

Abstract
Tumors with anaplastic lymphoma kinase (ALK) fusion rearrangements, including non-small-cell lung cancer and anaplastic large cell lymphoma, are highly sensitive to ALK tyrosine kinase inhibitors (TKIs), underscoring the notion that such cancers are addicted to ALK activity. Although mutations in ALK are heavily implicated in childhood neuroblastoma, response to the ALK TKI crizotinib has been disappointing. Embryonal tumors in patients with DNA repair defects such as Fanconi anemia (FA) often have a poor prognosis, because of lack of therapeutic options. Here we report a child with underlying FA and ALK mutant high-risk neuroblastoma responding strongly to precision therapy with the ALK TKI ceritinib. Conventional chemotherapy treatment caused severe, life-threatening toxicity. Genomic analysis of the initial biopsy identified germline FANCA mutations as well as a novel ALK-I1171T variant. ALK-I1171T generates a potent gain-of-function mutant, as measured in PC12 cell neurite outgrowth and NIH3T3 transformation. Pharmacological inhibition profiling of ALK-I1171T in response to various ALK TKIs identified an 11-fold improved inhibition of ALK-I1171T with ceritinib when compared with crizotinib. Immunoaffinity-coupled LC-MS/MS phosphoproteomics analysis indicated a decrease in ALK signaling in response to ceritinib. Ceritinib was therefore selected for treatment in this child. Monotherapy with ceritinib was well tolerated and resulted in normalized catecholamine markers and tumor shrinkage. After 7.5 mo treatment, the residual primary tumor shrunk, was surgically removed, and exhibited hallmarks of differentiation together with reduced Ki67 levels. Clinical follow-up after 21 mo treatment revealed complete clinical remission including all metastatic sites. Therefore, ceritinib presents a viable therapeutic option for ALK-positive neuroblastoma.
Keywords: abnormality of the enteric ganglia, anaplastic large-cell lymphoma, neuroblastoma
INTRODUCTION
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase (RTK) comprising an extracellular ligand-binding domain, a transmembrane domain and an intracellular tyrosine kinase domain (Iwahara et al. 1997; Morris et al. 1997). ALK was first described in 1994 as a fusion partner in the t(2;5) chromosomal translocation in anaplastic large cell lymphoma (ALCL) (Morris et al. 1994). Vertebrate ALK is activated by the recently described small secreted ALKAL (FAM150/AUG) ligands, which potently activate ALK signaling (Zhang et al. 2014; Guan et al. 2015; Reshetnyak et al. 2015; Mo et al. 2017; Fadeev et al. 2018). ALK activates multiple signaling pathways, such as the PI3K-AKT, CRKL-C3G, MEKK2/3-MEK5-ERK5, JAK-STAT, and MAPK pathways (Hallberg and Palmer 2016).
Mutation of full-length ALK has been well described in the neural crest derived pediatric cancer neuroblastoma (Carén et al. 2008; Chen et al. 2008; George et al. 2008; Janoueix-Lerosey et al. 2008; Mosse et al. 2008; Hallberg and Palmer 2013). Of the more than 35 mutations in ALK described, the majority are point mutations in the kinase domain, although deletions in the extracellular domain and translocations have also been reported (Okubo et al. 2012; Cazes et al. 2013; Hallberg and Palmer 2013; Bresler et al. 2014; Fransson et al. 2015). Within the kinase domain three hotspot residues, F1174, F1245, and R1275, represent most patient mutations (De Brouwer et al. 2010; Hallberg and Palmer 2013; Bresler et al. 2014). In addition to ALK mutations in treatment-naïve neuroblastoma, examination of ALK status in relapsed neuroblastoma samples has highlighted an increase in activating ALK mutations appearing later in the disease course (Martinsson et al. 2011; Schleiermacher et al. 2014; Eleveld et al. 2015).
In neuroblastoma, ALK signaling has been shown to act synergistically with MYCN to drive tumor development (Berry et al. 2012; Heukamp et al. 2012; Schonherr et al. 2012; Zhu et al. 2012; Cazes et al. 2014; Ueda et al. 2016), with combined occurrence of MYCN amplification and ALK mutations being associated with particularly bad prognosis (De Brouwer et al. 2010). ALK has led to the suggestion that inhibition of ALK with small molecule tyrosine kinase inhibitors (TKIs) may offer clinical benefit in neuroblastoma. The ALK TKI crizotinib was approved for clinical use in patients with ALK-positive non-small-cell lung cancer (NSCLC) in 2011 ([FDA] 26th of August 2011; Kwak et al. 2010), based on a robust response in this patient population. Although significant problems with resistance to ALK TKIs occur, a trial comparing crizotinib with chemotherapy concluded that crizotinib is superior in patients with previously treated, advanced ALK-positive NSCLC (Shaw et al. 2013). Similarly, treatment with crizotinib resulted in a strong response in a phase I crizotinib monotherapy trial of pediatric patients with ALK-fusion positive tumors, although patient responses in pediatric ALK mutant neuroblastoma were less encouraging (Mosse et al. 2013, 2017). It is not clear whether this relates to clinical factors unique to neuroblastoma or to issues of efficacy of inhibition of ALK by crizotinib. The clinical data thus far motivates exploration of alternative strategies in neuroblastoma, including ALK monotherapy with next-generation TKIs and combination strategies (Berry et al. 2012; Moore et al. 2014; Umapathy et al. 2014; Guan et al. 2016; Infarinato et al. 2016; Krytska et al. 2016).
Other ALK TKIs include ceritinib, brigatinib, alectinib, and lorlatinib (Christensen et al. 2007; Katayama et al. 2011, 2015; Sakamoto et al. 2011; Marsilje et al. 2013; Chia et al. 2014; Johnson et al. 2014) (for the latest details of clinical trials using ALK TKIs, please see Clinicaltrials.gov). These ALK inhibitors bind slightly differently within the ATP-binding pocket of the ALK kinase domain, show varying abilities to cross the blood–brain barrier, and have differing profiles of inhibition for the wild-type ALK kinase domain compared with the various ALK kinase mutants. These properties have important implications for potential treatment of ALK-positive neuroblastoma in which ALK mutations are present in treatment-naïve tumors. Ceritinib gained U.S. Food and Drug Administration approval in 2014 following accelerated review for the treatment of patients with ALK-positive (ALK+) metastatic NSCLC who have progressed on, or are intolerant to, crizotinib ([FDA] 29th of April 2014; Shaw et al. 2014; [FDA] 26th of May 2017).
Here we report the robust response of an ALK-positive neuroblastoma patient to ceritinib treatment. The patient received chemotherapy according to protocol after being diagnosed with high-risk neuroblastoma but displayed severe hematological failure early after initial treatment. This led to suspicion of Fanconi anemia (FA), which was confirmed by chromosomal breakage assessment and identification of FANCA gene mutations. FA is a rare recessive genetic disorder clinically characterized by congenital abnormalities and progressive bone marrow failure (Kutler et al. 2003b), although some patients may show only subtle symptoms or no phenotype at all (Neveling et al. 2009). FA is caused by mutations in one of at least 21 different FA genes encoding proteins that function in interstrand cross-link and double strand DNA repair (Mamrak et al. 2017). Because of the impaired DNA damage response, FA patients have significantly increased cancer susceptibility, particularly to acute myeloid leukemia, head and neck squamous cell carcinoma (Kutler et al. 2003a,b), and, more rarely, embryonic tumors such as Wilms tumor, hepatoblastoma, or neuroblastoma (Abbondanzo et al. 1986; Bissig et al. 2002; Berrebi et al. 2006; Kopic et al. 2011; Malric et al. 2015). The management of cancer in FA patients is challenging as defective DNA repair leads to hypersensitivity to both radiation and cross-linking agents such as mitomycin C and cisplatin with increased risk of developing severe toxicity (Walsh et al. 2017) (http://fanconi.org/explore/clinical-care-guidelines). In parallel to identification of FANCA gene mutations in this neuroblastoma patient, we also noted the presence of an ALK mutation at residue I1171.
Preclinical analysis showed that the mutation of ALK-I1171 to threonine (T), which has not previously been reported in neuroblastoma, generates a potent gain-of-function mutant. PC12 cells expressing ALK-I1171T display ligand-independent activation of ALK, neurite outgrowth and activation of downstream signaling. In addition, ALK-I1171T drives foci formation in NIH3T3 cells. We were able to identify ceritinib as a next-generation ALK TKI that effectively inhibits ALK-I1171T in a preclinical setting, and based on these experimental data, the patient was treated with ceritinib. A dramatic response was observed and 7.5 mo after initiation of treatment, tumor material was surgically removed, exhibiting reduced Ki67 levels and up-regulation of markers of differentiation. Based on this case, we suggest that ceritinib presents a viable therapeutic option for patients with ALK-positive neuroblastoma.
RESULTS
Patient Presentation
A 16-mo-old boy presenting with neurologic symptoms at the pediatric emergency unit was found to have metastatic neuroblastoma stage M (INRGSS) (Fig. 1). There was no previous family history of neuroblastoma. SNP-microarray analysis of genomic DNA from initial biopsy showed no MYCN-amplification but revealed several unfavorable segmental alterations (Fig. 2A). COJEC chemotherapy induction was started according to HR-NBL-1 SIOPEN (Ladenstein et al. 2017) and the patient received one A course (day 0; vincristine, carboplatin, etoposide), one B course (day 10; vincristine, cisplatin), and a partial C course (day 20; vincristine, etoposide, cyclophosphamide), before chemotherapy had to be discontinued because of severe bone marrow toxicity with anemia, neutropenia, and thrombocytopenia. For the ensuing 4 months, the patient was continuously hospitalized and suffered repeated episodes of septicemia, intestinal hemorrhage, and typhlitis as well as respiratory failure requiring intensive care. Because of the unusual severity of the toxicity, and also because the patient displayed moderate radial dysplasia with deviant thumbs bilaterally, suspicion of Fanconi anemia was raised. A clinical chromosome breakage assessment was found positive. A more detailed genomic analysis then revealed inherited mutations in the FANCA gene; one novel missense variant at genomic position Chr 16:89831348 (NM_000135.2; c.2728C>T, p.L910F) predicted to be deleterious and probably damaging by scale-invariant feature transform (SIFT) and PolyPhen-2 (PP2), respectively, and one splice site mutation at the acceptor site of intron 42 at genomic position Chr 16:89805118, (NM_000135.2; c.4261-2A>C) (Table 1; Fig. 2B). The L910F variant is not present in either ClinVar or HGMD and, to best of our knowledge, this is the first report of a L910F variant in association with FA. A third rare missense variant at Chr 16:89871709 (c.688G>A; p.V230I) was detected in the patient, although predicted to be benign by SIFT and PP2, indicating a nonpathogenic function. This is supported by a recent study showing that the V230I variant is localized to the nucleus and does not confer sensitivity to mitomycin C (Kimble et al. 2017). The fact that the child presents as a compound heterozygote for FANCA (p.L910F/c.4261-2A>C) is a likely cause for the above described toxicity (Table 1; Fig. 2B). Further use of chemotherapy, or irradiation, was advised against in this patient because of the DNA repair defects in Fanconi anemia. Because initial genomic analysis also identified an ALK-I1171T mutation in the patient (Table 1; Fig. 2C), ALK TKI therapy was considered and experimentally investigated preclinically for this patient.
