

Abstract
Overgrowth syndromes are a clinically heterogeneous group of disorders characterized by localized or generalized tissue overgrowth and varying degrees of developmental and intellectual disability. An expanding list of genes associated with overgrowth syndromes include the histone methyltransferase genes EZH2 and NSD1, which cause Weaver and Sotos syndrome, respectively, and the DNA methyltransferase (DNMT3A) gene that results in Tatton-Brown–Rahman syndrome (TBRS). Here, we describe a 5-year-old female with a paternally inherited pathogenic mutation in EZH2 (c.2050C>T, p.Arg684Cys) and a maternally inherited 505-kb duplication of uncertain significance at 2p23.3 (encompassing five genes, including DNMT3A) who presented with intrauterine growth restriction, slow postnatal growth, short stature, hypotonia, developmental delay, and neuroblastoma diagnosed at the age of 8 mo. Her father had tall stature, dysmorphic facial features, and intellectual disability consistent with Weaver syndrome, whereas her mother had short stature, cognitive delays, and chronic nonprogressive leukocytosis. It has been previously shown that EZH2 directly controls DNA methylation through physical association with DNMTs, including DNMT3A, with concomitant H3K27 methylation and CpG promoter methylation leading to repression of EZH2 target genes. Interestingly, NSD1 is involved in H3K36 methylation, a mark associated with transcriptional activation, and exhibits exquisite dosage sensitivity leading to overgrowth when deleted and severe undergrowth when duplicated in vivo. Although there is currently no evidence of dosage effects for DNMT3A, the co-occurrence of a duplication involving this gene and a pathogenic alteration in EZH2 in a patient with severe undergrowth is suggestive of a similar paradigm and further study is warranted.
Keywords: bilateral talipes equinovarus, delayed gross motor development, generalized neonatal hypotonia, hip dysplasia, microretrognathia, moderate expressive language delay, moderate global developmental delay, moderate intrauterine growth retardation, neuroblastoma, overgrowth, short stature
INTRODUCTION
Overgrowth syndromes are a heterogeneous group of disorders clinically defined by increased height and/or head circumference (Ko 2013; Tatton-Brown et al. 2017). These disorders may also present with intellectual disability, developmental delay, congenital anomalies, and predisposition to tumor development (Lapunzina 2005). The most common single-gene overgrowth disorders include Sotos syndrome (NSD1, MIM: 117550), Weaver syndrome (EZH2, MIM: 277590), and Tatton-Brown–Rahman syndrome (TBRS) (DNMT3A, MIM: 602769). These syndromes are characterized by overlapping clinical manifestations such as pre- and postnatal overgrowth and variable intellectual disability (Opitz et al. 1998; Vora and Bianchi 2009).
NSD1 (Nuclear receptor SET-domain containing protein 1), EZH2 (Enhancer of Zeste, drosophila, Homolog 2), and DNMT3A (DNA methyltransferase 3A) are epigenetic modifiers involved in chromatin remodeling and transcriptional regulation either by histone lysine methylation (NSD1 and EZH2) or DNA methylation (DNMT3A). Pathogenic alterations in other genes involved in epigenetic regulation of developmental processes have similarly been implicated in overgrowth disorders and include CHD8 (MIM: 610528), BRWD3 (MIM: 300553), SETD2 (MIM: 616831), HIST1H1E (MIM: 617537), EED (MIM: 605984) (Tatton-Brown et al. 2017). In addition, many of these genes appear to exhibit dosage sensitivity with gene expression levels being tightly regulated, and changes in copy number resulting in significant downstream epigenetic consequences (Bjornsson 2015).
In this research report, we describe a 5-year-old female with severe undergrowth and developmental delay who was found to carry a paternally inherited pathogenic EZH2 variant and a maternally inherited duplication encompassing DNMT3A. The proband's father, who previously did not have a clinical diagnosis, exhibited classic features of Weaver syndrome including significant overgrowth (both pre- and postnatally) with an adult height of >2 SD above the mean. The proband's mother had short stature (−2.3 SD below the mean), cognitive delays, and chronic nonprogressive leukocytosis. Growth restriction and short stature are highly atypical for Weaver syndrome, suggesting that the maternally inherited 505-kb duplication could have a modifying effect on the proband's phenotype.
Several lines of evidence support this hypothesis. First, EZH2 is sufficient to recruit DNMT3A and the two proteins have been shown to directly interact at target genomic loci (Viré et al. 2006). Alterations in either gene are associated with disorders of overgrowth with significant phenotypic overlap (Tatton-Brown et al. 2014). Furthermore, changes in histone lysine methylation result in changes in DNA methylation patterns, suggesting that these two types of epigenetic modifications are intimately associated (Moore et al. 2013; Rose and Klose 2014). Because other epigenetic regulators (NSD1) have shown evidence of dosage effects (i.e., loss of one copy leads to overgrowth and gain of an additional copy leads to growth restriction) (Dikow et al. 2013; Rosenfeld et al. 2013), we hypothesize that duplications involving DNMT3A could similarly result in growth retardation and short stature.
