

Abstract
Most common genetic risk variants associated with neuropsychiatric disease are noncoding and are thought to exert their effects by disrupting the function of cis regulatory elements (CREs), including promoters and enhancers. Within each cell, chromatin is arranged in specific patterns to expose the repertoire of CREs required for optimal spatiotemporal regulation of gene expression. To further understand the complex mechanisms that modulate transcription in the brain, we used frozen postmortem samples to generate the largest human brain and cell-type–specific open chromatin data set to date. Using the Assay for Transposase Accessible Chromatin followed by sequencing (ATAC-seq), we created maps of chromatin accessibility in two cell types (neurons and non-neurons) across 14 distinct brain regions of five individuals. Chromatin structure varies markedly by cell type, with neuronal chromatin displaying higher regional variability than that of non-neurons. Among our findings is an open chromatin region (OCR) specific to neurons of the striatum. When placed in the mouse, a human sequence derived from this OCR recapitulates the cell type and regional expression pattern predicted by our ATAC-seq experiments. Furthermore, differentially accessible chromatin overlaps with the genetic architecture of neuropsychiatric traits and identifies differences in molecular pathways and biological functions. By leveraging transcription factor binding analysis, we identify protein-coding and long noncoding RNAs (lncRNAs) with cell-type and brain region specificity. Our data provide a valuable resource to the research community and we provide this human brain chromatin accessibility atlas as an online database “Brain Open Chromatin Atlas (BOCA)” to facilitate interpretation.
Within the human brain, combinational binding of transcription factors at chromatin accessible cis regulatory elements (CREs), such as promoters and enhancers, orchestrates gene expression in different cell types and brain regions. Understanding the role of CREs in the human brain is of great interest, because the majority of common genetic risk variants associated with neuropsychiatric disease affect transcriptional regulatory mechanisms as opposed to protein structure and function (Maurano et al. 2012; Gusev et al. 2014; Roussos et al. 2014; Roadmap Epigenomics Consortium 2015; Fullard et al. 2017). Previous efforts to map CREs in human brain were limited either by their use of homogenate tissue, consisting of a mixture of markedly different cell types (Maurano et al. 2012; Andersson et al. 2014; Roadmap Epigenomics Consortium 2015) or focused on a single cortical region (Fullard et al. 2017).
To further understand the role of CREs in human brain function, we sought to generate a comprehensive map of open chromatin regions (OCRs). We applied ATAC-seq to postmortem nuclei extracted from two broad cell types—neuronal (NeuN+) and non-neuronal (NeuN−)—isolated from 14 discrete brain regions of five adult individuals. Chromatin accessibility varies enormously between neurons and non-neurons, and both show enrichment with known cell type markers. Although the pattern of open chromatin in non-neurons is largely invariable, significant variability in neuronal chromatin structure is observed across different brain regions with the most extensive differences seen between neurons of the cortical regions, hippocampus, thalamus, and striatum. We identify numerous cell-type– and region-specific OCRs. Transcription factor (TF) footprinting analysis infers cell type differences in the regulation of gene expression and identifies protein-coding and long noncoding RNAs (lncRNA) with cell type and brain region specificity. Moreover, cell- and region-specific differentially accessible OCRs are enriched for genetic variants associated with neuropsychiatric traits.
Overall, our findings emphasize the importance of conducting cell-type– and region-specific epigenetic studies to elucidate regulatory and disease-associated mechanisms in the human brain. Our data provide a valuable resource to the research community, and we provide our raw data and genome browser tracks to facilitate further studies of gene regulation in the human brain.
Results
Maps of chromatin accessibility in neuronal and non-neuronal nuclei across 14 brain regions
To map chromatin accessibility in neuronal and non-neuronal nuclei across 14 brain regions (Fig. 1; Supplemental Fig. S1), we combined fluorescence-activated nuclear sorting (FANS) followed by ATAC-seq on 122 nuclear preparations obtained from 14 brain regions of five control subjects (Supplemental Table S1). We processed these data bioinformatically (Supplemental Fig. S2), and multiple metrics, including genotype concordance, gender, and evaluation of cell types, did not indicate sample mislabeling or contamination (Supplemental Fig. S3). Quality control (QC) metrics (confirmed by visual inspection of the mapped reads) led to the exclusion of seven libraries, leaving a final total of 115 libraries (Supplemental Table S2; Supplemental Fig. S3A). Overall, we obtained 4.3 billion (average of 37.8 million) uniquely mapped reads after removing duplicate reads (mean 24%) and those aligning to the mitochondrial genome (mean 1%) (Supplemental Table S3). Samples within the same brain region and cell type were very strongly correlated (Pearson correlation, r = 0.913), indicating high reproducibility among the samples (Supplemental Fig. S4). To assess the quality of our data, we compared it to five publicly available data sets generated using more optimal starting material, such as fresh tissue and cell lines (Supplemental Fig. S5; Qu et al. 2015; Corces et al. 2016, 2017; Novakovic et al. 2016; Banovich et al. 2018). Our data compared favorably and showed the lowest fraction of mitochondrial reads and the highest amount of uniquely mapped, nonduplicated, paired-end reads.