Figure 1.

Patient tumor imaging. Disease at initial diagnosis and after targeted therapy in full clinical remission (A–D). (A) Whole-body MIBG-I123 scintigraphy showing extensive metastatic disease at diagnosis of high-risk neuroblastoma (upper panels). (Left panel) anterior view; (right panel) posterior view. Lower panels display head and neck region. (B) MIBG-scintigraphy after 21 mo of ceritinib therapy showing complete remission at all metastatic sites. (C) Brain CT scans at diagnosis showing a CNS metastasis (43 mm × 46.7 mm) posterior to the right orbit. (D) CT scans after 21 mo of ceritinib therapy showing complete metastatic response. (E–H) Primary tumor immediately before and after targeted therapy. (E) Magnetic resonance imaging (MRI) of primary abdominal tumor (indicated by arrows and measurements inserted) immediately before start of ceritinib targeted therapy (∼18.8 mL tumor volume), (F) MRI after 3 mo therapy (∼13.8 mL tumor volume), and (G) MRI after 6 mo therapy immediately prior to radical surgery (∼10.6 mL tumor volume). (H) Decrease of evaluable tumor volume after 3 and 6 mo of targeted therapy.
Figure 2.

Genomic analysis of the tumor sample. (A) Copy number profiling using Affymetrix HD SNP-microarray shows a genomic profile with 11q-deletion and 17q-gain indicative of high-risk neuroblastoma. (B) Two different likely pathogenic variants in the FANCA gene were detected through exome sequencing and confirmed in constitutional DNA of the patient. Read mapping is visualized in IGV. (C) Exome sequencing also detected a novel ALK I1171T mutation in the tumor that was confirmed to be somatic through Sanger sequencing. Electrophorogram of tumor in upper panel and from constitutional DNA in lower panel.
Table 1.
FANCA and ALK mutation details
| Gene | Description | Somatic | Cytoband | Pos (hg19/GRCh37) | Ref | Var | Coverage at mutation site | Var allele fraction | Variant function on protein | Change cDNA/protein | Predicted impact on protein function | Presence public databases | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SIFT | PP2 | LRT | dbSNP(132) | 1000 Genome | esp6500 all | Cosmic | ClinVar | HGMD | |||||||||||
| FANCA | Fanconi anemia group A protein | No | 16q24.3 | Chr 16:89805118 | T | G | 80× | 43% | Splicing | FANCA: NM_000135.2:exon43:c.4261-2A>C | - | - | - | - | - | - | - | - | CS04812 |
| FANCA | Fanconi anemia group A protein | No | 16q24.3 | Chr 16:89831348 | G | A | 185× | 49% | Nonsynonymous SNV | FANCA:NM_000135.2:exon28:c.C2728T:p.L910F | Deleterious | Probably damaging | Deleterious | - | - | - | 4483710 | - | - |
| FANCA | Fanconi anemia group A protein | No | 16q24.3 | Chr 16:89871709 | C | T | 81× | 62% | Nonsynonymous SNV | FANCA:NM_000135.2:exon7:c.G688A:p.V230I | Nondeleterious | Benign | Neutral | rs144560850 | - | 0.000231 | - | 321366 | - |
| ALK | ALK tyrosine kinase receptor | Yes | 2p23.2 | Chr 2:29445213 | A | G | 83× | 40% | Nonsynonymous SNV | ALK:NM_004304.4:exon22:c.T3512C:p.I1171T | - | Probably damaging | Deleterious | - | - | - | 4381100 | - | - |
Variant specific information including position, transcript-ID, coverage, and presence in publically available databases.
ALK-I1171T Is a Gain-of-Function ALK Mutation
Exome sequence analysis of a tumor biopsy sample revealed a heterozygous mutation of ALK (Chr 2:29445213) in exon 22; (NM_004304.4; c.3512T>C) leading to a missense mutation I117IT (Table 1; Fig. 2C). Sanger sequencing of tumor and patient's corresponding constitutional DNA indicated that the ALK variant was caused by a somatic event. I1171 is located in the ALK kinase domain in close proximity to F1174, one of the mutational hotspots of ALK in neuroblastoma that is localized in the αC-helix in the amino-terminal lobe of the kinase (Fig. 3A). Mutation of I1171 to either N/T or S has previously been described in ALK TKI-treated patients with ALK fusion-positive NSCLC (in the EML4-ALK and HIP1-ALK fusion proteins) (Katayama et al. 2014; Ou et al. 2014; Toyokawa et al. 2014), resulting in tumor resistance.
Figure 3.

Characterization of ALK-I1171T with cell culture systems. (A) Model of the ALK kinase domain (PDB #3LCS) using PyMol, which can be divided into the upper amino-terminal lobe and the lower carboxy-terminal lobe by the hinge region and ceritinib binding/ATP (red) binding pocket. Regulatory spine (R-spine) residues are shown in magenta. The spine is anchored in the αF-helix (D1311) and includes the DFG (F1271) and the HRD motif (H1247) of the C-lobe. R-spine residues in the amino-terminal lobe include the β4 strand (C1182) and the residue mutated in this patient in the αC-helix (I1171). Mutation of residue I1171 to threonine in PyMol, results in a small shift, which potentially drives a dynamic allostery that results in a gain-of-function ALK activity (upper insert: wild-type ALK kinase domain; lower insert: ALK-I1171T kinase domain). (B) ALK kinase activity and activation of downstream signaling pathways were visualized by western blot with antibodies against pALK (Y1604) and pERK1/2. Total ALK and pan-ERK were used as loading controls. Blots are representative of three independent experiments. (C) Neurite outgrowth of PC12 cells as a readout for ALK kinase activity was performed with wild-type ALK and ALK variants in the absence or presence of ALKAL1 ligand. Bars represent mean percentage ± STD of neurite-carrying cells among GFP-positive cells from three independent experiments. (D) Representative focus formation assays for NIH 3T3 cells transfected with wild-type ALK, ALK variants (ALK-I1171T and ALK-F1174L), or empty vector. (E) Inhibition profiling of ALK TKIs on ALK-I1171T. PC12 cells expressing ALK-I1171T were treated with serial dilution of ALK inhibitors as indicated. Phosphorylation of ALK was detected with pALK (Y1604) antibody and total ALK was used as loading control. (F) IC50 values of different ALK inhibitors were calculated with GraphPad Prism 6.0 by fitting data to a log (inhibitor concentration) versus normalized response (variable slope) equation and shown in the accompanying table. Values represent average ± STD from three independent experiments.
This is the first report of mutation of ALK-I1171T in neuroblastoma, although mutation of ALK-I1171N has been reported (Mosse et al. 2008; Bresler et al. 2014). Intriguingly, the residue I1171 contributes to the regulatory spine (Fig. 3A; Kornev and Taylor 2015), prompting us to experimentally validate its behavior in the context of the full-length ALK RTK. To evaluate the ALK-I1171T mutant we used a PC12 cell culture system for expression of either wild-type or ALK-I1171T. Wild-type ALK can be activated with the ALKAL1 ligand (FAM150A, AUGβ) (Guan et al. 2015; Reshetnyak et al. 2015), leading to extensive tyrosine phosphorylation of the receptor and activation of downstream targets, such as ERK1/2 (Fig. 3B). In contrast to wild-type ALK, we observed ligand-independent auto-trans-phosphorylation of the ALK-I1171T mutant together with activation of the downstream target ERK1/2 (Fig. 3B).
We next investigated whether the ALK-I1171T mutant was capable of stimulating neurite outgrowth in the PC12 cell line. PC12 cells are a clonal rat adrenal pheochromocytoma cell line with enteric cell origin, which differentiate and extend neurites upon extended ERK1/2 stimulation (Cowley et al. 1994). We and others have previously shown that activation of ALK triggers differentiation of PC12 cells into sympathetic-like neurons, a process characterized by extension of neurites (Fig. 3C; Motegi et al. 2004; Schonherr et al. 2011). Expression of wild-type ALK mediates only a low level of neurite outgrowth in this assay (Fig. 3C). In contrast, expression of the ALK-I1171T mutant results in robust neurite outgrowth to a level similar to that observed for the well characterized ALK-F1174L mutant used as positive control.
Finally, we investigated whether the human ALK-I1171T mutant displays transforming potential in NIH3T3 cells. Expression of either the positive control human ALK-F1174L or the ALK-I1171T mutant led to formation of foci of transformed cells over the background monolayer (Fig. 3D), whereas expression of the wild-type human ALK receptor was unable to mediate foci formation. Thus, ALK-I1171T exhibits intrinsic transforming activity in a focus formation assay, in addition to ligand-independent activation of downstream targets and neurite outgrowth.