RESULTS
Clinical Presentation
The proband is a 5-year-old female with a history of global developmental delay, neuroblastoma, congenital anomalies, and hypotonia. She was born at 37 wk by Cesarean section to a 31-year-old G2 (now P1) mother. The pregnancy was complicated by maternal depression, nicotine dependence, high caffeine consumption in early pregnancy, and acute gangrenous appendicitis with perforation at 12 6/7 wk gestation. Prenatal ultrasound showed shortened femur and arm lengths and a right clubfoot. At 36 wks and 3 d, ultrasound biometry fell at 8% of anticipated for gestational age, with both biparietal diameter (83 mm) and femur length (62 mm) measuring <2.5% of expected. Head circumference was measured at 308 mm (16th percentile) and abdominal circumference was 312 mm (29th percentile). The estimated total fetal weight was 2350 g. Labor was induced secondary to intrauterine growth restriction (IUGR). The proband's birth weight was 2.17 kg (<1st percentile, −2.61 SD) and she was 47 cm long (12th percentile, −1.15 SD). Her head circumference was 32.5 cm (12th percentile, −1.16 SD). Apgar scores were 6 and 8 at 1 and 5 min, respectively. At birth she did not cry or grimace even with stimulation and was cyanotic. Skin color and oxygen saturations improved quickly after blow-by oxygen for 1–2 min.
After 36 h, she had persistent mild hypoxemia with oxygen saturation between 80% and 90%. A chest X ray, EKG, and echocardiography were performed (Supplemental Fig. 1). The chest X ray and electrocardiogram were unremarkable except for possible left ventricular hypertrophy and a nonspecific T wave abnormality. Echocardiography showed a small ventricular septal defect with left-to-right shunt, a small patent ductus arteriosus with continuous left-to-right shunt, and a patent foramen ovale with left-to-right shunt. There was also mild tricuspid valve regurgitation. After 7 d, the patient had worsening desaturations (as low as 70%) and noisy breathing and required oxygen via nasal cannula. A polysomnography study at 2 wk of age was severely abnormal, suggestive of combined obstructive and central sleep apnea.
Some dysmorphic features and congenital anomalies had been noted, including microretrognathia, small mouth, hypertelorism, bilateral hip dysplasia, and right hip dislocation, right clubfoot deformity, and overlapping of toes 3 and 4 on the left foot. Some of the features were present only in early infancy, including generalized redundant and wrinkled skin, mostly of the extremities, and long fingers (Fig. 1A–C). The patient underwent bilateral osteotomy and jaw distraction because of her severe microretrognathia (Fig. 1D–G; Supplemental Fig. 2).
Figure 1.

Phenotypic features of the proband. (A) Shortened femur and arm lengths, a right clubfoot; (B,C) soft and doughy excessive skin at birth; (D) retrognathia resolved with osteotomy and detractor placement at 1 mo of age; (E,F) photos of the proband at 8 mo showing relative macrocephaly and obesity; and (G) a photo of the proband at age of 2 yr and 11 mo old.
At 7 mo, the proband was evaluated for persistent developmental delays and hypotonia. She had problems with head holding and rolling over and was noted to have significant gross motor delay and hypotonia. Head magnetic resonance imaging (MRI) findings were nonspecific, including some paucity of white matter with small periatrial zones of signal abnormality bilaterally that were more prominent on the left where there was some leukomalacia and delayed myelination of the white matter with some indistinctness at the gray–white junction.
At 8 mo, an abdominal ultrasound revealed mild pyelocaliectasis on the left and slightly enlarged liver and spleen. A repeat ultrasound for worsening hepatomegaly showed heterogeneity throughout the liver with several small, round, hypoechoic foci. Urine analysis of catecholamine metabolites showed increased excretions of homovanillic acid (HVA) and vanillylmandelic acid (VMA) with quantitative testing showing increased levels at 138.1 mg/g Cr (85.7 µmol/mmol Cr) (reference <35.0 mg/g Cr or <21.7 µmol/mmol Cr) and 477.5 mg/g Cr (272.6 µmol/mmol Cr) (reference <27.0 mg/g Cr or <15.4 µmol/mmol Cr), respectively. Subsequent MRI demonstrated a solid 2.5 cm right paraspinal mass in the upper chest and innumerable liver infiltrates. Radiological findings were most consistent with thoracic neuroblastoma with diffuse hepatic metastasis. Metaiodobenzylguanidine (MIBG) scan showed intense focal MIBG uptake in a right paraspinal mass in the upper chest, and heterogeneous uptake throughout the liver that correlated with biopsy-proven metastatic lesions. Tumor cytogenetic testing demonstrated no evidence of MYCN amplification and ∼85% of nuclei had three copies of Chromosome 2. The proband was diagnosed with intermediate-risk stage 4S thoracic neuroblastoma with involvement of the liver and the posterior mediastinum. She was enrolled in the Children's Oncology Group study ANBL0531 and received eight courses of chemotherapy, including carboplatin, cyclophosphamide, doxorubicin hydrochloride, etoposide, and filgrastim, which was well tolerated. Repeat MIBG scans showed a positive but incomplete response to therapy with disappearance of the right paraspinal mass but persistent increased MIBG uptake in the liver. She is currently doing well at 5 yr of age per mother's report.
At her 2-yr health evaluation, testing revealed speech and language development below 1-yr level. She showed short attention span and hyperactivity. She has received physical, occupational, and speech therapies. Although her birth length was low-normal, she started to fall off the growth chart since early infancy and continued to grow at around 2nd–3rd percentile. At the 2-yr evaluation her weight was 12.36 kg (72nd percentile, +2.23 SD), her height was 79.2 cm (1st percentile, −2.23 SD), her head circumference was 47 cm (45th percentile, −0.13 SD) (Supplemental Figs. 3–5).
