected 11 samples with scores distributed across the full spectrum of assay scores (5 cMCL, 5 nnMCL, and 1 Undetermined). For intralaboratory comparison, the RNA was run on the L-MCL16 assay in duplicate or triplicate, with each run performed on a different NanoString cartridge. For interlaboratory comparisons, RNA aliquots were analyzed in 3 independent laboratories in Barcelona (Spain), Scottsdale, Arizona, and Würzburg (Germany). The results were highly concordant across the duplicates/triplicates, as well as across the different laboratories (Figure 2). A greater variability in scores was observed in the samples predicted as nnMCL compared with cMCL. To evaluate the possible impact of this variability, we calculated the intra- plus interlaboratory standard deviation of each subgroup, which was 21.3 for the nnMCL subgroup and 6.1 for the cMCL subgroup. These deviations are low enough in comparison with the range of scores of the “Undetermined” subgroup (67 points) to ensure that the probability of a nnMCL sample being predicted as cMCL (or vice versa) is almost negligible.
Figure 2.

Intra- and interlaboratory reproducibility of the L-MCL16 assay. L-MCL16 scores of RNA from blood samples run in duplicate or triplicate in Barcelona (Spain) or from RNA aliquots run and analyzed in Scottsdale (AZ) and Würzburg (Germany). The x-axis corresponds to the average L-MCL16 score of the replicates of the same patient, whereas the y-axis corresponds to the score of each replicate. Samples with a score between the dashed lines are considered Undetermined (1 case).
Finally, to determine whether a lower RNA input could have an impact on the final L-MCL16 score, RNA from 6 PB samples was run on the assay with a reduced input of 100 ng of RNA. No significant effect was observed on the scores when they were compared with the 200-ng scores (95% confidence interval [CI], −1.08 to 2.85; P = .301; supplemental Figure 4). Next, serial-dilution experiments using RNA from a healthy PB sample (100%, 60%, 40%, 20%, 10%) were performed in 7 samples distributed across the full spectrum of scores. As expected, dilutions had a high impact, shifting the L-MCL16 score and CCND1 levels to the reference blood score as the proportion of tumor RNA decreased (supplemental Figure 5). Overall, we determined that ≥60% tumor cell content was required to reliably identify a cMCL sample. Given the high overexpression levels of CCND1 in all MCL cases, the dilutional effect of the sample was minor, and we could detect CCND1 overexpression in all cMCL and nnMCL cases, even with only 10% of tumor cells (supplemental Figure 5).
Different clinical and biological features in nnMCL and cMCL
The L-MCL16 assay was then applied to the pretreatment leukemic samples of 70 independent patients diagnosed with MCL (validation cohort). The assay assigned 39 cases (56%) to cMCL and 26 (37%) to nnMCL, and 5 (7%) were Undetermined (Figure 1). The clinicobiological features for each predicted MCL subtype are shown in Table 1. Importantly, there were statistically significant differences in the time from diagnosis to the time that the sample was obtained for the study between both subgroups, with 92% of cMCL samples obtained ≤1 year from diagnosis compared with only 58% of nnMCL samples. Given that most nnMCL patients have stable disease for long periods but eventually may progress, we determined whether the nnMCL expression signature was stable over time. We performed the analysis in sequential samples of 9 cases of nnMCL and 2 cases of cMCL. The nnMCL samples were from untreated patients at diagnosis and at follow-up (range 9-86 months). In 7 patients, the second sample was obtained at the time of clinical progression and prior to any treatment. At the time of the second sample, 2 patients remained untreated and asymptomatic 1 and 4 years later, whereas 4 patients developed splenomegaly but were still asymptomatic, and 3 patients required chemotherapy. For cMCL, we analyzed 2 cases with a PB and splenic sample at diagnosis and a second nodal or PB sample obtained at relapse after treatment 4 and 38 months later, respectively. The L-MCL16 prediction of the initial and sequential samples for all 11 patients was concordant (Figure 3). Additionally, there was no difference between the mean score of the initial and the sequential samples (95% CI, −13.5 to 122.9; P = .104). These findings emphasize the stability of the nnMCL and cMCL signatures, suggesting that they may represent a constitutive feature of the tumor cells.
Table 1.