Ceritinib Abrogates Growth in ALK-Addicted Neuroblastoma Cell Lines
Ceritinib is a small molecule ALK TKI that has been extensively explored for ALK-positive NSCLC and is FDA approved for clinical use in that setting. It has not yet been reported as being used clinically in the context of ALK mutations in neuroblastoma. Previous analysis of the I1171T mutation in NSCLC (arising in response to sequential crizotinib and alectinib treatment) noted that the I1171 mutation is relatively insensitive to inhibition by crizotinib and alectinib but was still sensitive to other ALK TKIs such as TAE684 and ceritinib (Katayama et al. 2014). We therefore assembled a panel of neuroblastoma cell lines harboring either mutated or wild-type ALK in combination with other genetic aberrations, CLB-GE, CLB-GA, CLB-BAR, CLB-PE, IMR32, Kelly, SK-N-AS, SK-N-DZ, and SK-N-BE (Table 2) to examine the effect of ceritinib on ALK activity and cell proliferation in a neuroblastoma setting (Kohl et al. 1984; Combaret et al. 1995; Schleiermacher et al. 2003; Cazes et al. 2013; Fransson et al. 2015; Umapathy et al. 2017). In this analysis we also compared the ability of ceritinib to inhibit proliferation of neuroblastoma cells with the well-characterized clinical ALK TKI crizotinib (Christensen et al. 2007), treating cells with increasing doses of either crizotinib or ceritinib. Proliferation of ALK-addicted cell lines such as CLB-BAR, CLB-GE, CLB-GA, and Kelly was inhibited in a dose-dependent manner by both crizotinib and ceritinib (Table 2). Of the neuroblastoma cell lines tested, growth of CLB-BAR and CLB-GE was inhibited in a manner similar to that previously shown for crizotinib (Schonherr et al. 2012), although the observed IC50 values differ up to twofold. IC50 values were similar for both ceritinib and crizotinib in CLB-GA and Kelly cells with values of ∼110 and 330 nM, respectively (Table 2). Importantly, we did not observe inhibition of proliferation in non-ALK-addicted cell lines such as CLB-PE, SK-N-AS, SK-N-BE, and SK-N-DZ, which carry other driver mutations. IMR32 cells, which express wild-type ALK and respond to stimulation with ALKAL ligands (Guan et al. 2015), are more sensitive to crizotinib than ceritinib, although IC50 values are high, more than 350 nM (Table 2). Thus, these preclinical results show that although activity of the ALK-dependent neuroblastoma cells tested here is inhibited by crizotinib, treatment with ceritinib has greater efficacy.
Table 2.
Indicated neuroblastoma cell lines were treated with increasing doses of either ceritinib or crizotinib for 72 h and cell viability was assessed by resazurin assay
| Cell line | ALK status | MYCN status | IC50 nM (ceritinib) | IC50 nM (crizotinib) |
|---|---|---|---|---|
| CLB-GE | F1174V, A | A | 131.3 ± 24 | 233.7 ± 18 |
| CLB-BAR | g-o-f Δexon 4-12 | A | 36.1 ± 6.2 | 54.9 ± 20 |
| KELLY | F1174L, A | A | 328.9 ± 36 | 333.3 ± 78 |
| CLB-GAR | R1275Q | NA | 102.3 ± 16 | 119.3 ± 45 |
| IMR32 | wt | A | >500 | 348.7 ± 45 |
| SK-N-BE | wt, NA | A | >1500 | >1000 |
| SK-N-DZ | wt | A | >800 | >4000 |
| SK-N-AS | wt, NA | NA | >3000 | 353.0 ± 27 |
| CLB-PE | wt | A | >15000 | >4000 |
NA, nonamplified; A, amplified; g-o-f Δexon 4-11, gain-of-function deletion of exons in the extracellular domain of ALK; wt, wild-type.
Ceritinib Effectively Inhibits the Crizotinib-Resistant ALK-I1171T Mutant in PC12 Cells
To determine which ALK TKI would be suitable in this clinical case we examined the ability of different ALK inhibitors to abrogate ALK-I1171T activity. As readout for ALK activity we used phosphorylation of ALK Y1604, which reflects ALK activation, in PC12 cells (Chand et al. 2013). PC12 cells expressing ALK-I1171T were treated with serial dilution of the ALK inhibitors crizotinib, ceritinib, lorlatinib, and brigatinib, and Y1604 phosphorylation levels were quantified. We observed that full-length ALK-I1171T was more resistant to crizotinib than other inhibitors as measured by Y1604 phosphorylation (Fig. 3E). The IC50 of crizotinib for ALK-I1171T in this analysis was 193 ± 57 nM, approximately 11 times that of ceritinib for ALK-I1171T (Fig. 3F). The next-generation ALK TKIs also showed strong anti-ALK-I1171T activity. The observed IC50 values of ALK-I1171T for brigatinib and lorlatinib were in a similar range (6.8–7.5 nM), approximately 1/28th of the crizotinib IC50 for ALK-I1171T (Fig. 3F). Taken together, our results indicate that ceritinib as well as the third-generation TKIs lorlatinib and brigatinib represent experimentally well-supported choices for treatment of tumor(s) harboring the ALK-I1171T mutation.
To extend our analysis we examined the activity of ceritinib to inhibit a range of gain-of-function ALK variants expressed in PC12 cells, using ALK Y1604 phosphorylation as readout (Supplemental Fig. S2; includes all loading ALK control blots for this experiment). In this analysis crizotinib abrogates wild-type ALK phosphorylation with an IC50 of ∼18 nM, which is similar to earlier reports (Schonherr et al. 2011; Siaw et al. 2016). In comparison, ceritinib blocks ALK Y1604 phosphorylation with a threefold lower IC50 level, of ∼5 nM. The IC50 values for crizotinib inhibition of ALK Y1604 phosphorylation of the various ALK mutations tested here are similar to earlier reports, highlighting a requirement for higher crizotinib dosage for effective inhibition of ALK-I1171N (Table 3; Schonherr et al. 2011; Guan et al. 2016; Siaw et al. 2016). ALK-G1269A has only been described in EML4-ALK fusions arising in NSCLC patients that have developed resistance to ALK TKI treatment (Doebele et al. 2012), but is included here as reference in the context of full-length ALK.
Table 3.
IC50 values for inhibition of ALK Y1604 phosphorylation by ceritinib or crizotinib in PC12 cells
| ALK mutation | Ceritinib (IC50 nM) | Normalized to IC50 of wt | Crizotinib (IC50 nM) | Normalized to IC50 of WT | Fold change |
|---|---|---|---|---|---|
| wt | 5.3 ± 0.2 | 1.0 | 17.8 ± 0.2 | 1 | 3.4 |
| G1128A | 19.8 ± 3.5 | 3.7 | 22.2 ± 3.4 | 1.2 | 1.1 |
| I1171N | 46.6 ± 16 | 8.8 | 139.5 ± 13.8 | 7.8 | 3.0 |
| F1174L | 16.9 ± 8 | 3.2 | 29.9 ± 4.8 | 1.7 | 1.8 |
| R1192P | 26.1 ± 1.2 | 4.9 | 28.1 ± 7.5 | 1.6 | 1.1 |
| F1245C | 24.6 ± 6.4 | 4.6 | 37.5 ± 7.7 | 2.1 | 1.5 |
| G1269A | 28.9 ± 3.6 | 5.5 | 113.5 ± 8.5 | 6.4 | 3.9 |
| R1275Q | 10.8 ± 3.5 | 2.0 | 30.3 ± 0.9 | 1.7 | 2.8 |
| Y1278S | 20.5 ± 0.1 | 3.9 | 69.5 ± 0.03 | 3.9 | 3.4 |
IC50 values were determined by quantification of Y1604 phosphorylation from the immunoblots in Supplemental Figure 1. Values represent mean ± SD from at least two independent experiments.
Our extensive examination of the inhibition profiles of both crizotinib and ceritinib on the different ALK mutations reveals one important exception—ALK-I1171T. We observe that inhibition of ALK-I1171T by ceritinib exhibits an IC50 value (17.4 ± 3.1 nM; Fig. 3F) that is not too far from that of wild-type ALK (5.3 ± 0.2 nM) (Table 3). This is in stark contrast to the ALK-I1171N mutation at the same residue, which displays resistance to both ceritinib (46.6 ± 0.16 nM) and crizotinib (139.5 ± 13.8 nM) when compared with wild-type ALK (Table 3). In summary, ceritinib was active in a range comparable to crizotinib as measured by inhibition of ALK phosphorylation/activation in PC12 cells with one exception, the ALK-I1171T mutant which was insensitive to crizotinib but efficiently inhibited by ceritinib.
Phosphoproteomic Profiling of Ceritinib in Different Neuroblastoma Cell Lines
Phosphoproteomic profiling has clinical value because it can be translated into prognostic biomarkers, which can be used to identify functional downstream targets as combinatorial treatment target options together with ceritinib. Here we determined the difference in the phosphoproteomic patterns upon treatment with ceritinib in ALK-addicted (CLB-BAR and CLB-GE) neuroblastoma cell lines with that of a non-ALK-addicted (SK-N-AS) neuroblastoma cell line (Fig. 4A). The CLB-BAR and CLB-GE cell lines harbor activating ALK mutations, are ALK/MYCN amplified, lack p53/RAS mutations, and display high levels of NF1 (Umapathy et al. 2017). The SK-N-AS cell line harbors an activating mutation in the NRAS gene and exhibits down-regulated NF1 expression leading to an activation of the RAS-MAPK pathway. SK-N-AS has a 11q deletion, is non-MYCN-amplified, has no p53/ALK mutations, and has undetectable levels of ALK protein (Umapathy et al. 2017). SK-N-AS was used as a control cell line for off-target effects of the inhibitors used, because ALK protein is undetectable in this cell line with no MYCN amplification.
Figure 4.