Family History
The proband's 36-year-old mother reported a history of loose joints and easy bruising. She was also noted to have short stature (height, 148.5 cm, 1st percentile, −2.3 SD) and mild cognitive deficits. She had been followed by hematology for a chronic, nonprogressive leukocytosis ranging from 12.0 to 20.8 × 109/L (reference range: 3.5–10.5 × 109/L) with a predominant neutrophilia. To the best of our knowledge she has not pursued recommended evaluations.
The proband's 41-year-old father had tall stature (height, 195.5 cm, 99th percentile, +2.6 SD; arm span of 200 cm; arm span to height ratio of 1.02), and Marfan syndrome was suspected in the past; however, he did not meet the clinical criteria (Fig. 2). There were no abnormalities noted on audiology, ophthalmology, and echocardiography examinations. X rays showed platyspondyly and mild metaphyseal and epiphyseal dysplasia. His plasma homocystine and insulin-like growth factor-1 (IGF-1) levels were normal. From the records, he was born at 7 mo at home after a precipitous onset of labor. His birth weight was 2.55 kg (4th percentile, −1.78 SD). After birth, he possibly had a hypoxic insult. He has history of developmental delays and was noted to have mild intellectual disability. He had two one-sided spontaneous pneumothoraces at 22 and 37 yr of age. On CT scan he had multiple apical blebs, right greater than left. He underwent right apical bullectomy and parietal pleurectomy after his second pneumothorax.
Figure 2.
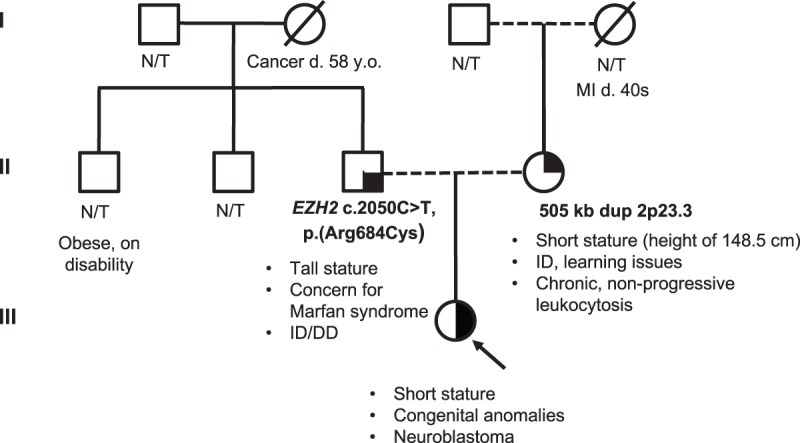
Family pedigree. Pedigree showing inheritance of the pathogenic variant in EZH2 from proband's father and duplication of 2p23.3 from the proband's mother. ID, intellectual disability; DD, developmental delay; MI, myocardial infarction; N/T, not tested.
Laboratory Testing
The proband had normal newborn screen, congenital disorders of glycosylation screen, quantitative plasma amino acids profiling, and peroxisomal disorders panel. Urinary catecholamine analysis revealed elevations of homovanillic acid (HVA) and vanillylmandelic acid (VMA), consistent with her history of neuroblastoma. Her thyroid function studies were normal.
Genetic Testing
The proband's previous testing consisted of Marfan and related disorders panel (Ambry Genetics) that included ACTA2 (MIM: 102620), CBS (MIM: 613381), FBN1 (MIM: 134797), FBN2 (MIM: 612570), MYH11 (MIM: 160745), COL3A1 (MIM: 120180), SLC2A10 (MIM: 606145), SMAD3 (MIM: 603109), TGFBR1 (MIM: 190181), and TGFBR2 (MIM: 190182); sequencing of the PHOX2B gene (MIM: 603851) (Children's Hospital of Philadelphia); and a Noonan syndrome panel (Prevention Genetics) that included PTPN11 (MIM: 176876), SOS1 (MIM: 182530), RAF1 (MIM: 164760), KRAS (MIM: 190070), SHOC2 (MIM: 602775), BRAF (MIM: 164757), and NRAS (MIM: 164790), all of which were negative.
The proband's karyotype was normal (46,XX) with no apparent structural abnormalities. Chromosomal microarray (CMA) analysis detected a 505-kb duplication (arr[hg19] 2p23.3(25,336,135–25,841,368)x3) that was inherited from her mother (Table 1; Fig. 2). The duplication was confirmed by interphase FISH using a probe within the duplicated interval (RP11–81M3). Metaphase FISH studies characterized the duplication as tandem.
Table 1.
Variant information for EZH2 and the 505-kb duplication
| Type | Genomic position, cDNA, and protein reference | Inheritance | Pathogenicity | Variant ID/accession | Additional information | |
|---|---|---|---|---|---|---|
| EZH2 | SNV | Chr 7(GRCh37): g.148506462G>A, NM_004456.4: c.2050C>T, NP_004447.2: p.Arg684Cys | Paternal | Pathogenic | 0000369006 (LOVD) | Exome sequencing:
|
| 505 kb | dup | arr[hg19] 2p23.3(25,336, 135-25,841,368)x3 | Maternal | Uncertain clinical significance | SCV000179757.4 (ClinVar) | Duplicated genes: POMC (MIM: 176830), MIR1301, DTNB (MIM: 602415), DNMT3A (MIM: 602769), and EFR3B (MIM: 616797) |
SNV, single-nucleotide variation; dup, duplication.