Figure 1.
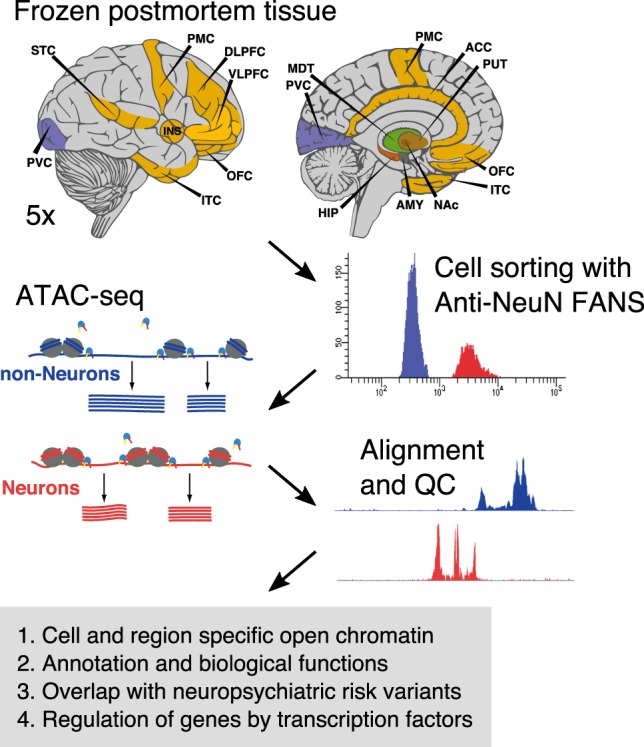
Schematic outline of the study design. Dissections from 14 brain regions of five control subjects were obtained from frozen human postmortem tissue. We combined fluorescence-activated nuclear sorting (FANS) with ATAC-seq, followed by downstream analyses, to identify cell-type–specific open chromatin regions. The brain regions and abbreviations are described in Supplemental Table S2.
We detected an average of 73,350 and 42,942 OCRs for neuronal and non-neuronal libraries, accounting for 1.05% and 0.709% of the genome, respectively (Supplemental Fig. S6A). Analysis of known neuronal and non-neuronal-specific genes indicate that our data identify OCRs in a cell-type–specific manner (Fig. 2A,B). The neuronal OCRs were more distal to transcription start sites (TSSs) compared to non-neuronal OCRs (Fig. 2C; Supplemental Fig. S6B). Further, there was a high overlap of OCRs within the neuronal and non-neuronal samples across the different brain regions, with >56.6% and 67.7% of OCRs found in two or more neuronal and non-neuronal samples, respectively. In general, promoter OCRs and non-neuronal OCRs were more frequently identified in multiple samples (Fig. 2D; Supplemental Fig. S7). Jointly, these findings suggest higher regional variability of OCRs and more distal regulation of genes in neurons compared to non-neurons.
Figure 2.

Comparisons between neuronal and non-neuronal OCRs of various brain regions. Representative cell-type–specific open chromatin tracks in the dorsolateral prefrontal cortex (DLPFC) and hippocampus at known neuron-specific (CAMK2A) (A) and non-neuron-specific (OLIG1 and OLIG2) genes (B). (C) Neuronal and non-neuronal OCRs show distinct distribution of genomic contexts. OCRs within 3 kb of a TSS were considered as promoter OCRs. (D) The distribution of the number of brain regions in which a consensus OCR was found, stratified by cell type and promoter/nonpromoter OCRs. OCRs within 3 kb of a TSS were considered as promoter OCRs. (E) Clustering of the individual samples (n = 115) using t-SNE. Brain regions are grouped in six broad areas: (AMY) amygdala; (HIP) hippocampus; (MDT) mediodorsal thalamus; (NCX) neocortex; (PVC) primary visual cortex; (STR) striatum. (F) Distribution of statistical dissimilarity (quantified based on the proportion of true tests, pi1) for inter- and intra-cell-type pairwise comparisons. Larger pi1 indicates a larger fraction of OCRs estimated to be different between samples based on pairwise comparisons. (G) Multidimensional scaling of brain regions and cell types (n = 28) using the pi1 estimates of statistical dissimilarity as distance. Same abbreviations as in E. The MDT non-neuronal group is immediately adjacent to, and partly obscured by, the leftmost non-neuronal striatum group.