Clinicobiological characteristics of the MCL patients in the validation cohort according to the L-MCL16 prediction
| Variable | Total | L-MCL16 prediction | P value (cMCL vs nnMCL) | ||
|---|---|---|---|---|---|
| cMCL | nnMCL | Undetermined | |||
| Number of cases (%) | 70 | 39 (56) | 26 (37) | 5 (7) | |
| Clinical data (at diagnosis) | |||||
| Male/female, n | 40/30 | 24/15 | 15/11 | 1/4 | .8 |
| Nodal presentation, n (%) | 28/67 (42) | 24/39 (62) | 4/24 (17) | 0/4 (0) | .001 |
| Splenomegaly, n (%) | 33/61 (54) | 21/38 (55) | 8/19 (42) | 4/4 (100) | .408 |
| LDH (>ULN), n (%) | 11/47 (23) | 11/30 (37) | 0/15 (0) | 0/2 (0) | .008 |
| MIPI high risk, n (%) | 21/40 (52) | 16/26 (62) | 5/13 (38) | 0/1 (0) | .196 |
| ECOG (≥2), n (%) | 6/40 (15) | 6/25 (24) | 0/14 (0) | 0/1 (0) | .071 |
| Lymphocytosis (L/mm3), median (range) | 9 000 (1 200-236 000) | 9 100 (1 200-149 700) | 9 050 (1 930-236 000) | 5750 (3800-7700) | .936 |
| Pathological and molecular data (at sampling) | |||||
| Mean CCND1 expression | 14.3 | 14.5 | 13.9 | 14.7 | .029 |
| Range CCND1 expression | 11.8-17.4 | 12.6-17.4 | 11.8-16.3 | 11.8-17.4 | |
| IGHV (<97%), n (%) | 27/68 (40) | 5/39 (13) | 20/24 (83) | 2/5 (40) | <.001 |
| 17p/TP53 alteration, n (%) | 24/68 (35) | 14/39 (36) | 9/24 (38) | 1/5 (20) | 1 |
| 9p/CDKN2A deletion, n (%) | 15/68 (22) | 13/39 (33) | 0/24 (0) | 2/5 (40) | .001 |
| 11q deletion, n (%) | 20/68 (29) | 17/39 (44) | 0/24 (0) | 3/5 (60) | <.001 |
| CNA, median (range) | 6.5 (0-52) | 10 (1-43) | 1 (0-45) | 12 (0-52) | <.001 |
| Treatment at diagnosis, n (%)* | <.001 | ||||
| High-dose therapy | 10/67 (15) | 8/38 (21) | 1/24 (4) | 1/5 (20) | |
| Immunochemotherapy | 13/67 (19) | 12/38 (32) | 1/24 (4) | 0/5 (0) | |
| Low-dose chemotherapy | 9/67 (13) | 8/38 (21) | 1/24 (4) | 0/5 (0) | |
| Observation | 35/67 (52) | 10/38 (26) | 21/24 (88)† | 4/5 (80) | |
| Follow-up data | |||||
| Median follow-up, mo | 43 | 35 | 88 | 30 | .019 |
| Mean time from diagnosis to sample (range), mo | 16.6 (0-185) | 2.8 (0-36) | 36 (0-185) | 22.8 (0-92) | .002 |
| Dead patients, n (%) | 24/70 (34) | 16/39 (41) | 7/26 (27) | 1/5 (20) | .296 |
| Treated at 3 y from diagnosis, % (95% CI) | 65 (51-75) | 88 (70-96) | 31 (9-48) | 47 (0-79) | <.001 |
| Treated at 3 y from sampling, % (95% CI) | 71 (57-80) | 89 (73-96) | 44 (19-62) | 47 (0-79) | <.001 |
| 3-y OS, diagnosis, % (95% CI) | 78 (69-89) | 69 (55-86) | 92 (81-100) | 80 (52-100) | .006 |
| 3-y OS, sampling, % (95% CI) | 72 (61-85) | 68 (53-86) | 79 (62-100) | 80 (52-100) | .379 |
ECOG, Eastern Cooperative Oncology Group; LDH, lactate dehydrogenase; MIPI, mantle cell lymphoma International Prognostic Index; ULN, upper level of normal.
High-dose therapy includes cytarabine-based immunochemotherapy and/or autologous stem cell transplantation, immunochemotherapy includes rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP)–like regimens, and low-dose therapy includes alkylating agents alone or in combination.