Ceritinib phosphoproteomic profile in neuroblastoma cell lines. (A) Three neuroblastoma cell lines (CLB-BAR, CLB-GE, and SK-N-AS) were cultured in control conditions or in the presence of ceritinib. Cells were harvested for tyrosine and serine/threonine phosphoproteomic analysis after 60 min. The pie chart indicates the number of targeted sites. (B) Overview of all ALK tyrosine phosphorylation sites with log2 fold change values (FC; treated/untreated) in CLB-BAR and CLB-GE. Note that no signal was measured in SK-N-AS cells, reflecting the known absent expression of ALK. (C) Log2 FC values of all analyzed phosphosites indicating the number of proteins found to contain phosphorylated (red) or dephosphorylated (green) sites for the three cell lines analyzed. Dotted lines indicate thresholds to determine differential phosphorylation. (D) Venn diagrams show the correlation between phosphorylated (red) and dephosphorylated (green) proteins in the different cell lines. Sixty proteins were found to be dephosphorylated upon ceritinib treatment in the ALK-addicted CLB-BAR and CLB-GE lines but not in the SK-N-AS cell line. (E) These proteins were mapped to the InWeb_ InBioMap protein–protein interaction network. For visualization purposes, only direct interactions between the identified proteins are shown. Node sizes are proportional to the number of connections, whereas node colors indicate different log2 FC values as indicated in the color scale legend on the top left. (F) Reactome gene set enrichment analysis on all 60 proteins that were identified to be dephosphorylated. The 10 most significantly enriched pathways are shown and ranked on FDR values.
To identify sites with altered phosphorylation associated with ALK activity, we utilized an immunoaffinity-coupled LC-MS/MS approach by treating neuroblastoma cell lines with ceritinib. Prior to MS, lysates were examined for ALK, ERK, and AKT activity, which have previously been shown to be reduced upon ALK TKI treatment for 1 h (Supplemental Fig. S3). We observed reduced phosphorylation levels of ALK and ERK in response to ceritinib, validating response to inhibitor treatment (Supplemental Fig. S3). Phosphoproteomic analysis of neuroblastoma cell lines (CLB-BAR, CLB-GE, and SK-N-AS) identified 2223 phosphothreonine sites, 2583 phosphotyrosine sites, and 5013 phosphoserine residue sites in 3345 different proteins (Fig. 4A). All previously observed 11 ALK phosphotyrosine sites were identified and showed drastically reduced phosphorylation signals in both ALK-addicted neuroblastoma cell lines (i.e., CLB-BAR and CLB-GE) after ceritinib treatment. The strongest dephosphorylation signals were measured at ALK tyrosine sites 1096, 1278, 1282, and 1283 (Fig. 4B). The latter three sites were previously reported as important residues in the ALK activation loop (Donella-Deana et al. 2005; Rush et al. 2005; Rikova et al. 2007; Tartari et al. 2008; Wang et al. 2010; Sattu et al. 2013; Guan et al. 2017). As expected, no phosphorylation signal was observed in the control SK-N-AS cell line. Furthermore, this strong dephosphorylation response was only observed for ALK and not for any other RTK (Supplemental Fig. S4). In CLB-BAR, 182 proteins were dephosphorylated and 52 proteins were phosphorylated upon treatment with ceritinib (Fig. 4C). Similarly, in CLB-GE, 200 proteins were dephosphorylated and 120 proteins were phosphorylated (Fig. 4C). Treatment with ceritinib in SK-N-AS revealed only 35 proteins that were dephosphorylated and 20 proteins that were phosphorylated. Few similarities were found between the RAS-addicted SK-N-AS cell line and the ALK-addicted CLB-BAR/GE cell lines and, although phosphorylation responses were specific between all cell lines with little overlap, 60 proteins were dephosphorylated in both CLB-BAR and CLB-GE but not in SK-N-AS (Fig. 4D; Supplemental Table S1). Assuming that these 60 proteins represent the most specific response to ceritinib, they were used for further analysis. To illustrate downstream phosphorylation targets of ALK signaling activity that are inhibited upon treatment with ceritinib in ALK-addicted neuroblastoma cells, we mapped the dephosphorylated proteins to a protein–protein interaction network (Fig. 4E). This network analysis highlighted FRS2, IRS2, ERK1/2, AKT family members, SHC family members, SOS1, and GAB1/2 as key components of the underlying signaling pathways. Many of these proteins are known to be active in insulin, NGF, and FGFR receptor signaling pathways, as confirmed by a gene set enrichment analysis (Fig. 4F). Similar results were found when independently focusing on the 182 and 200 dephosphorylated proteins of the CLB-BAR and CLB-GE cell lines respectively (Supplemental Figs. S5, S6).
Patient Response to Ceritinib
Based on the experimental data presented here that indicated a potentially poor response to crizotinib, as well as evidence for FA prohibiting use of conventional chemotherapy or irradiation, the patient was started on ceritinib monotherapy as soon as severe acute toxicity had subsided (initiated 9.5 mo after diagnosis, indicated by *; Fig. 5A). Initial dosage was 225 mg once daily (450 mg/m2 body surface area) for a period of 10.5 mo, after which dosage was increased to 540 mg/m2 body surface area. Hematological counts were stabilized at normal levels (Fig. 5A). After the first 4-wk cycle, elevated urinary catecholamines had decreased to levels below the upper reference limit (Fig. 5B). Other than mild gastrointestinal side effects, no toxicity was observed, and the child was able to lead a normal life (Kim et al. 2016). After 6.5 mo of ceritinib treatment, the primary tumor had decreased in size (−43.6% according to MRI with volumetric measurements; Fig. 1) and the remaining CNS metastasis showed response by MRI (Supplemental Fig. S1), although not as sensitive as CT scans performed at diagnosis and after 21 mo of treatment showing complete metastatic remission (Fig. 1). The remaining primary was radically removed surgically together with the left adrenal gland at 7.5 mo from start of ceritinib treatment. The tumor was subjected to immunohistochemical analysis with both proliferation and differentiation markers. The treated tumor displayed a large proportion of calcification and nonmalignant stroma, reduced cell proliferation as measured by Ki-67 positivity (5% vs. 27% before treatment), and hallmarks of differentiation as assessed with NB84 and NFP (Fig. 6). The overall morphology of the primary tumor indicated differentiation in response to treatment, resembling a ganglioneuroblastoma, and was rich in schwannian stroma containing scattered foci of tumor cells of varying degrees of maturation to ganglion cells. Complete clinical evaluation with CT and MRI 1 yr after surgery and 21 mo after initiation of ceritinib treatment showed no residual tumor in the abdomen and complete resolution of metastases at all sites (Fig. 1). MIBG-I123 scintigraphy showed normal findings, indicating completely resolved tumor activity (Fig. 1B). After 34 mo of continuous ceritinib treatment, the child is in continuous complete remission (Park et al. 2017).
Figure 5.

Hematological counts and catecholamine metabolites in urine. (A) After conventional high-risk neuroblastoma chemotherapy was started, the patient suffered severe bone marrow toxicity with protracted anemia and thrombocytopenia requiring frequent transfusions (each peak indicates posttransfusion measurement). When hematological counts stabilized, ceritinib treatment could be started (*). Hemoglobin and platelet counts remained stable during TKI treatment, allowing for surgery (#) with radical removal of the differentiated and calcified decreased primary tumor. (B) Catecholamine metabolites (dopamine in black, homovanillic acid [HVA] in red, and vanillylmandelic acid [VMA] in green) as neuroblastoma markers in the patient's urine (molar concentrations/creatinine concentration). During ceritinib therapy (*), elevated urine catecholamine metabolites returned to normal (below respective dashed line), indicating a biologically inactive residual tumor. At surgery (#) normal levels had been reached.
Figure 6.
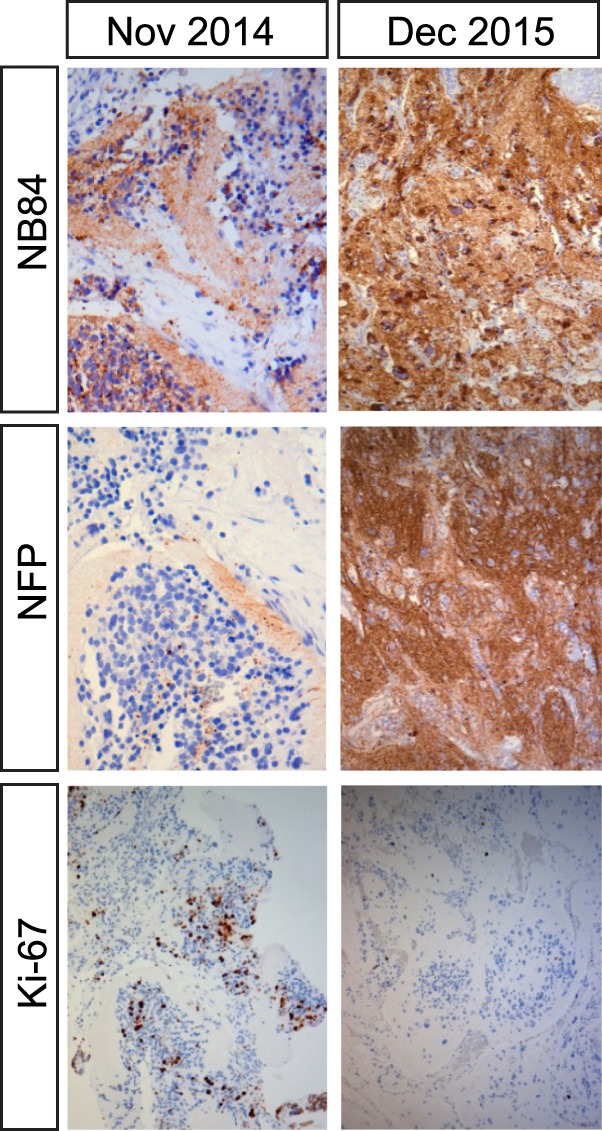
Immunohistochemical analysis of tumor material. Tumor sample taken in November 2014 was stroma-poor, comprising of small, round, primitive-appearing cells positive for the Ki-67 proliferation marker in up to 27%. Postceritinib treatment resected tumor displayed few Ki-67 positive cells and increased expression of NB84 and NFP, which are markers for differentiated neuroblastoma.