Cell type and regional differences in chromatin accessibility
To quantitatively analyze differences among cell types and brain regions, we generated a consensus set of 300,444 OCRs by taking the union of peaks called in the individual cells/brain regions (Methods). We next quantified how many reads overlapped each. Covariate analyses (Methods) revealed that, besides cell type and brain region, fraction of reads within peaks (FRiP) explains a large proportion of variation in our data (Supplemental Fig. S8). After covariate correction, all variables besides cell type and brain region explained <1% of variance. t-SNE-based clustering using the adjusted read counts clearly separated neuronal from non-neuronal samples (Fig. 2E). In addition, we also observed a more modest separation among neuronal samples into neo- and subcortical regions (hippocampus, striatum, and thalamus), indicating that regional differences are more prominent in neurons.
To further assess differences in cell type and/or brain region, we performed pairwise comparisons among all samples and quantified the level of statistical significance based on the proportion of true tests, pi1. The pi1 (which equals to 1 − pi0) is an estimate of the fraction of OCRs that are differentially accessible between two groups; “1” corresponds to all OCRs estimated to have differential accessibility, whereas “0” corresponds to none of the OCRs having differential accessibility. This yielded results comparable to the t-SNE-based clustering: Among the pairwise comparisons, those between neuronal and non-neuronal cells (inter-cell-type comparison) showed a large pi1 (median = 0.59, SD = 0.10). For the intra-cell-type comparisons, there was, on average, a higher pi1 among pairs of neuronal samples than pairs of non-neuronal samples (median = 0.27, SD = 0.19 versus median = 0.064, SD = 0.10) (Fig. 2F). Furthermore, multidimensional scaling of the samples, based on the pi1 estimates as the distance metric, showed a clear distinction between neurons and non-neurons in the first dimension (Fig. 2G). Here, the neuronal samples displayed distinct clustering among different regions of the brain in the second dimension. Within the neocortex, the primary visual cortex has the most unique profile. The hippocampus and amygdala clusters showed a more similar profile with the neocortical regions when compared to the mediodorsal thalamus and striatum (putamen and nucleus accumbens). These findings are in agreement with those identified by gene expression analysis of homogenate tissue (Kang et al. 2011) and suggest a significant neuronal contribution to the regional variability described in the previous study (Kang et al. 2011).
To define cell-type– and brain region–specific OCRs, we next performed differential chromatin accessibility analysis. For the brain region analysis, we only considered neuronal samples due to the comparably minor variance seen in non-neuronal samples. The cell type analysis identified 221,957 neuronal and 46,299 non-neuronal differential OCRs at false discovery rate (FDR) of 5% (Supplemental Figs. S9, S10A; Supplemental Table S4). Regional analysis identified neuronal OCRs specific to neocortex (61,410), primary visual cortex (22,248), hippocampus (11,535), mediodorsal thalamus (42,560), and striatum (97,707) at FDR 5% (Supplemental Figs. S9, S10B; Supplemental Table S4). Due to the complementary nature of the two approaches, in the following sections we used these cell-specific (neuronal and non-neuronal) and region-specific (neocortex, primary visual cortex, hippocampus, striatum, and mediodorsal thalamus) OCRs in parallel with all OCRs identified in each brain region and cell type.
Overlap with existing epigenomic annotations, cell/region-specific genes, and biological processes
We compared the OCRs with existing epigenetic data from the NIH Roadmap Epigenomics Mapping Consortium (REMC) (Ernst and Kellis 2015; Roadmap Epigenomics Consortium 2015), considering both DNase-seq (Supplemental Fig. S11) and chromatin states (Supplemental Fig. S12) from homogenate brain tissue, brain-derived cells, and nonbrain tissues (referred as “Other”). Overall, we identified a higher overlap between our OCRs and REMC brain-related DNase-seq and active chromatin states. Notably, we saw a comparatively higher overlap with non-neuronal-specific OCRs than those of neurons (Fig. 3A). This may be an indication that many neuron-specific regulatory elements are not captured when studying homogenate tissue due to an abundance of non-neurons relative to neurons.
Figure 3.

Overlap with other epigenomes and biological functions. (A) Overlap between DNase-seq OCRs and promoter/primary enhancer states of 127 epigenomes from REMC and neuronal and non-neuronal OCRs identified by ATAC-seq. Samples from REMC are split into three groups: brain tissue, brain-derived cells, and nonbrain tissues (referred to as “Other”). The full results for the individual REMC samples are shown in Supplemental Figures S11 and S12. (B) Overlap between cell- and region-specific open chromatin (ATAC-seq) and gene sets representing biological processes and pathways. Only those that were within the top five most significant gene sets in one or more ATAC-seq categories are shown. Pathways were clustered by the Jaccard index using the WardD method based on the overlap between the genes in the different gene sets and not the enrichments. This was done to show how enrichments varied by cell type and region in terms of related pathways. (#) FDR < 0.001; (·) FDR < 0.05; (Bi) BIOCARTA; (GO) gene ontology; (KG) KEGG; (Re) REACTOME. In this analysis, the region-specific OCRs were derived from neuronal samples only.