Two patients underwent splenectomy.
Figure 3.

L-MCL16 scores of RNA from sequential samples of 9 nnMCL patients and 2 cMCL patients. The x-axis corresponds to the time (in years) between the sample collections, and the y-axis corresponds to the L-MCL16 scores. The arrows start at the first sample and are directed to the sequential ones. The signature was stable over time in all cases.
Interestingly, patients predicted to have nnMCL by the L-MCL16 assay had significant clinical and biological differences compared with those having cMCL at diagnosis (Table 1). None of the nnMCL patients had a high Eastern Cooperative Oncology Group score (≥2), which was present in 24% of cMCL patients (P = .07). Similarly, a nodal presentation was only seen in 17% of nnMCL cases vs 62% of cMCL cases (P = .001), and none of the nnMCL patients had high lactate dehydrogenase (LDH) levels compared with 37% of the cMCL patients (P = .008). Both subgroups also received different treatment. Although 88% of nnMCL patients did not receive any treatment at diagnosis, 74% of cMCL patients received chemotherapy-based treatment. Additionally, at 3 years from diagnosis, only 31% (95% CI, 9-48%) of nnMCL patients had been treated, whereas 88% (95% CI, 70-96%) of cMCL patients had received treatment.
nnMCL and cMCL also differed with regard to several genetic and molecular characteristics analyzed at the time of sampling (Table 1; Figure 4). nnMCL had more frequently mutated IGHVs than cMCL (83% vs 13%), an absence of 9p/CDKN2A and 11q/ATM deletions (compared with 33% and 44%, respectively, in cMCL), and a very low number of chromosomal alterations (median of 1 in nnMCL vs 10 CNA per case in cMCL) (all comparisons P < .001). However, 17p/TP53 alterations were distributed similarly in nnMCL and cMCL (38% and 36%, respectively).
Figure 4.

Molecular features of the 70 leukemic MCL samples in the validation cohort. Features are shown as rows, and samples are shown as columns. Samples are ordered according to the L-MCL16 score. IGHV mutations are in yellow, and 17p/TP53, 9p/CDKN2A, and 11q alterations are in green. The number of CNAs in each sample is shown in the bar graph.
Prognostic impact of the L-MCL16 assay
Because we observed that the L-MCL16 assay prediction was stable over time, we evaluated the outcome of the 2 predicted subgroups of patients from the time of diagnosis and from the sampling time. Patients with nnMCL had a significantly longer TTT than did those with cMCL, from the time of diagnosis (patients treated at 3 years, 31% vs 88%; P < .001) and from the sampling time (patients treated at 3 years, 44% vs 89%; P < .001) (Table 1; Figure 5A; supplemental Figure 6A). Similarly, the OS from the time of diagnosis was significantly longer for nnMCL patients than for cMCL patients, with 3-year survival of 92% and 69%, respectively (P = .006) (Table 1; Figure 5B). However, the difference in OS did not reach statistical significance when it was evaluated from the sampling time (supplemental Figure 6B). Of note, nodal presentation or LDH levels at diagnosis in the entire validation series were not associated with shorter OS (P > .1).
Figure 5.

Prognostic impact of the L-MCL16 assay in the validation cohort. KM curves of TTT (A) and OS (B) from diagnosis date of the nnMCL and cMCL subgroups identified by the L-MCL16 assay. KM curves of the OS from sampling date according to the presence of 17p/TP53 and/or 9p/CDKN2A alterations (C) and number of CNAs (D) for the nnMCL and cMCL subgroups. The number of CNAs was associated with OS as a continuous variable.
We then evaluated the biological factors that may influence the OS in both subgroups of MCL from the sampling time, given that the genetic alterations could be acquired during the progression of the disease. Considering all patients in the validation series, the presence of 17p/TP53 and/or 9p/CDKN2A alterations and the increasing number of CNAs were associated with a significantly shorter OS (P = .002 and P <. 0001, respectively) (supplemental Figure 7). Interestingly, in cMCL, the 17p/TP53 and/or 9p/CDKN2A alterations and the increased number of CNAs were associated with poor OS (P = .038 and P = .025, respectively), whereas in nnMCL, the increased number of CNAs predicted shorter OS (P = .005), but 17p/TP53 alterations did not reach statistical significance (Figure 5C,D). Of note, there was no relationship between the L-MCL16 score and the number of CNAs or TP53/CDKN2A alterations in either MCL subtype (P > .05).