DISCUSSION
Here we report the case of a metastatic high-risk neuroblastoma that was treated according to HR_NBL-1 SIOPEN protocol and developed severe toxicity including protracted neutropenia, anemia, and thrombocytopenia that required intensive clinical care for 4 mo. During this time genetic analyses revealed inherited biallelic sequence variants in FANCA. This confirmed the initial suspicion of FA as likely cause of the observed toxicity and a potential contributing factor to tumor development. Genomic analysis also detected a somatic ALK-I1171T mutation that has not previously been described in neuroblastoma. This presented a clinical option to treat the child, whose FA status precluded further chemotherapy or irradiation, with targeted inhibition of ALK. Therefore an extensive preclinical experimental investigation of the ALK-I1171T mutant was performed.
ALK-I1171T exhibits robust gain-of-function activity in several different systems, such as triggering neurite outgrowth in PC12 cells, activation of validated downstream ALK targets, and transformation competence in NIH 3T3 cells. A critical part of this investigation was defining the sensitivity of ALK-I1171T to available ALK TKIs, including crizotinib, ceritinib, brigatinib, and lorlatinib. Of these, the next-generation inhibitors ceritinib, brigatinib, and lorlatinib, which have been explored in preclinical neuroblastoma models (Guan et al. 2016; Wood et al. 2016), were effective in inhibition. Most importantly, crizotinib was a very poor inhibitor of ALK-I1171T activity and does not efficiently cross the blood–brain barrier. This is in agreement with data from NSCLC patients, where resistance mutations in ALK fusions have been reported at I1171 (Katayama et al. 2014; Ou et al. 2014, 2015; Toyokawa et al. 2014). We also performed a preclinical LC-MS/MS analysis of ceritinib activity in neuroblastoma confirming a strong dephosphorylation response at all tyrosine residues that are known to be involved in the ALK activation loop, but not in any other RTK, indicating the specificity of the response. The downstream signaling response included FRS2, ERK1/2, AKT family members, SHC family members, and GAB1/2 among others and was similar to insulin, NGF, and FGFR signaling pathways. Based on the collective preclinical information, the child was included in the compassionate use individual patient program for ceritinib.
The patient responded strongly to ceritinib with minimal side effects allowing the child to lead a normal life. To date, the patient is in complete clinical remission after continuous ceritinib monotherapy treatment for 34 mo. It is unclear to what extent the efficacy of ceritinib treatment may have been influenced by the initial chemotherapy treatment, which induced partial remission. One possibility that should be considered is that the initial chemotherapy treatment sensitized the tumor to the subsequent ALK TKI treatment. A preclinical report investigating combination of crizotinib with commonly used chemotherapy showed synergistic cytotoxicity and increased apoptosis in neuroblastoma cell lines with ALK aberrations, supporting this scenario (Krytska et al. 2016). However, it is important to note that at initiation of ceritinib treatment the patient displayed significant active disease, as shown by persistent elevated urine catecholamine markers, metastatic disease, and remaining primary tumor tissue, which all responded strongly and rapidly to ALK TKI therapy with finally complete clinical remission at all sites.
In conclusion, we have shown the importance of comprehensive genetic profiling combined with preclinical investigation, which was conducted during a forced drug holiday of a neuroblastoma patient later discovered to present with FA caused by FANCA mutations and an ALK mutation. A compassionate use protocol with ceritinib has shown a clear benefit for the patient who is in complete clinical remission with clearance of all metastatic sites 34 mo from start of targeted ceritinib treatment. After the initial 7.5 mo of treatment with ceritinib, the primary tumor had decreased in size and was removed surgically. The removed tumor displayed a large amount of calcification and nonmalignant stroma, reduced proliferation index, and hallmarks of differentiation. Here we show that monotherapy with next-generation ALK TKIs can be effective against metastatic high-risk neuroblastoma carrying an activating ALK mutation, confirming ceritinib as a viable therapeutic option for ALK-positive neuroblastoma patients.
METHODS
Patient
A 16-mo-old boy presented at the pediatric emergency unit with neurologic symptoms including facial palsy, impaired balance, and strabismus. There was no past medical history of note except for examination by a hand surgeon and a dysmelia team because of slightly deviant thumbs, which had been present since birth. At the emergency visit, exophthalmus and an abdominal mass were also noted, and imaging using MIBG scintigraphy, CT scans, and MRI revealed a metastatic neuroblastoma with metastases in bone, lungs, and intracranially (Fig. 1; Supplemental Fig. S1) but with no neuroblastoma detected in the bone marrow. Biological workup of the diagnostic biopsy showed a number of unfavorable chromosomal aberrations, including 11q loss, 17q gain, 2q loss, 4p loss,7q, and 9q gain (Fig. 2). Treatment was initiated according to the high-risk neuroblastoma protocol HR-NBL-1 SIOPEN (Ladenstein et al. 2017) (ethical permit 02-294). In response to this treatment, the patient developed protracted neutropenia, anemia, and thrombocytopenia, necessitating repeated transfusions of erythrocytes and platelets for almost 6 mo (Fig. 5). Furthermore the patient experienced repeated episodes of severe septicemia and typhlitis requiring intensive care. Based on the suspicion of underlying FA (Walsh et al. 2017), chromosomal breakage assessment was perfomed and found positive. Further genomic analysis revealed inherited germline mutations in the FANCA gene, confirming FA diagnosis (Table 1; Fig. 2B). Detailed genomic analysis further identified a somatic ALK-I1171T mutation in the patient, raising the possibility of ALK TKI treatment (Fig. 2C; Table 1). Following preclinical investigation of the ALK-I1171T mutation, a clinical multidisciplinary conference, an Institutional Ethical Board meeting, and a Clinical Ethical Board meeting endorsed application for a Medical Products Agency (MPA) license (according to LVFS 2008:1). In addition, written informed consent from the patient's guardians was obtained and the patient was included in the Novartis compassionate use Individual Patient Program for ceritinib (CLDK378A2003M). Sampling and analyses of tumor tissues and linkage to clinical information were performed according to the ethical permit 2009/1369-31/1. Written informed consent was obtained from the patient's guardians allowing scientific research publication.
Immunohistochemistry
Staining for NB84 (Novocastra, NCL-NB84), NF (DAKO, 2F11), and Ki-67 (DAKO, MIB-1) was performed using a DAKO Autostainer. Slides were rehydrated with Xylene followed by a series of alcohol dilutions and a rinse in buffer (DAKO 8007). PTLINK (DAKO) was used for enzyme antigen retrieval. Endogenous enzyme block was performed for 5 min with EnVision FLEX Peroxidase-Blocking Reagent (DAKO). The antibodies (diluted at DAKO Company) were incubated for 20 min at room temperature. After a 5-min rinse in buffer, the labeled polymer, EnVision FLEX/HRP (DAKO), was applied and incubated for 20 min with two additional rinses before the slides were incubated with substrate-chromogen, Substrate Working Solution (DAKO), for 10 min. Slides were ultimately rinsed and counterstained in EnVision FLEX Hematoxylin prior to mounting.
Genomics Profile with SNP Array
Microarray analyses of DNA from the tumor samples were performed using Affymetrix Human Cytoscan High Density arrays essentially as described earlier (Fransson et al. 2016). For primary data analysis, the GDAS software (Affymetrix) was used, whereas genomic profiles were generated using CNAG (Copy Number Analyzer for AffymetrixGeneChip Mapping arrays) version 3.3 (Genome Laboratory, Tokyo University; http://www.genome.umin.jp; 11).
Detection of ALK Mutation with Massive Parallel DNA Sequencing
DNA was extracted from tumor sample using standard procedures and evaluated through fluorometric quantitation and DNA integrity assessment on Agilent Tapestation (Agilent) prior to exome sequencing. Exome sequencing was performed at GATC (GATC) through paired-end sequencing (101 bp read length) on Illumina platforms after enrichment with Agilent SureSelect Human All Exon v5 enrichment kit giving an average coverage of 85× (sequencing details are supplied in Supplemental Information file “Exome sequencing”). Alignment against hg19 was performed using BWA with GATK local realignment followed by SNV calling using SNPeff and Annovar annotation. Visualization of mapped reads and manual QC of called variants was done through IGV (Robinson et al. 2011). Only variants located at regions covered by at least 10 unique reads were kept. A systematic filtering approach was used to identify critical variants by removal of common variants (e.g., showing an allele frequency of >0.005 in the 1000 Genomes or exome variant server) as well as excluding all synonymous variants or variants in noncoding regions except those affecting canonical splice sites. PolyPhen-2 (PP2) and SIFT were used for prediction of functional relevance of called SNVs. Sanger sequencing of the patient's constitutional and tumor DNA verified presence of a somatic ALK mutation in the tumor. Sanger sequencing of ALK exons was performed as described previously (Carén et al. 2008). All genomic positions are specified according hg19/GRCh37.
Generation of Human ALK Mutant Constructs in PC12 Cells
The ALK-I1171T mutation was generated based on pcDNA3-ALK-WT (NM_004304.3) by Eurofins Genomics. ALK-F1174L has been described previously (Martinsson et al. 2011). All mutations generated in the kinase domain were confirmed by sequencing from both directions.
Neurite Outgrowth Assay
PC12 (2 × 106) cells were cotransfected with 0.5 µg of empty pcDNA3 vector, pcDNA3-ALK-WT, pcDNA3-ALK-I1171T, or pcDNA3-ALK-F1174L, respectively, together with 0.5 µg of pEGFP-N1 (Clonetech) as indicated by electroporation using Amaxa electroporator (Amaxa Biosystems) and Ingenio electroporation solution (Mirus Bio LCC). After electroporation, cells were kept in minimum essential medium (MEM) supplemented with 7% horse serum and 3% fetal bovine serum and seeded into 24-well plates. For samples stimulated with ALK ligand, purified ALKAL1 (FAM150A) protein was added to the well with a final concentration of 1 µg/ml (Guan et al. 2015). After 48 h of incubation, the fraction of green fluorescent protein (GFP)-positive and neurite-carrying cells versus GFP-positive cells was observed under a Zeiss Axiovert 40 CFL microscope. To be judged as a neurite-carrying cell, the neurite of the cell should be at least twice the diameter of a normal cell body. Experiments were performed in triplicate, and each sample within an experiment was performed in triplicate.