To examine the overlap with cell- and region-specific genes, as well as genes involved in various biological processes, we next used the approach from GREAT (Methods; McLean et al. 2010). Using cell-type–specific genes (Zhang et al. 2014; Zeisel et al. 2015), we identified an overlap between neuronal OCRs and genes of pyramidal cells and interneurons, whereas non-neuronal OCRs overlapped with oligodendrocyte and astrocyte specific genes (Supplemental Fig. S13).
Similarly, we explored the overlap with genes displaying region-specific expression profiles (Supplemental Fig. S14; Hawrylycz et al. 2012) and showed that region-specific OCRs overlapped predominantly with genes expressed in the same brain region. Although this analysis describes high-order enrichment of OCRs with region-specific genes, regional and cell-type specificity of chromatin accessibility is readily visualized at the gene level. As representative examples, we considered genes with preferential expression in cortical regions (SATB2, GJD4, STX1A, and CALHM1), mediodorsal thalamus (CHRNA2 and PLCD4), and striatum (DRD2, ADORA2A, and RGS9) (Supplemental Fig. S15).
Finally, we agnostically examined the overlap with biological processes and pathways (Fig. 3B; Supplemental Fig. S16). In this analysis, neuron-specific OCRs overlapped ion channels and a range of brain-related functions, whereas non-neuronal OCRs overlapped with terms relating, among others, to the NOTCH pathway, gliogenesis, and ensheathment of neurons.
Overlap of open chromatin with neuropsychiatric traits
We used an LD-score partitioned heritability approach (Finucane et al. 2015) to assess the overlap of OCRs with genetic variants associated with 15 neuropsychiatric and unrelated traits. We found significant enrichment only for neuropsychiatric traits (Fig. 4A; Supplemental Fig. S17; Supplemental Table S5). For the cell- and region-specific OCRs, for example, neuronal- and striatal-specific OCRs were enriched for schizophrenia-associated variants, whereas neocortical- and striatal-specific OCRs were enriched for variants correlated with educational attainment. Further exploration of OCRs identified in each brain region and cell type showed that neuronal OCRs in hippocampus, nucleus accumbens, and superior temporal cortex provide the most significant enrichment with schizophrenia risk variants (Fig. 4B). These findings are in agreement with a recent study highlighting striatal medium spiny neurons and hippocampal C1A pyramidal neurons in schizophrenia (Skene et al. 2018) and DRD2, an antipsychotic drug target, being highly expressed in medium spiny neurons (Schizophrenia Working Group of the Psychiatric Genomics Consortium 2014; Skene et al. 2018). By applying the LD-score partitioned heritability approach to OCRs from homogenate brain or other tissues, we observed the strongest enrichment of schizophrenia risk variants with neuronal ATAC-seq and homogenate fetal brain OCRs (Supplemental Fig. S18; Supplemental Table S5), which is consistent with the neurodevelopmental hypothesis of schizophrenia (Rapoport et al. 2012).
Figure 4.

Overlap between genetic variants associated with various complex traits and identified OCRs assayed using LD-score partitioned heritability. (A) Overlap between cell-type– and region-specific OCRs and genetic risk variants of various traits. The region-specific OCRs are based only on neuronal samples. (B) Overlap between all OCRs identified in 14 brain regions by two cell types and schizophrenia genetic risk variants. OCRs were in all cases padded with 1000 bp to also capture adjacent genetic variants. (Chronotype) whether one is a morning or an evening person; (·) nominally significant; (#) significant after FDR correction of multiple testing across all traits and OCRs sets.
Classifying brain sample epigenomes using machine learning
We applied a support vector machine approach to identify OCR signatures that predict cell type and brain region in ATAC-seq samples of unknown origin (Supplemental Table S6). For accurate classification of cell type (neuron versus non-neuron) alone, cell type and cortical/subcortical regions, and cell type and five different brain regions defined based on the differential chromatin accessibility analysis, signatures of 3, 115, and 252 OCRs were needed, respectively (Supplemental Figs. S19A–D, S20; Supplemental Tables S7, S8). To corroborate our finding, we tested our models on independent ATAC-seq data sets (Egervari et al. 2017; Fullard et al. 2017). Here, the cell type classifier (3 OCR signature) achieved perfect accuracy in distinguishing neuronal and non-neuronal samples (Supplemental Fig. S19E; Supplemental Table S7). The cell type and cortical/subcortical classifier (115 OCR signature) attained an overall accuracy of 90% (Supplemental Fig. S19F; Supplemental Table S7). However, the classification of non-neuronal samples into cortical and subcortical groups seemed more challenging, yielding an accuracy of 86% versus an accuracy of 96% obtained for neurons. This difference was also evident using the cell-type and five-brain-region model (252 OCR signature). Although overall accuracy using the validation data set attained 85% (Supplemental Fig. S19F; Supplemental Table S7), neuronal and non-neuronal subgroups were classified with accuracies of 92% and 79%, respectively. The difficulties in classifying the brain region of non-neuronal samples mirror the previously observed small inter-region differences in MDS clustering and pi1 estimates and provide evidence for lesser regional variability among non-neuronal cells.