Most patients with cMCL (82%) were treated at the time of diagnosis or shortly thereafter. However, 70% of nnMCL patients had not been treated 1 year after sampling. An increased number of CNAs, but not the presence of 17p/TP53 alterations, predicted shorter TTT in these patients (P = .03; supplemental Figure 8).
We also analyzed our previously described MCL35 proliferation score in this cohort of leukemic patients.25 The assay identified 3 risk groups with low, standard, and high proliferation; only 16% of the cases were included in the latter 2 groups, whereas this proportion was 55% in the MCL35 series24 (supplemental Figure 9A). Given the low number of cases in the standard/high group, we analyzed the MCL35 score as a continuous variable. In our validation series, a higher score was significantly associated with shorter OS (P = .043; supplemental Figure 9B). In addition, it predicted a poor outcome in nnMCL (P = .042) but not in cMCL (supplemental Figure 9C).
Discussion
In this study, we developed a novel gene-expression assay that identifies the 2 molecular subtypes of MCL: conventional and leukemic nonnodal. Previous studies have identified the different clinical and biological characteristics of these subtypes. However, a robust diagnostic assay for their identification in clinical practice was not yet available. The application of this assay in an independent cohort of leukemic MCL confirmed the differences in tumors classified as cMCL and nnMCL. The combination of this molecular assay with the analysis of the genomic complexity and TP53/CDKN2A alterations of the tumors identified subsets of patients with significantly different outcomes.
We designed the assay for leukemic MCL samples, because all patients with nnMCL have lymphocytosis, and most of them do not have lymphadenopathy or other accessible tissues for diagnosis. On the other hand, up to 75% of patients with cMCL may have lymphocytosis and, therefore, may also benefit from a precise diagnosis with this assay.34 Although we could recognize the cMCL signatures in some tissue samples, we could not expand the study because lymph node biopsies of nnMCL with enough tumor cells were not available. In addition, the genes of our assay were selected from purified leukemic samples, and previous studies have shown the modulation of gene expression between lymph node and PB MCL cells.35 One limitation of the assay is the need for ≥60% of tumor cells for a reliable discrimination of the 2 subtypes. This tumor cell content is similar to the requirement of our recently designed MCL35 proliferation assay for nodal MCL.25 Although the percentages of lymphocytes in most leukemic MCL are above this number, some cases may need an initial step of tumor cell purification. In our study, only 21% of the cases would have needed this purification step. The diagnosis of leukemic MCL with limited access to tissue diagnosis is challenging and, not uncommonly, these cases are misclassified as other leukemic lymphoid neoplasms.2 The current assay, with the added diagnostic detection of elevated cyclin D1 mRNA, may be a useful tool for the diagnosis of leukemic MCL and assignment into the 2 molecular subtypes.
The analytical validity of the assay was confirmed by the high intra- and interlaboratory reproducibility, similar to our proliferative MCL35 assay.25 The selected 16 most informative genes that differentiated cMCL and nnMCL included genes previously described as being overexpressed in cMCL, as well as new genes overexpressed in indolent nnMCL (CD200, BTLA, and SLAMF1). Flow cytometry studies had shown CD200 expression in SOX11-negative MCL3 and MCL with indolent clinical behavior.11 Most previous studies in leukemic and nodal MCL have used only SOX11 as a discriminatory or prognostic biomarker.14,15,36,37 Although the expression levels of SOX11 are clearly different in most nnMCL and cMCL cases, there were some cases with borderline SOX11 levels that required the analysis of other genes of the signature to be properly classified. This observation may explain, in part, the limitations in subclassifying MCL based only on the detection of a single marker.
The identification of cMCL and nnMCL in the independent validation series confirmed the different clinical and biological features of these 2 subtypes of MCL, particularly the nodal presentation, high LDH, unmutated IGHVs, 9p/CDKN2A and 11q/ATM deletions, and high genomic complexity of cMCL. Although these features were significantly different in both subtypes, most of them had a certain overlap between cMCL and nnMCL and, therefore, could not be used as a single diagnostic parameter. These findings emphasize the value of the L-MCL16 assay for the biological identification of these MCL subtypes and its possible interest in future clinical and biological studies delineating these subtypes.