Transformation Assay
NIH 3T3 cells (5 × 104) were seeded into collagen-coated 12-well plates and transfected with 0.55 µg of pcDNA3 vector, pcDNA3-ALK-WT, pcDNA3-ALK-I1171T, or pcDNA3-ALK-F1174L, respectively, using Lipofectamine 2000 according to the manufacturer's protocol (Invitrogen). Twenty-four hours later, three-fifths of the cells from each well were transferred to wells in six-well plates and kept in DMEM supplemented with 10% FBS and 0.5 mg/ml G418 until the cells reached confluence. Thereafter, cells were kept in DMEM supplemented with 5% FBS and 0.25 mg/ml G418 for another 10 d, with replacement of medium every second day. Plates were washed with PBS and air-dried, and fixed with methanol for 20 min, and cells were stained with 0.2% crystal violet in 20% ethanol for 20 min, followed by three short rinses in water.
Cell Culture, Viability, Lysis, and Immunoblotting
Cell viability was assessed as relative redox metabolic activity using aresazurin-based assay. Cell lines used were CLB-BAR, CLB-GE, CLB-GA, Kelly, SK-N-AS, SK-N-BE, SK-N-DZ, IMR32, and CLB-PE neuroblastoma cells (Umapathy et al. 2017). Cells (4 × 104) were plated on collagen-coated 48-well plates and treated with either ceritinib or crizotinib with indicated concentrations. Cell viability was determined after 72 h with 55 µM resazurin (Sigma-Aldrich). After 3 h at 37°C, the amount of metabolized resazurin was analyzed as relative fluorescence with an Infinit200 plate reader (TEKAN). Results were from one of three representative experiments, with each experiment being performed in triplicate. GraphPad Prism 6.0 was used to calculate IC50 values by fitting data to a log (inhibitor concentration) versus normalized response (variable slope) equation.
PC12 cells were transfected with 0.5 µg of empty pcDNA3 vector, pcDNA3-ALK-WT, or pcDNA3-ALK variants as specified, respectively by electroporation as described above. Cells were serum-starved for 36 h before lysis. For samples stimulated with ALK ligand, they were treated with 1 µg/ml of purified ALKAL1 protein for 30 min prior to lysis. Cells were washed twice with ice-cold PBS prior to harvest in lysis buffer (25 mmol/L Tris-Cl, pH7.5, 150 mmol/L NaCl, 1% (v/v) Triton X-100, 1 mmol/L DTT, protease inhibitor cocktail tablet [Roche]). Cell lysates were cleared by centrifugation at 14,000 rpm for 15 min at 4°C. Samples were boiled in 1× SDS sample buffer and analyzed by immunoblotting. Primary antibodies used for immunoblotting were: anti-pan-ERK (1:10,000), purchased from BD Transduction Laboratories, anti-pALK (Y1604) and anti-pERK1/2 (T202/Y204) from Cell Signaling Technology. Monoclonal antibody 135 (anti-ALK) was produced in the Hallberg laboratory against the extracellular domain of ALK as described (Moog-Lutz et al. 2005; Schonherr et al. 2012). Horseradish peroxidase–conjugated secondary antibodies goat anti-rabbit IgG and goat anti-mouse IgG (1:5,000) were from Thermo Scientific.
ALK Inhibitor Profiles on ALK Variants
PC12 cells (2 × 106) were transfected with 1.0 µg of pcDNA3-ALK-WT, or pcDNA3-ALK variants as described (Guan et al. 2016; Siaw et al. 2016). Cells from three electroporations were pooled together, mixed, and equally seeded into nine wells of one 24-well plate. After 24–36 h culture, cells were treated with serial dilutions of the indicated inhibitors for 1 h before lysis. Cells were washed with cold 1× PBS and lysed with 1× SDS sample buffer and samples were boiled for 5 min at 95°C. Phospho-ALK (Y1604) antibody was used to detect ALK phosphorylation, and ALK mAb135 was used to detect total ALK. The intensity of pALK (Y1604) and total ALK bands was quantified with Image Studio Lite 3.1 software. Data were normalized to the 0 nM inhibitor samples. GraphPad Prism 6.0 was used to calculate IC50 values by fitting data to a log (inhibitor concentration) versus normalized response (variable slope) equation.
Phosphoproteomics
Tyrosine and serine/threonine phosphorylation profiling of neuroblastoma cells (CLB-BAR, CLB-GE, and SK-N-AS) in the presence or absence of ceritinib (200 nM for 60 min) was undertaken by immunoaffinity purification with P-Tyr-100 or P-Ser/Thr-antibodies accordingly (Cell Signaling Technology). Cells were lysed in lysis buffer (20 mM Hepes pH 8.0, 9 M UREA, 1 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate and 1 mM β-glycerol-phosphate) and ∼20 mg protein per sample subjected to LC-MS/MS. This was performed as previously described (Sattu et al. 2013). Phosphorylation responses were quantified for each site (tyrosine, serine, or threonine) under analysis as log2 fold change (FC) values of ceritinib treated/untreated control counts. If a specific site was measured in multiple peptides, its median log2 FC value was used for analysis. Protein sites were considered differentially phosphorylated when their absolute log2 FC values were >1.5. A gene set enrichment analysis (GSEA) was performed using Fisher's exact test and false discovery rate correction using the Benjamini–Hochberg method (Benjamini and Hochberg 1995). Reactome pathway information was downloaded from the Molecular Signatures Database v5.2 (Subramanian et al. 2005). The differentially phosphorylated proteins were mapped to the InWeb_InBioMap protein–protein interaction (PPI) network (Li et al. 2017). For the analysis of related TRKs, a list of 40 different TRKs was downloaded from HGNC (Gray et al. 2015).
ADDITIONAL INFORMATION
Data Deposition and Access
The detected DNA variants in FANCA and ALK have been submitted to ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession numbers SCV000777897–SCV000777901. Raw sequencing data could not be deposited because of lack of patient consent.
Ethics Statement
Treatment was initiated according HR-NBL-1 SIOPEN protocol (permit 02-294). A Clinical Ethical Board meeting and an endorsed license application from the MPA license (according to LVFS 2008:1) supported LDK378 compassionate use. Written informed consent from the patient's guardians was obtained and the patient was included in the Novartis compassionate use program for ceritinib (CLDK378A2003M). Tumor analyses and linkage to clinical information were performed according to the ethical permit 2009/1369-31/1. Written informed consent was obtained from the patient's guardians allowing the scientific research publication and are filed with the case notes.
Acknowledgments
This work has been supported by grants from the Swedish Cancer Society (TMCAN2015/794; BHCAN15/775; RHP CAN15/391; PK CAN16/3625), the Swedish Childhood Cancer Foundation (TM PR2016-0147; BH 2015-80, and 2014-150; RHP 2015-96; PK PR2014-0084; JG 2016-0011; SF 15-0061; DT 12-009 and 12-002), the Swedish Research Council (RHP 2015-04466; BH 521-2012-2831; TM 521-2014-3031; PK 2014-3036); the Swedish Foundation for Strategic Research (RB13-0204, www.nnbcr.se), and the Göran Gustafsson Foundation (RHP2016). D.T. was supported by a PhD-Student program (NC2012-0026), J.G. was supported by a Post-doctoral Research Fellow Position (TJ2016-0088), and S.F. was supported by a Research Assistant Fellowship (14-0064), by the Swedish Childhood Cancer Foundation. The CLB-BAR, CLB-GE, CLB-GA, and CLB-PE cell lines used in this study were kindly provided by Valérie Combaret under an MTA agreement with the Center Léon Bérard laboratory (Lyon, France). Novartis supplied ceritinib free of charge within the compassionate use program “Ceritinib IPP for ALK-Positive IMT, ALCL, and Neuroblastoma Pediatric Patients.”
Competing Interest Statement
The authors have declared no competing interest.
Referees
Rogier Versteeg
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Abbondanzo SL, Manz HJ, Klappenbach RS, Gootenberg JE. 1986. Hepatocellular carcinoma in an 11-year-old girl with Fanconi's anemia. Report of a case and review of the literature. Am J Pediatr Hematol Oncol 8: 334–337. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Methodol 57: 289–300. [Google Scholar]
- Berrebi D, Lebras MN, Belarbi N, Couturier J, Fattet S, Faye A, Peuchmaur M, de Lagausie P. 2006. Bilateral adrenal neuroblastoma and nephroblastoma occurring synchronously in a child with Fanconi's anemia and VACTERL syndrome. J Pediatr Surg 41: e11–e14. [DOI] [PubMed] [Google Scholar]
- Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, Pei D, Sharma B, Vetharoy WR, Hallsworth A, et al. 2012. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell 22: 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissig H, Staehelin F, Tolnay M, Avoledo P, Richter J, Betts D, Bruder E, Kühne T. 2002. Co-occurrence of neuroblastoma and nephroblastoma in an infant with Fanconi's anemia. Hum Pathol 33: 1047–1051. [DOI] [PubMed] [Google Scholar]
- Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, Laudenslager M, Rappaport EF, Wood AC, McGrady PW, et al. 2014. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 26: 682–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carén H, Abel F, Kogner P, Martinsson T. 2008. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem J 416: 153–159. [DOI] [PubMed] [Google Scholar]
- Cazes A, Louis-Brennetot C, Mazot P, Dingli F, Lombard B, Boeva V, Daveau R, Cappo J, Combaret V, Schleiermacher G, et al. 2013. Characterization of rearrangements involving the ALK gene reveals a novel truncated form associated with tumor aggressiveness in neuroblastoma. Cancer Res 73: 195–204. [DOI] [PubMed] [Google Scholar]
- Cazes A, Lopez-Delisle L, Tsarovina K, Pierre-Eugène C, De Preter K, Peuchmaur M, Nicolas A, Provost C, Louis-Brennetot C, Daveau R, et al. 2014. Activated Alk triggers prolonged neurogenesis and Ret upregulation providing a therapeutic target in ALK-mutated neuroblastoma. Oncotarget 5: 2688–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chand D, Yamazaki Y, Ruuth K, Schönherr C, Martinsson T, Kogner P, Attiyeh EF, Maris J, Morozova O, Marra MA, et al. 2013. Cell culture and Drosophila model systems define three classes of anaplastic lymphoma kinase mutations in neuroblastoma. Dis Model Mech 6: 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, et al. 2008. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 455: 971–974. [DOI] [PubMed] [Google Scholar]
- Chia PL, Mitchell P, Dobrovic A, John T. 2014. Prevalence and natural history of ALK positive non-small-cell lung cancer and the clinical impact of targeted therapy with ALK inhibitors. Clin Epidemiol 6: 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, Yamazaki S, Alton GR, Mroczkowski B, Los G. 2007. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther 6: 3314–3322. [DOI] [PubMed] [Google Scholar]
- Combaret V, Turc-Carel C, Thiesse P, Rebillard AC, Frappaz D, Haus O, Philip T, Favrot MC. 1995. Sensitive detection of numerical and structural aberrations of Chromosome 1 in neuroblastoma by interphase fluorescence in situ hybridization. Comparison with restriction fragment length polymorphism and conventional cytogenetic analyses. Int J Cancer 61: 185–191. [DOI] [PubMed] [Google Scholar]
- Cowley S, Paterson H, Kemp P, Marshall CJ. 1994. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell 77: 841–852. [DOI] [PubMed] [Google Scholar]
- De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, Lakeman A, Vandesompele J, Hoebeeck J, Van Maerken T, et al. 2010. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res 16: 4353–4362. [DOI] [PubMed] [Google Scholar]
- Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WA, et al. 2012. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non–small cell lung cancer. Clin Cancer Res 18: 1472–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donella-Deana A, Marin O, Cesaro L, Gunby RH, Ferrarese A, Coluccia AM, Tartari CJ, Mologni L, Scapozza L, Gambacorti-Passerini C, et al. 2005. Unique substrate specificity of anaplastic lymphoma kinase (ALK): development of phosphoacceptor peptides for the assay of ALK activity. Biochemistry 44: 8533–8542. [DOI] [PubMed] [Google Scholar]
- Eleveld TF, Oldridge DA, Bernard V, Koster J, Daage LC, Diskin SJ, Schild L, Bentahar NB, Bellini A, Chicard M, et al. 2015. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 47: 864–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadeev A, Mendoza-Garcia P, Irion U, Guan J, Pfeifer K, Wiessner S, Serluca F, Singh AP, Nüsslein-Volhard C, Palmer RH. 2018. ALKALs are in vivo ligands for ALK family receptor tyrosine kinases in the neural crest and derived cells. Proc Natl Acad Sci 115: E630–E638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA. 26th of August 2011. U.F.a.D.A. FDA approves Xalkori with companion diagnostic for a type of late-stage lung cancer. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm269856.htm.