The duplicated interval contains five known genes: POMC (MIM: 176830), MIR1301, DTNB (MIM: 602415), DNMT3A (MIM: 602769), and EFR3B (MIM: 616797). Although missense, nonsense, splice site, and indel variants in DNMT3A have been described in Tatton-Brown–Rahman syndrome (TBRS) (Tatton-Brown et al. 2014), complete gene duplications have not been described in the literature to date. Missense mutations, synonymous polymorphisms, and deletions of the POMC have been identified in early onset obesity (Krude et al. 1998; Feng et al. 2003; Farooqi et al. 2006; Lee et al. 2006). However, no link between duplication of POMC and adiposity has been reported, and therefore increased BMI of the proband cannot be solely explained by duplication of POMC. Other duplicated genes (MIR1301, DTNB, and EFR3B) have not been shown to be associated with growth regulation. Furthermore no clinical phenotypes associated with duplication of any of these genes have been described. Therefore, this duplication was classified as being of uncertain clinical significance.
Whole-exome sequencing (Baylor Genetics) revealed a single pathogenic heterozygous missense variant in EZH2 (Chr 7(GRCh37): g.148506462G>A, NM_004456.4: c.2050C>T, NP_004447.2: p.Arg684Cys) previously reported as disease-causing of autosomal dominant Weaver syndrome (Table 1; Fig. 2). The following quality metrics were achieved: >70% of reads aligned to target, >95% target bases covered at >20×, 85% target base covered at >40×, mean coverage of target bases >100×. This variant was inherited from the father, who exhibits many of the features of Weaver overgrowth syndrome, including increased pre- and postnatal height and weight, relative macrocephaly, large ears, dysmorphic facial features, skeletal anomalies, developmental delay, and mild intellectual disability. This variant has also been reported somatically in acute myeloid leukemia and other malignancies (Simon and Lange 2008; Ernst et al. 2010).
In addition, the proband was diagnosed with neuroblastoma at 8 mo of age. A number of different tumor types have been described in individuals with Weaver syndrome, including neuroblastoma (Basel-Vanagaite 2010). One male child with a c.2044G>A (p.Ala682Thr) pathogenic variant in EZH2 was diagnosed with neuroblastoma and acute lymphoblastic leukemia at 13 mo of age and a female with a c.458A>G (p.Tyr153Cys) pathogenic variant was diagnosed with a neuroblastoma at 4 yr of age (Tatton-Brown et al. 2013). It has been also demonstrated that several tumor types, including neuroblastoma, are characterized by high EZH2 activation (EZH2 is a proto-oncogene) (Cohen et al. 2013). To our knowledge, this is the first reported case of neuroblastoma in a patient with the recurrent c.2050C>T (p.Arg684Cys) pathogenic variant.
DISCUSSION
In this case report, we describe a 5-year-old child with a paternally inherited pathogenic p.Arg684Cys variant in EZH2 and maternally inherited 505-kb duplication at 2p23.3 encompassing five genes, including DNMT3A. The proband displayed some overlapping features with Weaver syndrome such as hypotonia and soft doughy skin at birth, micrognathia, global developmental delay, relative macrocephaly, and neuroblastoma. However, the proband had IUGR and short stature with height in the 12th percentile (−1.15 SD) at birth and in the <1st percentile (−2.23 SD) by 2 yr of age with no evidence of overgrowth (Supplemental Figs. 3–5). Her father has a tall stature (+2.6 SD), dysmorphic facial features, and intellectual disability consistent with Weaver syndrome. This may suggest that 2p23.3 duplication does not completely mask the effect of EZH2 pathogenic variant.
The p.Arg684Cys variant detected in the proband and her father has been seen in several unrelated individuals with Weaver syndrome (Table 2; Tatton-Brown et al. 2011, 2013; Cohen et al. 2016) and falls within the SET domain (catalytic Drosophila proteins Su(var)3–9, Enhancer-of-zeste, and Trithorax). The p.Arg684 residue does not directly participate in cofactor or substrate binding; however, its side chain is structurally important in homologous SET domains (Antonysamy et al. 2013). The p.Arg684Cys variant, among others, showed reduced histone methyltransferase activity in vitro, suggesting that decreased H3K27me3 may play a role in the expression of disease phenotypes (Cohen et al. 2016). Individuals with the p.Arg684Cys (and related p.Arg684His) variant consistently showed increased height, ranging from 2.9 to 5.4 SD above the mean (Table 2; Tatton-Brown et al. 2011; Cohen et al. 2016). Therefore, short stature seems to be highly atypical for Weaver syndrome, in which most individuals are >2.1 SD above the mean for height at birth (range −0.5 SD to +4.9 SD) (Tatton-Brown et al. 2013).
Table 2.