Our study also shows the stability of the nnMCL and cMCL signatures over time, in spite of the progression of the disease. Based on this stability, we could also confirm the significantly better outcome and, particularly, the longer TTT of nnMCL when estimated from the time of diagnosis. However, a number of patients with nnMCL and stable disease were only referred to our center at the time of progression, and this was the only sample available. Although the TTT from the sampling time was still significantly longer for nnMCL, OS was similar in both MCL subtypes, suggesting that nnMCL had acquired biological factors impacting further evolution. To determine parameters that could influence this evolution, we studied the proliferation signature with our MCL35 assay.25 Although we could recognize 3 subgroups with high, standard, and low proliferation in the entire series and in the 2 MCL subtypes, the prognostic power was not as robust as previously found in nodal samples. These results are not totally unexpected, because leukemic MCL cells seem to downregulate the expression of proliferation genes in leukemic samples compared with their respective nodal counterpart,35 indicating that, contrary to nodal samples, leukemic cells may not be suitable for evaluating the prognostic value of proliferation in MCL. Intriguingly, a recent study has shown that the MCL35 proliferation score and other strong MCL prognostic factors, such as MCL International Prognostic Index and Ki67, did not predict outcome in nodal samples from patients with indolent disease who were managed with a watchful waiting attitude.20 These findings highlight the need for additional prognostic parameters that may identify indolent MCL where clinical outcomes may not be compromised by an observational approach.
Aberrations in TP53 and CDKN2A were recognized early as very detrimental factors in the evolution of MCL,38-42 and these findings have been recently confirmed in patients treated with intensive immunochemotherapy regimens.43-45 On the other hand, increasing numbers of chromosomal alterations and complex karyotypes have also been associated with the adverse evolution of MCL.46,47 TP53/CDKN2A aberrations are frequently associated with complex karyotypes, but the independent prognostic relevance of these parameters has not been evaluated. Genomic complexity and TP53/CDKN2A aberrations were associated with significantly poor outcome in our entire validation cohort and in cMCL. Genomic complexity also predicted shorter OS and TTT in nnMCL. Although TP53/CDKN2A aberrations were also associated with worse outcome in these patients, they did not reach statistical significance, probably due to the low number of patients and events. Nevertheless, some nnMCL patients had TP53 aberrations without increased CNA, and they had stable disease for 6-38 months without the need for therapy. These findings are similar to the independent prognostic value of genomic complexity over TP53 aberrations observed in CLL.48-50
In conclusion, the novel molecular assay developed for leukemic MCL reliably identifies the 2 cMCL and nnMCL subtypes and confirms the different clinical and biological characteristics of these patients. The combination of this assay with the analysis of the genomic complexity identifies subsets of patients with different outcomes; therefore, it may provide useful biological information for management decisions in these patients.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank other members of the Leukemia Lymphoma Molecular Profiling Project (LLMPP) for helpful discussions, the Hematopathology Collection for sample procurement, and the Genomics Core Facility of the Institut d’Investigacions Biomèdiques August Pi i Sunyer for technical help.
This research was funded by the Spanish Ministerio de Economía y Competitividad (Grant SAF2015-64885-R), a National Institutes of Health National Cancer Institute Strategic Partnering to Evaluate Cancer Signatures grant (5U01CA157581-05), Generalitat de Catalunya Suport Grups de Recerca Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR) 2017-SGR-1142 and 2014-SGR-378, Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III PI14/00571 and PI17/01061, Fundació La Marató de TV3 TV3-Cancer/2013410, and the European Regional Development Fund “Una manera de fer Europa,” Centres de Recerca de Catalunya (CERCA) Programme/Generalitat de Catalunya. E.C. is an Academia Researcher of the “Institució Catalana de Recerca i Estudis Avançats” of the Generalitat de Catalunya. The genotyping service was carried out at Centro Nacional de Genotipado-PRB2-Instituto de Salud Carlos III (CGEN-PRB2-ISCIII) (Grant PTI3/0001, Instituto de Salud Carlos III-Subdirección General de Evaluación y Fomento de la Investigación-Fondo Europeo de Desarrollo Regional ISCIII-SGEFI/FEDER).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: G.C., L.M.R., S.B., and E.C. designed the study. G.C. performed the statistical and clinical analyses. G.C., P.J., A.N., C.R., M.P., D.M.-G., S.D., H.R.-W., G.W.W., and S.B. performed and interpreted molecular studies. E.G., B.E., A.S., A.F., A.M., M.A., A.L.-G., and E.C. provided cases and collected clinical and pathological data. E.G. coordinated all clinical information. E.S.J., J.M.C., R.D.G., J.D., G.O., L.M.S., A.R., D.W.S., L.M.R., and E.C. contributed to the definition of the MCL proliferation signature. G.C., S.B., and E.C. wrote the manuscript. S.B. and E.C. directed and supervised the research. All authors approved the final manuscript.