- FDA. 29th of April 2014. FDA approves Zykadia for late-stage lung cancer. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&applno=205755.
- FDA. 26th of May 2017. FDA broadens ceritinib indication to previously untreated ALK-positive metastatic NSCLC. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm560873.htm.
- Fransson S, Hansson M, Ruuth K, Djos A, Berbegall A, Javanmardi N, Abrahamsson J, Palmer RH, Noguera R, Hallberg B, et al. 2015. Intragenic anaplastic lymphoma kinase (ALK) rearrangements: translocations as a novel mechanism of ALK activation in neuroblastoma tumors. Genes Chromosomes Cancer 54: 99–109. [DOI] [PubMed] [Google Scholar]
- Fransson S, Östensson M, Djos A, Javanmardi N, Kogner P, Martinsson T. 2016. Estimation of copy number aberrations: comparison of exome sequencing data with SNP microarrays identifies homozygous deletions of 19q13.2 and CIC in neuroblastoma. Int J Oncol 48: 1103–1116. [DOI] [PubMed] [Google Scholar]
- George RE, Sanda T, Hanna M, Frohling S, Luther W II, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, et al. 2008. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 455: 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray KA, Yates B, Seal RL, Wright MW, Bruford EA. 2015. Genenames.org: the HGNC resources in 2015. Nucleic Acids Res 43: D1079–D1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J, Umapathy G, Yamazaki Y, Wolfstetter G, Mendoza P, Pfeifer K, Mohammed A, Hugosson F, Zhang H, Hsu AW, et al. 2015. FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. Elife 4: e09811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J, Tucker ER, Wan H, Chand D, Danielson LS, Ruuth K, El Wakil A, Witek B, Jamin Y, Umapathy G, et al. 2016. The ALK inhibitor PF-06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and MYCN. Dis Model Mech 9: 941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J, Yamazaki Y, Chand D, van Dijk JR, Ruuth K, Palmer RH, Hallberg B. 2017. Novel mechanisms of ALK activation revealed by analysis of the Y1278S neuroblastoma mutation. Cancers (Basel) 9: E149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallberg B, Palmer RH. 2013. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer 13: 685–700. [DOI] [PubMed] [Google Scholar]
- Hallberg B, Palmer RH. 2016. The role of the ALK receptor in cancer biology. Ann Oncol 27(Suppl 3): iii4–iii15. [DOI] [PubMed] [Google Scholar]
- Heukamp LC, Thor T, Schramm A, De Preter K, Kumps C, De Wilde B, Odersky A, Peifer M, Lindner S, Spruessel A, et al. 2012. Targeted expression of mutated ALK induces neuroblastoma in transgenic mice. Sci Transl Med 4: 141ra191. [DOI] [PubMed] [Google Scholar]
- Infarinato NR, Park JH, Krytska K, Ryles HT, Sano R, Szigety KM, Li Y, Zou HY, Lee NV, Smeal T, et al. 2016. The ALK/ROS1 inhibitor PF-06463922 overcomes primary resistance to crizotinib in ALK-driven neuroblastoma. Cancer Discov 6: 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T. 1997. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 14: 439–449. [DOI] [PubMed] [Google Scholar]
- Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, et al. 2008. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455: 967–970. [DOI] [PubMed] [Google Scholar]
- Johnson TW, Richardson PF, Bailey S, Brooun A, Burke BJ, Collins MR, Cui JJ, Deal JG, Deng YL, Dinh D, et al. 2014. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J Med Chem 57: 4720–4744. [DOI] [PubMed] [Google Scholar]
- Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, Shakespeare WC, Iafrate AJ, Engelman JA, Shaw AT. 2011. Therapeutic strategies to overcome crizotinib resistance in non–small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci 108: 7535–7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama R, Friboulet L, Koike S, Lockerman EL, Khan TM, Gainor JF, Iafrate AJ, Takeuchi K, Taiji M, Okuno Y, et al. 2014. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin Cancer Res 20: 5686–5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama R, Lovly CM, Shaw AT. 2015. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: a paradigm for precision cancer medicine. Clin Cancer Res 21: 2227–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, Camidge DR, Vansteenkiste J, Sharma S, De Pas T, Riely GJ, et al. 2016. Activity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ASCEND-1): updated results from the multicentre, open-label, phase 1 trial. Lancet Oncol 17: 452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimble DC, Lach FP, Gregg SQ, Donovan FX, Flynn EK, Kamat A, Young A, Vemulapalli M, Thomas JW, Mullikin JC, et al. 2017. A comprehensive approach to identification of pathogenic FANCA variants in Fanconi anemia patients and their families. Hum Mutat 39: 237–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl NE, Gee CE, Alt FW. 1984. Activated expression of the N-myc gene in human neuroblastomas and related tumors. Science 226: 1335–1337. [DOI] [PubMed] [Google Scholar]
- Kopic S, Eirich K, Schuster B, Hanenberg H, Varon-Mateeva R, Rittinger O, Schimpl G, Schindler D, Jones N. 2011. Hepatoblastoma in a 4-year-old girl with Fanconi anaemia. Acta Paediatr 100: 780–783. [DOI] [PubMed] [Google Scholar]
- Kornev AP, Taylor SS. 2015. Dynamics-driven allostery in protein kinases. Trends Biochem Sci 40: 628–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krytska K, Ryles HT, Sano R, Raman P, Infarinato NR, Hansel TD, Makena MR, Song MM, Reynolds CP, Mosse YP. 2016. Crizotinib synergizes with chemotherapy in preclinical models of neuroblastoma. Clin Cancer Res 22: 948–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutler DI, Auerbach AD, Satagopan J, Giampietro PF, Batish SD, Huvos AG, Goberdhan A, Shah JP, Singh B. 2003a. High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Arch Otolaryngol Head Neck Surg 129: 106–112. [DOI] [PubMed] [Google Scholar]
- Kutler DI, Singh B, Satagopan J, Batish SD, Berwick M, Giampietro PF, Hanenberg H, Auerbach AD. 2003b. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood 101: 1249–1256. [DOI] [PubMed] [Google Scholar]
- Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB, et al. 2010. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363: 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladenstein R, Potschger U, Pearson ADJ, Brock P, Luksch R, Castel V, Yaniv I, Papadakis V, Laureys G, Malis J, et al. 2017. Busulfan and melphalan versus carboplatin, etoposide, and melphalan as high-dose chemotherapy for high-risk neuroblastoma (HR-NBL1/SIOPEN): an international, randomised, multi-arm, open-label, phase 3 trial. Lancet Oncol 18: 500–514. [DOI] [PubMed] [Google Scholar]
- Li T, Wernersson R, Hansen RB, Horn H, Mercer J, Slodkowicz G, Workman CT, Rigina O, Rapacki K, Staerfeldt HH, et al. 2017. A scored human protein–protein interaction network to catalyze genomic interpretation. Nat Methods 14: 61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malric A, Defachelles AS, Leblanc T, Lescoeur B, Lacour B, Peuchmaur M, Maurage CA, Pierron G, Guillemot D, d'Enghien CD, et al. 2015. Fanconi anemia and solid malignancies in childhood: a national retrospective study. Pediatr Blood Cancer 62: 463–470. [DOI] [PubMed] [Google Scholar]
- Mamrak NE, Shimamura A, Howlett NG. 2017. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev 31: 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsilje TH, Pei W, Chen B, Lu W, Uno T, Jin Y, Jiang T, Kim S, Li N, Warmuth M, et al. 2013. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 56: 5675–5690. [DOI] [PubMed] [Google Scholar]
- Martinsson T, Eriksson T, Abrahamsson J, Caren H, Hansson M, Kogner P, Kamaraj S, Schonherr C, Weinmar J, Ruuth K, et al. 2011. Appearance of the novel activating F1174S ALK mutation in neuroblastoma correlates with aggressive tumor progression and unresponsiveness to therapy. Cancer Res 71: 98–105. [DOI] [PubMed] [Google Scholar]
- Mo ES, Cheng QN, Reshetnyak AV, Schlessinger J, Nicoli S. 2017. Alk and Ltk ligands are essential for iridophore development in zebrafish mediated by the receptor tyrosine kinase Ltk. Proc Natl Acad Sci 114: 12027–12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moog-Lutz C, Degoutin J, Gouzi JY, Frobert Y, Brunet-de Carvalho N, Bureau J, Creminon C, Vigny M. 2005. Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. J Biol Chem 280: 26039–26048. [DOI] [PubMed] [Google Scholar]