Regulatory effects of transcription factor binding on gene expression
To explore gene regulation in the brain, we performed footprinting analysis using PIQ (Sherwood et al. 2014) to infer transcription factor (TF) binding within the OCRs for cell type and region, independently. This approach utilized 431 TF binding motifs representing 807 TFs aggregated from a meta-database (Methods; Weirauch et al. 2014). We estimated a regulatory score for the impact of each TF on gene expression by weighing each TF binding site by the probability of that site being bound and the distance to the TSS. We found the overall regulatory score of a gene (sum of regulatory scores across all TFs for a given gene) to correlate markedly with gene expression (range of Spearman's rho: 0.318–0.523) (Supplemental Fig. S21), which is greater than the null (estimated based on permutation analysis: mean = −1.1 × 10−4, 95% CI range = −2 × 10−4; −1.5 × 10−5). The correlation is higher for brain-derived expression compared to whole blood, and this difference is more prominent in neuronal ATAC-seq libraries (Supplemental Fig. S21A).
We next explored cell type (neuronal and non-neuronal) and brain region (cortical and subcortical) regulatory differences among samples at the gene level (protein-coding, lncRNA, and miRNA) by using a regulatory divergence score. This score takes into account both the difference in regulatory burden between samples and the regulatory divergence (defined as one minus the correlation of the gene regulation) (Methods; Qu et al. 2015).
For protein-coding genes, this approach highlighted, among others, HOOK1 and KCNB2 for neuronal cells and SOX8 and HES1 for non-neuronal cells (Fig. 5A). The top 500 most neuronal and top 500 most non-neuronal genes were enriched in biologically relevant pathways. Similar analysis identified multiple protein-coding genes, including PPP1R1B, DRD2, and CACNG4 for subcortical neurons, and NRN1 and SERTM1 for cortical neurons (Fig. 5A). PPP1R1B had the twelfth highest subcortical regulatory divergence score. OCRs in the promoter and upstream enhancer region of PPP1R1B are only present in neurons of the striatum (Fig. 5B). PPP1R1B (also known as DARPP-32; dopamine and cAMP-regulated phosphoprotein) shows high expression in the dopaminoceptive medium spiny projection neurons (MSNs) of the striatum. Although frequently used as a marker of MSNs, PPP1R1B is actually widely active throughout the forebrain and in the Purkinje cells of the cerebellum (Ouimet et al. 1984; Brené et al. 1994). To validate the function of the PPP1R1B regulatory elements identified by ATAC-seq, we constructed a vector extending from 4.5 kb upstream of the TSS through the 5′ end of Exon 2 (Supplemental Fig. S22) engineered to express EGFP downstream from an Internal Ribosomal Entry Site (IRES). Pronuclear injection of this transgenic vector into mice yielded nine transgene-positive animals. Histological examination of the brain at 2 mo of age showed that seven of nine expressed EGFP in the majority of dorsal and ventral MSNs, and in the piriform cortex, a site of endogenous PPP1R1B (DARPP-32) expression (Fig. 5C–G). None showed expression outside of these regions, including in other regions with endogenous PPP1R1B expression.
Figure 5.

Identification of cell- and region-specific regulation of protein-coding genes. (A) Ranking of protein-coding genes based on their regulatory divergence score averaged across all neuronal versus all non-neuronal samples (left) and cortical neuronal samples versus subcortical neuronal samples (right). The regulatory divergence score is a combined measure for the difference in the regulatory burden for each gene, multiplied by how different the regulatory landscape is surrounding the gene (Methods). A gene set enrichment analysis using general gene sets and the top 500 most specific genes for either cell type/region using a one-sided Fisher's exact test was performed—the top three gene sets with P-values corrected for multiple testing using FDR are indicated. SOX8, AC009041.2, and LMF1 are all located in the same genetic locus. (B) Regional plot in the PPP1R1B locus showing OCRs. The promoter OCR and putative proximal enhancer OCRs are highlighted (dashed box). (C) The identified human PPP1R1B upstream OCR along with Exon 1, Intron 1, and the 5′ end of Exon 2 were used to direct expression of EGFP in transgenic mice. Expression identified with anti-PPP1R1B and DAB is restricted to the dorsal (dStr) and ventral striatum (vStr) (dorsal > ventral) and their projections (globus pallidus [gp] and substantia nigra [sn]) and the piriform cortex (pc). The black box indicates the region shown at higher magnification using immunofluorescence in D–G: (D) anti-EGFP (green); (E) anti-PPP1R1B (DARPP-32) (red); (F) DAPI (blue); (G) a merged image. EGFP is expressed exclusively in PPP1R1B positive neurons.