Conflict-of-interest disclosure: E.C. has received research funding from and has been an expert witness for Gilead Sciences. E.S.J., J.M.C., R.D.G., J.D., G.O., G.W.W., L.M.S., A.R., D.W.S., L.M.S., and E.C. are named inventors on 2 patents filed by the National Cancer Institute: “Methods for selecting and treating lymphoma types” licensed to NanoString Technologies and “Evaluation of mantle cell lymphoma and methods related thereof.” The remaining authors declare no competing financial interests.
This study was performed under the auspices of the US government and is in the public domain.
Correspondence: Elias Campo, Hematopathology Section, Hospital Clinic of Barcelona, Villarroel 170, 08036-Barcelona, Spain; ecampo@clinic.ub.es.
REFERENCES
- 1.Campo E, Rule S. Mantle cell lymphoma: evolving management strategies. Blood. 2015;125(1):48-55. [DOI] [PubMed] [Google Scholar]
- 2.Fernàndez V, Salamero O, Espinet B, et al. . Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res. 2010;70(4):1408-1418. [DOI] [PubMed] [Google Scholar]
- 3.Espinet B, Ferrer A, Bellosillo B, et al. . Distinction between asymptomatic monoclonal B-cell lymphocytosis with cyclin D1 overexpression and mantle cell lymphoma: from molecular profiling to flow cytometry. Clin Cancer Res. 2014;20(4):1007-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orchard J, Garand R, Davis Z, et al. . A subset of t(11;14) lymphoma with mantle cell features displays mutated IgVH genes and includes patients with good prognosis, nonnodal disease. Blood. 2003;101(12):4975-4981. [DOI] [PubMed] [Google Scholar]
- 5.Martin P, Chadburn A, Christos P, et al. . Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol. 2009;27(8):1209-1213. [DOI] [PubMed] [Google Scholar]
- 6.Swerdlow SH, Campo E, Pileri SA, et al. . The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Royo C, Navarro A, Clot G, et al. . Non-nodal type of mantle cell lymphoma is a specific biological and clinical subgroup of the disease. Leukemia. 2012;26(8):1895-1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navarro A, Clot G, Royo C, et al. . Molecular subsets of mantle cell lymphoma defined by the IGHV mutational status and SOX11 expression have distinct biologic and clinical features. Cancer Res. 2012;72(20):5307-5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Queirós AC, Beekman R, Vilarrasa-Blasi R, et al. . Decoding the DNA methylome of mantle cell lymphoma in the light of the entire B cell lineage. Cancer Cell. 2016;30(5):806-821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ondrejka SL, Lai R, Smith SD, Hsi ED. Indolent mantle cell leukemia: a clinicopathological variant characterized by isolated lymphocytosis, interstitial bone marrow involvement, kappa light chain restriction, and good prognosis. Haematologica. 2011;96(8):1121-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu Z, Sun Y, Schlette EJ, et al. . CD200 expression in mantle cell lymphoma identifies a unique subgroup of patients with frequent IGHV mutations, absence of SOX11 expression, and an indolent clinical course. Mod Pathol. 2018;31(2):327-336. [DOI] [PubMed] [Google Scholar]
- 12.Puente XS, Jares P, Campo E. Chronic lymphocytic leukemia and mantle cell lymphoma: Crossroads of genetic and microenvironment interactions. Blood. 2018;131(21):2283-2296. [DOI] [PubMed] [Google Scholar]
- 13.Soldini D, Valera A, Solé C, et al. . Assessment of SOX11 expression in routine lymphoma tissue sections: characterization of new monoclonal antibodies for diagnosis of mantle cell lymphoma. Am J Surg Pathol. 2014;38(1):86-93. [DOI] [PubMed] [Google Scholar]
- 14.Nygren L, Baumgartner Wennerholm S, Klimkowska M, Christensson B, Kimby E, Sander B. Prognostic role of SOX11 in a population-based cohort of mantle cell lymphoma. Blood. 2012;119(18):4215-4223. [DOI] [PubMed] [Google Scholar]