Summary of growth phenotypes related to a pathogenic variant in EZH2
| Reference | Sex | Mutation | Protein alteration | Inheritance | Height (SD) | OFC (SD) | Learning disability | Malignancy |
|---|---|---|---|---|---|---|---|---|
| This report (proband)a | F | c.2050C>T | p.R684C | Paternal | (−)2.2 | (−)0.1 | Moderate | Neuroblastoma |
| This report (father) | M | c.2050C>T | p.R684C | N/R | (+)2.6 | N/R | Mild | None |
| Tatton-Brown et al. 2011 | F | c.2050C>T | p.R684C | De novo | (+)2.9 | N/R | Mild | None |
| Tatton-Brown et al. 2011 | F | c.2050C>T | p.R684C | N/R | (+)3.5 | (+)1.5 | N/R | None |
| Tatton-Brown et al. 2011 | M | c.2050C>T | p.R684C | De novo | (+)2.7 | (+)2.6 | Mild | None |
| Tatton-Brown et al. 2011 | M | c.2050C>T | p.R684C | N/R | (+)5.4 | (+)1.7 | Mild | None |
| Tatton-Brown et al. 2013 | M | c.2050C>T | p.R684C | De novo | (+)3.8 | (+)1.4 | Moderate | None |
| Tatton-Brown et al. 2013 | M | c.2051G>A | p.R684H | N/R | (+)2.4 | (+)3.4 | Mild | None |
| Cohen et al. 2016 | M | c.2050C>T | p.R684C | De novo | (+)3 | (−)0.3 | Significant delays | None |
F, female; M, male; N/R, not reported; SD, standard deviation; OFC, occipital frontal circumference.
avalues at the 2-yr evaluation.
Interestingly, both mother and proband shared a 505 kb microduplication of uncertain significance involving 2p23.3 (arr[hg19] 2p23.3(25,336,135–25,841,368)x3)and had an overlapping phenotype, including short stature and cognitive deficits. The identified microduplication includes DNMT3A which encodes a de novo DNA methyltransferase. Loss-of-function (LoF) variants in this gene, including missense variants, canonical splice site variants, frameshift indels, and large deletions, have been shown to cause TBRS, which is characterized by somatic overgrowth and intellectual disability (Tatton-Brown et al. 2014; Xin et al. 2017).
It is well established that histone modifications play a role in targeting of DNA methylation in the genome, which is essential for normal development and cellular function (Cedar and Bergman 2009; Rose and Klose 2014). EZH2, NSD1, and SETD2 are all histone methyltransferases that are responsible for trimethylation of lysine 27 of histone H3 (H3K27met3), mono- and dimethylation of lysine 36 of histone 3 (H3K36me1 and H3K36me2), and trimethylation of lysine 36 of histone 3 (H3K36me3), respectively (Wagner and Carpenter 2012). DNMT3A has been shown to recognize the H3K36me3 mark (Dhayalan et al. 2010) and interact with EZH2 within the Polycomb repressive complex 1/2 (PRC1/2) (Viré et al. 2006). The H3K36me3 mark guides DNA methylation and enhances DNMT3A activity. Although EZH2 is sufficient for recruitment and targeting DNMT3A, de novo DNA methylation is likely context-dependent as H3K27me3 also correlates with decreased DNA methylation in embryonic stem cells (Fouse et al. 2008; Rush et al. 2009; Dhayalan et al. 2010).
Pathogenic alterations in EZH2, DNMT3A, NSD1, and SETD2 result in overgrowth syndromes suggesting that they act either as corepressors of genes that promote growth (EZH2 and DNMT3A) or coactivators of genes involved in growth arrest (NSD1 and SETD2) (Yuan et al. 2011; Luscan et al. 2014). Given the opposing activities of these components in normal human growth, disease pathogenesis is likely due to an imbalance of chromatin states (Fahrner and Bjornsson 2014; Bjornsson 2015). For example, decreased EZH2 activity (generating a repressive histone mark) leads to an imbalance in the opposing machinery (NSD1, activating histone mark) resulting in methylation disturbances at the histone and DNA levels that have downstream effects on expression of growth related genes (Yuan et al. 2011). The EZH2 pathogenic variant (p.Arg684Cys) observed in this study has been shown to impair histone methyltransferase activity and result in decreased H3K27me3 (Cohen et al. 2016). This could lead to an imbalance in activating (increase in H3K36 methylation) and repressive (decrease in H3K27 methylation) histone marks (Yuan et al. 2011; Musselman et al. 2012a,b). The growth restriction phenotype observed in the proband raises the possibility that increased DNMT3A expression resulting from the gene duplication could alter transcriptional programming via increased DNA methylation in the setting of decreased H3K27 methylation (Fig. 3), which in turn may mask the overgrowth phenotype of the EZH2 pathogenic variant.
Figure 3.

A hypothetical mechanism linking the Weaver-associated EZH2 variant (p.R684C) and DNMT3A duplication with the observed growth phenotype in the proband. (A) Normal growth regulation in a wild-type background; (B) overgrowth in the context of the p.R684C EZH2 variant that shows impaired histone methyltransferase activity; (C) growth restriction in the context of increased DNMT3A expression due to gene duplication together with the p.R684C EZH2 variant.
There is growing body of evidence suggesting that expression of epigenetic regulators is tightly controlled. For example, NSD1 has shown evidence of dosage effects: LoF mutations and microdeletions encompassing NSD1 lead to Sotos overgrowth syndrome, whereas duplications involving NSD1 are associated with severe undergrowth (Chen et al. 2006; Kirchhoff et al. 2007; Franco et al. 2010; Zhang et al. 2011; Rosenfeld et al. 2013; Dikow et al. 2013; Novara et al. 2014; Sachwitz et al. 2017). Analysis of peripheral blood from patients with Sotos syndrome showed a global decrease in H3K36 methylation was accompanied by altered global DNA methylation patterns (Choufani et al. 2015). One might expect an opposing pattern of histone and DNA methylation in individuals with NSD1 duplications could lead to the growth restriction; however, the exact mechanism at play is unclear.