We performed similar regulatory divergence analysis using a recent lncRNA gene assembly (Hon et al. 2017) and identified potential differentially regulated lncRNAs with cell type (neuronal and non-neuronal) and brain region (cortical and subcortical) specificity (Fig. 6A). We applied the same data used to create the aforementioned assembly and confirmed the cell type and brain region specificity of identified lncRNAs in closely related tissues. For example, non-neuronal and subcortical lncRNAs are more abundant in expression profiles derived from white matter (Neuron Projection Bundle in Fig. 6A) and striatum, respectively. Furthermore, cell type and regional specificity was validated by qPCR gene expression studies for two lncRNAs from each group (neuronal, non-neuronal, cortical, and subcortical) (Fig. 6B; Supplemental Table S9). Finally, we examined the regulation of microRNA genes in a similar manner (Supplemental Fig. S23). This analysis identified a number of differentially expressed miRNAs, including mir-124-1 for neurons, let-7a-3 for non-neurons, mir-148a for subcortical neurons, and mir-3139 for cortical neurons.
Figure 6.

Identification of cell- and region-specific regulation of lncRNA. (A) Top ranking of lncRNA genes based on their regulatory divergence score averaged across all neuronal versus all non-neuronal samples (left) and cortical neuronal samples versus subcortical neuronal samples (right). The regulatory divergence score is a combined measure of the difference in the regulatory burden for each gene multiplied by how different the regulatory landscape is surrounding that gene (Methods). lncRNA genes were obtained from the FANTOM CAT Robust category, from which only genes from the category “far from protein-coding genes” were retained. Genes with coding status “uncertain” were excluded. (Bottom) Heatmaps of whether gene expression (CAGE) identified genes associated with the given anatomical structure. “Neuron Projection Bundle” includes samples from the corpus callosum and the optic nerve, which are depleted in neuronal nuclei. Red indicates a high gene density, and blue indicates a low gene density. Numbers in parentheses indicate the number of lncRNAs associated with the ontology. (B) qPCR validation of cell-type–specific (left) and brain region–specific (right) lncRNA identified by a regulatory divergence analysis based on ATAC-seq data. Shown are fold differences in expression for neuronal (positive values) to non-neuronal (negative values) gene expression (left) and cortical (positive values) to subcortical (negative values) (right). Error bars indicate standard deviation. (PFC) prefrontal cortex; (STR) striatum; (*) P < 0.05; (**) P < 0.01; (***) P < 0.005.
Transcription factors underlying cell and regional differences
To infer TFs that underlie the regulatory differences between cell types and brain regions, we calculated the fold-change enrichment in the corresponding peaks compared to the background of all peaks (Fig. 7; Supplemental Fig. S24). Because TFs within a given TF family share binding motifs (Weirauch et al. 2014), it is difficult to determine those family members that are biologically relevant in a given context. We note, however, that a number of studies support our findings: basic helix-loop-helix (bHLH) TFs in neurons (Lee 1997); RFX1 in neurons and the hippocampus (Ma et al. 2006), and the RORA/RORB nuclear receptor TFs in the dorsal thalamus (Ino 2004). In addition, a recent study has shown that neocortical expression of the bHLH TFs, TWIST1, and TWIST2 may be unique to primates, and both genes have human-specific expressions in the neocortex compared to macaque and chimpanzee (Sousa et al. 2017). Together with our finding of enriched exposure of TWIST1 and TWIST2 binding sites in the human neocortex, this implicates these sites in the regulation of primate and human-specific neocortical genes.
Figure 7.

The top 10 transcription factor binding motifs showing the highest fold enrichment of footprinted binding sites within peaks specific to a given cell type or brain region compared to all peaks. The region-specific TFs are based only on neuronal samples. TF binding motifs are grouped by TF family, and line width indicates the log2-transformed fold enrichment. All shown enrichments were statistically significant after correcting for multiple testing in a one-sided binomial test. Similar plots of TF binding motif enrichments stratified by genomic context are shown in Supplemental Figure S24.
Discussion
The generation of a cell-type– and brain region–specific atlas of open chromatin enabled exploration of gene regulation in the adult human brain with previously unattained detail. Differential accessibility analyses and machine learning inferred cell-type– and brain region–specific signatures of open chromatin. Compared to non-neuronal populations, open chromatin regions in neurons were found to be more extensive, to be more distal to TSS, to show a smaller overlap with previously reported OCRs from bulk brain tissue, to show greater regional variability, and to show significant enrichment in generic risk variants of various neuropsychiatric traits. Enrichment analysis highlighted an overlap of open chromatin with previously reported genes showing cell-type– and region-specific expression and further implicated cell- and region-specific molecular pathways.