- 15.Nordström L, Sernbo S, Eden P, et al. . SOX11 and TP53 add prognostic information to MIPI in a homogenously treated cohort of mantle cell lymphoma--a Nordic Lymphoma Group study. Br J Haematol. 2014;166(1):98-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tucker D, Rule S. Novel agents in mantle cell lymphoma. Expert Rev Anticancer Ther. 2017;17(6):491-506. [DOI] [PubMed] [Google Scholar]
- 17.Dreyling M, Amador V, Callanan M, et al. ; European Mantle Cell Lymphoma Network. Update on the molecular pathogenesis and targeted approaches of mantle cell lymphoma: summary of the 12th Annual Conference of the European Mantle Cell Lymphoma Network. Leuk Lymphoma. 2015;56(4):866-876. [DOI] [PubMed] [Google Scholar]
- 18.Dreyling M, Ferrero S; European Mantle Cell Lymphoma Network. Personalized medicine in lymphoma: is it worthwhile? The mantle cell lymphoma experience. Haematologica. 2015;100(6):706-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin P, Leonard J. Is there a role for “watch and wait” in patients with mantle cell lymphoma? Semin Hematol. 2011;48(3):189-193. [DOI] [PubMed] [Google Scholar]
- 20.Abrisqueta P, Scott DW, Slack GW, et al. . Observation as the initial management strategy in patients with mantle cell lymphoma. Ann Oncol. 2017;28(10):2489-2495. [DOI] [PubMed] [Google Scholar]
- 21.Eve HE, Furtado MV, Hamon MD, Rule SA. Time to treatment does not influence overall survival in newly diagnosed mantle-cell lymphoma. J Clin Oncol. 2009;27(32):e189-e190, author reply e191. [DOI] [PubMed] [Google Scholar]
- 22.Rosenwald A, Wright G, Wiestner A, et al. . The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell. 2003;3(2):185-197. [DOI] [PubMed] [Google Scholar]
- 23.Hoster E, Rosenwald A, Berger F, et al. . Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: results from randomized trials of the European Mantle Cell Lymphoma Network. J Clin Oncol. 2016;34(12):1386-1394. [DOI] [PubMed] [Google Scholar]
- 24.Klapper W, Hoster E, Determann O, et al. ; European MCL Network. Ki-67 as a prognostic marker in mantle cell lymphoma-consensus guidelines of the pathology panel of the European MCL Network. J Hematop. 2009;2(2):103-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scott DW, Abrisqueta P, Wright GW, et al. ; Lymphoma/Leukemia Molecular Profiling Project. New molecular assay for the proliferation signature in mantle cell lymphoma applicable to formalin-fixed paraffin-embedded biopsies. J Clin Oncol. 2017;35(15):1668-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Navarro A, Clot G, Martínez-Trillos A, et al. . Improved classification of leukemic B-cell lymphoproliferative disorders using a transcriptional and genetic classifier. Haematologica. 2017;102(9):e360-e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCarthy BA, Yancopoulos S, Tipping M, et al. . A seven-gene expression panel distinguishing clonal expansions of pre-leukemic and chronic lymphocytic leukemia B cells from normal B lymphocytes. Immunol Res. 2015;63(1-3):90-100. [DOI] [PubMed] [Google Scholar]
- 28.McCall MN, Bolstad BM, Irizarry RA. Frozen robust multiarray analysis (fRMA). Biostatistics. 2010;11(2):242-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beà S, Valdés-Mas R, Navarro A, et al. . Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci USA. 2013;110(45):18250-18255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. [DOI] [PubMed] [Google Scholar]
- 31.Kim KI, Simon R. Probabilistic classifiers with high-dimensional data. Biostatistics. 2011;12(3):399-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bates D, Maechler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67(1):1-48. [Google Scholar]
- 33.Schemper M, Smith TL. A note on quantifying follow-up in studies of failure time. Control Clin Trials. 1996;17(4):343-346. [DOI] [PubMed] [Google Scholar]