Interestingly, there is some emerging evidence suggesting that EZH2 and DNMT3A may exhibit dosage sensitivity as well. A recent report of an individual with suspected overgrowth and a 1.2-Mb deletion involving EZH2 and several other genes supports haploinsufficiency as the underlying disease mechanism in Weaver syndrome (Suri and Dixit 2017). Similarly, 2p23 microdeletions involving DNMT3A have been observed in individuals with phenotypes consistent with TBRS, suggesting that haploinsufficiency of this gene could also result in overgrowth (Shoukier et al. 2012; Bloch et al. 2014; Okamoto et al. 2016).
In conclusion, the functional association between DNMT3A and EZH2, their shared involvement in disorders of growth, and the possibility of a dynamic interaction between impaired EZH2 histone methylation and DNMT3A duplication are intriguing observations. Based on the concordance of the growth restriction phenotype observed in the proband and her mother, we speculate that the effect of increased DNMT3A dosage could be dominant to overgrowth promoting effect of the EZH2 mutation, and further work is needed to elucidate the mechanisms responsible.
METHODS
Ethical Compliance
The subject and her parents provided written informed consent. The protocol was approved by the institutional review board of the Mayo Clinic.
Chromosomal Microarray
CMA was performed using a custom oligonucleotide microarray (Agilent 180K probe platform) representing a uniform design developed through the International Standards for Cytogenomic Arrays consortium. The genome-wide functional resolution of this array is approximately 100 kilobases (kb), with higher resolution in targeted genes and regions of known clinical significance. All data was analyzed and reported initially using the March 2006 NCBI human genome build 36.1 [hg18].
Whole-Exome Sequencing Methodology
Blood samples were collected from the proband and her parents, and genomic DNA was extracted. Clinical whole-exome sequencing (WES) was performed at the Baylor Miraca Genetics Laboratories at Baylor College of Medicine (BCM) (Yang et al. 2013). Briefly, genomic DNA was fragmented by sonication followed by ligation to Illumina multiplexing paired-end (PE) adapters. The adapter-ligated DNA was PCR-amplified using primers containing sequencing barcodes. The pre-capture library was enriched by hybridizing to biotin labeled, solution-based exome capture reagent VCRome 2.1 (Roche NimbleGen) and 100-bp paired end reads were generated using the Illumina HiSeq 2000 platform. Data generated was converted to FastQ format using Illumina CASAVA 1.8 software and mapped to Genome Reference Consortium human genome build 37 (hg19) using Burrows–Wheeler Aligner (BWA). Variant calling was performed using Atlas-SNP and Atlas-indel (Human Genome Sequencing Center (HGSC), BCM). Annotation was performed using HGSC-SNP-anno and HGSC-indel-anno (HGSC, BCM). As a quality control measure, the individual's DNA is also analyzed by a SNP-array (Illumina HumanExome-12v1 array). A filtering process removed synonymous variants, intronic variants, and benign variants with a minor allele frequency (MAF) >1% in the exome sequencing project (ESP) 5400 exomes or the 1000 genomes project control databases and were interpreted according to ACMG guidelines and in accordance with the subject's clinical phenotype. The focused report contains results of genes related to the subject's clinical phenotype. Analysis in this case specifically included review of variants in genes associated with IUGR, delayed motor milestones, delayed speech, hypotonia, micrognathia, short stature, congenital heart disease, hip dysplasia, clubfoot, and neuroblastoma. Confirmatory Sanger sequencing was performed on the proband and her parents for variants in genes associated with disorders consistent with the subject's phenotype and to determine the mode of inheritance.
ADDITIONAL INFORMATION
Data Deposition and Access
The 505-kb duplication of uncertain significance at 2p23.3 was deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession number SCV000179757.4. The EZH2 variant was deposited in LOVD (http://www.lovd.nl/3.0/home) and can be found under Variant ID #0000369006. Whole-exome sequencing data are not publicly available because patient consent could not be obtained.
Ethics Statement
The proband and her parents provided written informed consent to research protocol # 12-009346 approved by the Mayo Clinic Institutional Review Board for this study and publication of this paper.
Acknowledgments
We would like to thank the subject and her family for participating in this study. We would also like to thank the Mayo Clinic Center for Individualized Medicine (CIM) for supporting this research through the CIM Investigative and Functional Genomics Program.
Author Contributions
K.P. and P.R.B. drafted the manuscript. All authors contributed to the critical revision of the manuscript for important intellectual content.