- Moore NF, Azarova AM, Bhatnagar N, Ross KN, Drake LE, Frumm S, Liu QS, Christie AL, Sanda T, Chesler L, et al. 2014. Molecular rationale for the use of PI3K/AKT/mTOR pathway inhibitors in combination with crizotinib in ALK-mutated neuroblastoma. Oncotarget 5: 8737–8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. 1994. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science 263: 1281–1284. [DOI] [PubMed] [Google Scholar]
- Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, Witte DP. 1997. ALK, the Chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin's lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 14: 2175–2188. [DOI] [PubMed] [Google Scholar]
- Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, et al. 2008. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455: 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, Rolland D, Balis FM, Maris JM, Weigel BJ, et al. 2013. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children's Oncology Group phase 1 consortium study. Lancet Oncol 14: 472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosse YP, Voss SD, Lim MS, Rolland D, Minard CG, Fox E, Adamson P, Wilner K, Blaney SM, Weigel BJ. 2017. Targeting ALK With crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a Children's Oncology Group Study. J Clin Oncol 35: 3215–3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motegi A, Fujimoto J, Kotani M, Sakuraba H, Yamamoto T. 2004. ALK receptor tyrosine kinase promotes cell growth and neurite outgrowth. J Cell Sci 117: 3319–3329. [DOI] [PubMed] [Google Scholar]
- Neveling K, Endt D, Hoehn H, Schindler D. 2009. Genotype–phenotype correlations in Fanconi anemia. Mutat Res 668: 73–91. [DOI] [PubMed] [Google Scholar]
- Okubo J, Takita J, Chen Y, Oki K, Nishimura R, Kato M, Sanada M, Hiwatari M, Hayashi Y, Igarashi T, et al. 2012. Aberrant activation of ALK kinase by a novel truncated form ALK protein in neuroblastoma. Oncogene 31: 4667–4676. [DOI] [PubMed] [Google Scholar]
- Ou SH, Klempner SJ, Greenbowe JR, Azada M, Schrock AB, Ali SM, Ross JS, Stephens PJ, Miller VA. 2014. Identification of a novel HIP1-ALK fusion variant in non-small-cell lung cancer (NSCLC) and discovery of ALK I1171 (I1171N/S) mutations in two ALK-rearranged NSCLC patients with resistance to Alectinib. J Thorac Oncol 9: 1821–1825. [DOI] [PubMed] [Google Scholar]
- Ou SH, Greenbowe J, Khan ZU, Azada MC, Ross JS, Stevens PJ, Ali SM, Miller VA, Gitlitz B. 2015. I1171 missense mutation (particularly I1171N) is a common resistance mutation in ALK-positive NSCLC patients who have progressive disease while on alectinib and is sensitive to ceritinib. Lung Cancer 88: 231–234. [DOI] [PubMed] [Google Scholar]
- Park JR, Bagatell R, Cohn SL, Pearson AD, Villablanca JG, Berthold F, Burchill S, Boubaker A, McHugh K, Nuchtern JG, et al. 2017. Revisions to the International neuroblastoma response criteria: a consensus statement from the National Cancer Institute clinical trials planning meeting. J Clin Oncol 35: 2580–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reshetnyak AV, Murray PB, Shi X, Mo ES, Mohanty J, Tome F, Bai H, Gunel M, Lax I, Schlessinger J. 2015. Augmentor α and β (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: hierarchy and specificity of ligand-receptor interactions. Proc Natl Acad Sci 112: 15862–15867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al. 2007. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131: 1190–1203. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, Zhang H, Zha XM, Polakiewicz RD, Comb MJ. 2005. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol 23: 94–101. [DOI] [PubMed] [Google Scholar]
- Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, Oikawa N, Tsukuda T, Ishii N, Aoki Y. 2011. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 19: 679–690. [DOI] [PubMed] [Google Scholar]
- Sattu K, Hochgräfe F, Wu J, Umapathy G, Schönherr C, Ruuth K, Chand D, Witek B, Fuchs J, Li PK, et al. 2013. Phosphoproteomic analysis of ALK downstream signaling pathways identifies STAT3 as a functional target of activated ALK in neuroblastoma cells. FEBS J 280: 5269–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleiermacher G, Janoueix-Lerosey I, Combaret V, Derre J, Couturier J, Aurias A, Delattre O. 2003. Combined 24-color karyotyping and comparative genomic hybridization analysis indicates predominant rearrangements of early replicating chromosome regions in neuroblastoma. Cancer Genet Cytogenet 141: 32–42. [DOI] [PubMed] [Google Scholar]
- Schleiermacher G, Javanmardi N, Bernard V, Leroy Q, Cappo J, Rio Frio T, Pierron G, Lapouble E, Combaret V, Speleman F, et al. 2014. Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol 32: 2727–2734. [DOI] [PubMed] [Google Scholar]
- Schonherr C, Ruuth K, Yamazaki Y, Eriksson T, Christensen J, Palmer RH, Hallberg B. 2011. Activating ALK mutations found in neuroblastoma are inhibited by Crizotinib and NVP-TAE684. Biochem J 440: 405–413. [DOI] [PubMed] [Google Scholar]
- Schonherr C, Ruuth K, Kamaraj S, Wang CL, Yang HL, Combaret V, Djos A, Martinsson T, Christensen JG, Palmer RH, et al. 2012. Anaplastic lymphoma kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene 31: 5193–5200. [DOI] [PubMed] [Google Scholar]
- Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, et al. 2013. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 368: 2385–2394. [DOI] [PubMed] [Google Scholar]
- Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, Camidge DR, Vansteenkiste J, Sharma S, De Pas T, et al. 2014. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med 370: 1189–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siaw JT, Wan H, Pfeifer K, Rivera VM, Guan J, Palmer RH, Hallberg B. 2016. Brigatinib, an anaplastic lymphoma kinase inhibitor, abrogates activity and growth in ALK-positive neuroblastoma cells, Drosophila and mice. Oncotarget 7: 29011–29022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartari CJ, Gunby RH, Coluccia AM, Sottocornola R, Cimbro B, Scapozza L, Donella-Deana A, Pinna LA, Gambacorti-Passerini C. 2008. Characterization of some molecular mechanisms governing autoactivation of the catalytic domain of the anaplastic lymphoma kinase. J Biol Chem 283: 3743–3750. [DOI] [PubMed] [Google Scholar]
- Toyokawa G, Hirai F, Inamasu E, Yoshida T, Nosaki K, Takenaka T, Yamaguchi M, Seto T, Takenoyama M, Ichinose Y. 2014. Secondary mutations at I1171 in the ALK gene confer resistance to both crizotinib and alectinib. J Thorac Oncol 9: e86–e87. [DOI] [PubMed] [Google Scholar]
- Ueda T, Nakata Y, Yamasaki N, Oda H, Sentani K, Kanai A, Onishi N, Ikeda K, Sera Y, Honda ZI, et al. 2016. ALK(R1275Q) perturbs extracellular matrix, enhances cell invasion and leads to the development of neuroblastoma in cooperation with MYCN. Oncogene 35: 4447–4458. [DOI] [PubMed] [Google Scholar]
- Umapathy G, El Wakil A, Witek B, Chesler L, Danielson L, Deng X, Gray NS, Johansson M, Kvarnbrink S, Ruuth K, et al. 2014. The kinase ALK stimulates the kinase ERK5 to promote the expression of the oncogene MYCN in neuroblastoma. Sci Signal 7: ra102. [DOI] [PubMed] [Google Scholar]
- Umapathy G, Guan J, Gustafsson DE, Javanmardi N, Cervantes-Madrid D, Djos A, Martinsson T, Palmer RH, Hallberg B. 2017. MEK inhibitor trametinib does not prevent the growth of anaplastic lymphoma kinase (ALK)-addicted neuroblastomas. Sci Signal 10: eaam7550. [DOI] [PubMed] [Google Scholar]
- Walsh MF, Chang VY, Kohlmann WK, Scott HS, Cunniff C, Bourdeaut F, Molenaar JJ, Porter CC, Sandlund JT, Plon SE, et al. 2017. Recommendations for childhood cancer screening and surveillance in DNA repair disorders. Clin Cancer Res 23: e23–e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Wu F, Zhang J, McMullen T, Young LC, Ingham RJ, Li L, Lai R. 2010. Serine phosphorylation of NPM-ALK, which is dependent on the auto-activation of the kinase activation loop, contributes to its oncogenic potential. Carcinogenesis 32: 146–153. [DOI] [PubMed] [Google Scholar]
- Wood AC, Krytska K, Ryles HT, Infarinato NR, Sano R, Hansel TD, Hart LS, King FJ, Smith TR, Ainscow E, et al. 2016. Dual ALK and CDK4/6 inhibition demonstrates synergy against neuroblastoma. Clin Cancer Res 23: 2856–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Pao LI, Zhou A, Brace AD, Halenbeck R, Hsu AW, Bray TL, Hestir K, Bosch E, Lee E, et al. 2014. Deorphanization of the human leukocyte tyrosine kinase (LTK) receptor by a signaling screen of the extracellular proteome. Proc Natl Acad Sci 111: 15741–15745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Lee JS, Guo F, Shin J, Perez-Atayde AR, Kutok JL, Rodig SJ, Neuberg DS, Helman D, Feng H, et al. 2012. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell 21: 362–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.