- 34.Ferrer A, Salaverria I, Bosch F, et al. . Leukemic involvement is a common feature in mantle cell lymphoma. Cancer. 2007;109(12):2473-2480. [DOI] [PubMed] [Google Scholar]
- 35.Saba NS, Liu D, Herman SE, et al. . Pathogenic role of B-cell receptor signaling and canonical NF-κB activation in mantle cell lymphoma. Blood. 2016;128(1):82-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meggendorfer M, Kern W, Haferlach C, Haferlach T, Schnittger S. SOX11 overexpression is a specific marker for mantle cell lymphoma and correlates with t(11;14) translocation, CCND1 expression and an adverse prognosis. Leukemia. 2013;27(12):2388-2391. [DOI] [PubMed] [Google Scholar]
- 37.Kuo PY, Leshchenko VV, Fazzari MJ, et al. . High-resolution chromatin immunoprecipitation (ChIP) sequencing reveals novel binding targets and prognostic role for SOX11 in mantle cell lymphoma. Oncogene. 2015;34(10):1231-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hernandez L, Fest T, Cazorla M, et al. . p53 gene mutations and protein overexpression are associated with aggressive variants of mantle cell lymphomas. Blood. 1996;87(8):3351-3359. [PubMed] [Google Scholar]
- 39.Greiner TC, Moynihan MJ, Chan WC, et al. . p53 mutations in mantle cell lymphoma are associated with variant cytology and predict a poor prognosis. Blood. 1996;87(10):4302-4310. [PubMed] [Google Scholar]
- 40.Pinyol M, Hernandez L, Cazorla M, et al. . Deletions and loss of expression of p16INK4a and p21Waf1 genes are associated with aggressive variants of mantle cell lymphomas. Blood. 1997;89(1):272-280. [PubMed] [Google Scholar]
- 41.Espinet B, Salaverria I, Beà S, et al. . Incidence and prognostic impact of secondary cytogenetic aberrations in a series of 145 patients with mantle cell lymphoma. Genes Chromosomes Cancer. 2010;49(5):439-451. [DOI] [PubMed] [Google Scholar]
- 42.Halldórsdóttir AM, Lundin A, Murray F, et al. . Impact of TP53 mutation and 17p deletion in mantle cell lymphoma. Leukemia. 2011;25(12):1904-1908. [DOI] [PubMed] [Google Scholar]
- 43.Delfau-Larue MH, Klapper W, Berger F, et al. ; European Mantle Cell Lymphoma Network. High-dose cytarabine does not overcome the adverse prognostic value of CDKN2A and TP53 deletions in mantle cell lymphoma. Blood. 2015;126(5):604-611. [DOI] [PubMed] [Google Scholar]
- 44.Eskelund CW, Dahl C, Hansen JW, et al. . TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood. 2017;130(17):1903-1910. [DOI] [PubMed] [Google Scholar]
- 45.Aukema SM, Hoster E, Rosenwald A, et al. . Expression of TP53 is associated with the outcome of MCL independent of MIPI and Ki-67 in trials of the European MCL Network. Blood. 2018;131(4):417-420. [DOI] [PubMed] [Google Scholar]
- 46.Beà S, Ribas M, Hernández JM, et al. . Increased number of chromosomal imbalances and high-level DNA amplifications in mantle cell lymphoma are associated with blastoid variants. Blood. 1999;93(12):4365-4374. [PubMed] [Google Scholar]
- 47.Sarkozy C, Terré C, Jardin F, et al. . Complex karyotype in mantle cell lymphoma is a strong prognostic factor for the time to treatment and overall survival, independent of the MCL international prognostic index. Genes Chromosomes Cancer. 2014;53(1):106-116. [DOI] [PubMed] [Google Scholar]
- 48.Ouillette P, Collins R, Shakhan S, et al. . Acquired genomic copy number aberrations and survival in chronic lymphocytic leukemia. Blood. 2011;118(11):3051-3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Delgado J, Salaverria I, Baumann T, et al. . Genomic complexity and IGHV mutational status are key predictors of outcome of chronic lymphocytic leukemia patients with TP53 disruption. Haematologica. 2014;99(11):e231-e234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herling CD, Klaumünzer M, Rocha CK, et al. . Complex karyotypes and KRAS and POT1 mutations impact outcome in CLL after chlorambucil-based chemotherapy or chemoimmunotherapy. Blood. 2016;128(3):395-404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.