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Antonysamy S, Condon B, Druzina Z, Bonanno JB, Gheyi T, Zhang F, MacEwan I, Zhang A, Ashok S, Rodgers L, et al. 2013. Structural context of disease-associated mutations and putative mechanism of autoinhibition revealed by X-ray crystallographic analysis of the EZH2-SET domain. PLoS One 8: e84147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basel-Vanagaite L. 2010. Acute lymphoblastic leukemia in Weaver syndrome. Am J Med Genet A 152A: 383–386. [DOI] [PubMed] [Google Scholar]
- Bjornsson HT. 2015. The Mendelian disorders of the epigenetic machinery. Genome Res 25: 1473–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch M, Leonard A, Diplas AA, Pepermans X, Emanuel BS, Santa Rocca M, Revencu N, Sznajer Y. 2014. Further phenotype description, genotype characterization in patients with de novo interstitial deletion on 2p23.2-24.1. Am J Med Genet A 164A: 1789–1794. [DOI] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. 2009. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 10: 295–304. [DOI] [PubMed] [Google Scholar]
- Chen C-P, Lin S-P, Lin C-C, Chen Y-J, Chern S-R, Li Y-C, Hsieh L-J, Lee C-C, Pan C-W, Wang W. 2006. Molecular cytogenetic analysis of de novo dup(5)(q35.2q35.3) and review of the literature of pure partial trisomy 5q. Am J Med Genet A 140: 1594–1600. [DOI] [PubMed] [Google Scholar]
- Choufani S, Cytrynbaum C, Chung BHY, Turinsky AL, Grafodatskaya D, Chen YA, Cohen ASA, Dupuis L, Butcher DT, Siu MT, et al. 2015. NSD1 mutations generate a genome-wide DNA methylation signature. Nat Commun 6: 10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AL, Piccolo SR, Cheng L, Soldi R, Han B, Johnson WE, Bild AH. 2013. Genomic pathway analysis reveals that EZH2 and HDAC4 represent mutually exclusive epigenetic pathways across human cancers. BMC Med Genomics 6: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen ASA, Yap DB, Lewis MES, Chijiwa C, Ramos-Arroyo MA, Tkachenko N, Milano V, Fradin M, McKinnon ML, Townsend KN, et al. 2016. Weaver syndrome–associated EZH2 protein variants show impaired histone methyltransferase function in vitro. Hum Mutat 37: 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A. 2010. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem 285: 26114–26120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikow N, Maas B, Gaspar H, Kreiss-Nachtsheim M, Engels H, Kuechler A, Garbes L, Netzer C, Neuhann TM, Koehler U, et al. 2013. The phenotypic spectrum of duplication 5q35.2-q35.3 encompassing NSD1: is it really a reversed Sotos syndrome? Am J Med Genet A 161A: 2158–2166. [DOI] [PubMed] [Google Scholar]
- Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, et al. 2010. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 42: 722–726. [DOI] [PubMed] [Google Scholar]
- Fahrner JA, Bjornsson HT. 2014. Mendelian disorders of the epigenetic machinery: tipping the balance of chromatin states. Annu Rev Genomics Hum Genet 15: 269–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi IS, Drop S, Clements A, Keogh JM, Biernacka J, Lowenbein S, Challis BG, O'Rahilly S. 2006. Heterozygosity for a POMC-null mutation and increased obesity risk in humans. Diabetes 55: 2549–2553. [DOI] [PubMed] [Google Scholar]
- Feng N, Adler-Wailes D, Elberg J, Chin JY, Fallon E, Carr A, Frazer T, Yanovski JA. 2003. Sequence variants of the POMC gene and their associations with body composition in children. Obes Res 11: 619–624. [DOI] [PubMed] [Google Scholar]
- Fouse SD, Shen Y, Pellegrini M, Cole S, Meissner A, Van Neste L, Jaenisch R, Fan G. 2008. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, Polycomb binding and histone H3 lys4/lys27 trimethylation. Cell Stem Cell 2: 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco LM, de Ravel T, Graham BH, Frenkel SM, Van Driessche J, Stankiewicz P, Lupski JR, Vermeesch JR, Cheung SW. 2010. A syndrome of short stature, microcephaly and speech delay is associated with duplications reciprocal to the common Sotos syndrome deletion. Eur J Hum Genet 18: 258–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhoff M, Bisgaard A-M, Bryndorf T, Gerdes T. 2007. MLPA analysis for a panel of syndromes with mental retardation reveals imbalances in 5.8% of patients with mental retardation and dysmorphic features, including duplications of the Sotos syndrome and Williams–Beuren syndrome regions. Eur J Med Genet 50: 33–42. [DOI] [PubMed] [Google Scholar]
- Ko JM. 2013. Genetic syndromes associated with overgrowth in childhood. Ann Pediatr Endocrinol Metab 18: 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krude H, Biebermann H, Luck W, Horn R, Brabant G, Grüters A. 1998. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet 19: 155–157. [DOI] [PubMed] [Google Scholar]
- Lapunzina P. 2005. Risk of tumorigenesis in overgrowth syndromes: a comprehensive review. Am J Med Genet C Semin Med Genet 137C: 53–71. [DOI] [PubMed] [Google Scholar]
- Lee YS, Challis BG, Thompson DA, Yeo GSH, Keogh JM, Madonna ME, Wraight V, Sims M, Vatin V, Meyre D, et al. 2006. A POMC variant implicates β-melanocyte-stimulating hormone in the control of human energy balance. Cell Metab 3: 135–140. [DOI] [PubMed] [Google Scholar]
- Luscan A, Laurendeau I, Malan V, Francannet C, Odent S, Giuliano F, Lacombe D, Touraine R, Vidaud M, Pasmant E, et al. 2014. Mutations in SETD2 cause a novel overgrowth condition. J Med Genet 51: 512–517. [DOI] [PubMed] [Google Scholar]