We utilized the open chromatin patterns to infer transcription factor binding and to impute downstream gene regulation and expression. Despite limitations in predicting transcription factor binding, and ambiguity in subsequently linking its OCR to the gene(s) it regulates (Sherwood et al. 2014; Dixon et al. 2015; Maurano et al. 2015), we found a convincing correlation with gene expression studies. Using this regulatory analysis, we predicted cell- and region-specific protein-coding genes, lncRNAs, and microRNAs. As an example, we identified, and functionally validated, regulatory elements of the striatal, neuronal gene PPP1R1B. In addition, we predicted and experimentally validated cell type and regional patterns of lncRNA expression. Finally, we identified cell- and region-specific TFs based on the enrichment of their cognate binding motifs, which overlap with previous literature. We acknowledge, however, that footprinting analysis based on ATAC-seq data is limited due to the widespread sharing of recognition motifs between TFs (Weirauch et al. 2014), and future studies using other approaches such as ChIP-seq for specific TFs can complement our observations.
The most distinct brain region based on the neuronal OCRs was the striatum (putamen and nucleus accumbens). An explanation for this could be that, in contrast to the other assayed brain regions, the majority of neurons here are GABAergic medium spiny neurons (Kemp and Powell 1971). All experiments were performed on nuclei extracted from frozen postmortem brain specimens. Following thawing of the samples, the cell membrane is lost and, with it, many cell-type–specific antigens that would facilitate separation of different cell types by FANS. Future studies targeting additional neuronal subtypes using single-cell approaches or by cytometric separation into secondary cell subtypes could further elucidate gene regulation across the brain.
In conclusion, our findings indicate the utility of our open chromatin atlas in studying the regulation of gene expression in the brain and the impact of neuropsychiatric disease risk variants. We provide to the research community an atlas of chromatin accessibility in human brain as an online database “Brain Open Chromatin Atlas (BOCA)” to facilitate interpretation and future studies.
Methods
Brain tissue specimens from 14 brain regions of five controls with no history of psychiatric disorder and drug use were processed using a FACSAria flow cytometer to neuronal (NeuN+) and non-neuronal (NeuN−) nuclei. The Assay for Transposase Accessible Chromatin followed by sequencing (ATAC-seq) was performed using an established protocol (Buenrostro et al. 2013) and sequenced on HiSeq 2500 (Illumina) obtaining 2 × 50 paired-end reads. Reads from each sample were aligned on hg19 (GRCh37; see Supplemental Methods for a note about reference assembly) reference genome using the STAR aligner (Dobin et al. 2013) (v2.5.0). We excluded reads that (1) mapped to more than one locus using SAMtools (Li et al. 2009); (2) were duplicated using PICARD (v2.2.4); and (3) mapped to the mitochondrial genome. We merged the BAM files of samples from the same brain region and cell type and subsampled to a uniform depth. We subsequently called peaks using the model-based Analysis of ChIP-seq (MACS, v2.1) (Zhang et al. 2008) and created a joint set of peaks requiring each peak to be called in at least one of the merged BAM files. After removing peaks overlapping the blacklisted genomic regions, 300,444 peaks remained. We subsequently quantified read counts of all the individual nonmerged samples within these peaks using the featureCounts function in RSubread (v.1.15.0) (Liao et al. 2014).
We used the voomWithQualityWeights function from the limma package (Liu et al. 2015) to model the normalized read counts, including fraction of reads within peaks as covariates. We performed differential chromatin accessibility analysis by fitting weighted least-squares linear regression models for the effect of cell type (neuronal and non-neuronal) and/or brain region. P-values were adjusted for multiple hypothesis testing using false discovery rate (FDR) ≤5%. The protein interaction quantitation (PIQ) framework (Sherwood et al. 2014) was used to predict transcription factor binding sites from the genome sequence. To integrate functional annotations and GWAS results, we used the LD-score partitioned heritability (Finucane et al. 2015) approach. More details are described in the Supplemental Material.
Data access
The data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE96949. We further provide the online database “Brain Open Chromatin Atlas (BOCA)” as UCSC tracks and download links at our webpage (http://icahn.mssm.edu/boca).