- Moore LD, Le T, Fan G. 2013. DNA methylation and its basic function. Neuropsychopharmacology 38: 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman CA, Avvakumov N, Watanabe R, Abraham CG, Lalonde M-E, Hong Z, Allen C, Roy S, Nuñez JK, Nickoloff J, et al. 2012a. Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nat Struct Mol Biol 19: 1266–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman CA, Lalonde M-E, Côté J, Kutateladze TG. 2012b. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol 19: 1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novara F, Stanzial F, Rossi E, Benedicenti F, Inzana F, Di Gregorio E, Brusco A, Graakjaer J, Fagerberg C, Belligni E, et al. 2014. Defining the phenotype associated with microduplication reciprocal to Sotos syndrome microdeletion. Am J Med Genet A 164A: 2084–2090. [DOI] [PubMed] [Google Scholar]
- Okamoto N, Toribe Y, Shimojima K, Yamamoto T. 2016. Tatton-Brown–Rahman syndrome due to 2p23 microdeletion. Am J Med Genet A 170: 1339–1342. [DOI] [PubMed] [Google Scholar]
- Opitz JM, Weaver DW, Reynolds JF. 1998. The syndromes of Sotos and Weaver: reports and review. Am J Med Genet 79: 294–304. [DOI] [PubMed] [Google Scholar]
- Rose NR, Klose RJ. 2014. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim Biophys Acta 1839: 1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Kim KH, Angle B, Troxell R, Gorski JL, Westemeyer M, Frydman M, Senturias Y, Earl D, Torchia B, et al. 2013. Further evidence of contrasting phenotypes caused by reciprocal deletions and duplications: duplication of NSD1 causes growth retardation and microcephaly. Mol Syndromol 3: 247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush M, Appanah R, Lee S, Lam LL, Goyal P, Lorincz MC. 2009. Targeting of EZH2 to a defined genomic site is sufficient for recruitment of Dnmt3a but not de novo DNA methylation. Epigenetics 4: 404–414. [DOI] [PubMed] [Google Scholar]
- Sachwitz J, Meyer R, Fekete G, Spranger S, Matulevičienė A, Kučinskas V, Bach A, Luczay A, Brüchle NO, Eggermann K, et al. 2017. NSD1 duplication in Silver–Russell syndrome (SRS): molecular karyotyping in patients with SRS features. Clin Genet 91: 73–78. [DOI] [PubMed] [Google Scholar]
- Shoukier M, Schröder J, Zoll B, Burfeind P, Freiberg C, Klinge L, Kriebel T, Lingen M, Mohr A, Brockmann K. 2012. A de novo interstitial deletion of 2p23.3-24.3 in a boy presenting with intellectual disability, overgrowth, dysmorphic features, skeletal myopathy, dilated cardiomyopathy. Am J Med Genet A 158A: 429–433. [DOI] [PubMed] [Google Scholar]
- Simon JA, Lange CA. 2008. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res 647: 21–29. [DOI] [PubMed] [Google Scholar]
- Suri T, Dixit A. 2017. The phenotype of EZH2 haploinsufficiency-1.2-Mb deletion at 7q36.1 in a child with tall stature and intellectual disability. Am J Med Genet A 173: 2731–2735. [DOI] [PubMed] [Google Scholar]
- Tatton-Brown K, Hanks S, Ruark E, Zachariou A, Duarte SDV, Ramsay E, Snape K, Murray A, Perdeaux ER, Seal S, et al. 2011. Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget 2: 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton-Brown K, Murray A, Hanks S, Douglas J, Armstrong R, Banka S, Bird LM, Clericuzio CL, Cormier-Daire V, Cushing T, et al. 2013. Weaver syndrome and EZH2 mutations: clarifying the clinical phenotype. Am J Med Genet A 161: 2972–2980. [DOI] [PubMed] [Google Scholar]
- Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, Del Vecchio Duarte S, Zachariou A, Hanks S, O'Brien E, Aksglaede L, et al. 2014. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet 46: 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton-Brown K, Loveday C, Yost S, Clarke M, Ramsay E, Zachariou A, Elliott A, Wylie H, Ardissone A, Rittinger O, et al. 2017. Mutations in epigenetic regulation genes are a major cause of overgrowth with intellectual disability. Am J Hum Genet 100: 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden J-M, et al. 2006. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439: 871–874. [DOI] [PubMed] [Google Scholar]
- Vora N, Bianchi DW. 2009. Genetic considerations in the prenatal diagnosis of overgrowth syndromes. Prenat Diagn 29: 923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EJ, Carpenter PB. 2012. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol 13: 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin B, Cruz Marino T, Szekely J, Leblanc J, Cechner K, Sency V, Wensel C, Barabas M, Therriault V, Wang H. 2017. Novel DNMT3A germline mutations are associated with inherited Tatton-Brown–Rahman syndrome. Clin Genet 91: 623–628. [DOI] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al. 2013. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med 369: 1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Xu M, Huang C, Liu N, Chen S, Zhu B. 2011. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J Biol Chem 286: 7983–7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Lu X, Beasley J, Mulvihill JJ, Liu R, Li S, Lee J-Y. 2011. Reversed clinical phenotype due to a microduplication of Sotos syndrome region detected by array CGH: microcephaly, developmental delay and delayed bone age. Am J Med Genet A 155A: 1374–1378. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.