Supplementary Material
Acknowledgments
Data on coronary artery disease/myocardial infarction have been contributed by CARDIoGRAMplusC4D investigators. We additionally thank the International Genomics of Alzheimer's Project (IGAP) for providing GWAS summary results. The investigators within IGAP contributed to the design and implementation of IGAP and/or provided data but did not participate in analysis or writing of this report. IGAP was made possible by the generous participation of the control subjects, the patients, and their families. Next-generation sequencing was performed at the New York Genome Center. FANS was performed at the Mount Sinai Flow Cytometry CoRE, and mouse transgenics were performed at the Mount Sinai Mouse Genetics CoRE facility. This work was supported by the National Institutes of Health (National Institute on Aging, R01AG050986 to P.R.; National Institute of Mental Health, R01MH109677 to P.R.; National Institute of Neurological Disorders and Stroke, R01NS100529 to M.E.E.; and National Institute on Drug Abuse, R01DA015446 to Y.L.H.), Brain Behavior Research Foundation (National Alliance for Research on Schizophrenia and Depression, 20540 to P.R.), Alzheimer's Association (NIRG-340998 to P.R.), New York State Stem Cell Science (N13G-169 to M.E.E.), and the Veterans Affairs (Merit Grant BX002395 to P.R.). This study was additionally funded by The Lundbeck Foundation, Denmark (Grant No. R102-A9118). Further, this work was supported in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai. The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Author contributions: J.F.F., M.E.H., M.E.E., Y.L.H., and P.R. contributed to experimental and study design and planning analytical strategies. G.E. and Y.L.H. dissected and provided brain specimens. J.F.F. conducted FANS, ATAC-seq, and generated sequencing libraries. J.F.F. and S.M.R. performed the qPCR experiments. M.-D.C. and M.E.E. designed and conducted the PPP1R1B experiments. M.E.H. carried out primary data analyses. J.B. and J.M. carried out machine learning analysis. M.E.H. and P.R. performed the transcription factor, clustering, and differential accessibility analyses. P.R. conducted the qPCR analysis. J.F.F., M.E.H., J.B., M.E.E., Y.L.H., and P.R. wrote the manuscript. All authors contributed to data interpretation and approved the final version of the text.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.232488.117.
References
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T. 2014. An atlas of active enhancers across human cell types and tissues. Nature 507: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banovich NE, Li YI, Raj A, Ward MC, Greenside P, Calderon D, Tung PY, Burnett JE, Myrthil M, Thomas SM, et al. 2018. Impact of regulatory variation across human iPSCs and differentiated cells. Genome Res 28: 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brené S, Lindefors N, Ehrlich M, Taubes T, Horiuchi A, Kopp J, Hall H, Sedvall G, Greengard P, Persson H. 1994. Expression of mRNAs encoding ARPP-16/19, ARPP-21, and DARPP-32 in human brain tissue. J Neurosci 14: 985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, et al. 2016. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet 48: 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. 2017. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14: 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W. 2015. Chromatin architecture reorganization during stem cell differentiation. Nature 518: 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egervari G, Landry J, Callens J, Fullard JF, Roussos P, Keller E, Hurd YL. 2017. Striatal H3K27 acetylation linked to glutamatergic gene dysregulation in human heroin abusers holds promise as therapeutic target. Biol Psychiatry 81: 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. 2015. Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat Biotechnol 33: 364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P-R, Anttila V, Xu H, Zang C, Farh K. 2015. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 47: 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullard JF, Giambartolomei C, Hauberg ME, Xu K, Voloudakis G, Shao Z, Bare C, Dudley JT, Mattheisen M, Robakis NK, et al. 2017. Open chromatin profiling of human postmortem brain infers functional roles for non-coding schizophrenia loci. Hum Mol Genet 26: 1942–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Lee SH, Trynka G, Finucane H, Vilhjálmsson BJ, Xu H, Zang C, Ripke S, Bulik-Sullivan B, Stahl E, et al. 2014. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet 95: 535–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, Van De Lagemaat LN, Smith KA, Ebbert A, Riley ZL. 2012. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489: 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon CC, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJ, Gough J, Denisenko E, Schmeier S, Poulsen TM, Severin J. 2017. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 543: 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ino H. 2004. Immunohistochemical characterization of the orphan nuclear receptor RORα in the mouse nervous system. J Histochem Cytochem 52: 311–323. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AM, Pletikos M, Meyer KA, Sedmak G, et al. 2011. Spatio-temporal transcriptome of the human brain. Nature 478: 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp JM, Powell T. 1971. The structure of the caudate nucleus of the cat: light and electron microscopy. Philos Trans R Soc Lond B Biol Sci 262: 383–401. [DOI] [PubMed] [Google Scholar]
- Lee JE. 1997. Basic helix-loop-helix genes in neural development. Curr Opin Neurobiol 7: 13–20. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. [DOI] [PubMed] [Google Scholar]
- Liu R, Holik AZ, Su S, Jansz N, Chen K, Leong HS, Blewitt ME, Asselin-Labat ML, Smyth GK, Ritchie ME. 2015. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res 43: e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K, Zheng S, Zuo Z. 2006. The transcription factor regulatory factor X1 increases the expression of neuronal glutamate transporter type 3. J Biol Chem 281: 21250–21255. [DOI] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. 2012. Systematic localization of common disease-associated variation in regulatory DNA. Science 337: 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Haugen E, Sandstrom R, Vierstra J, Shafer A, Kaul R, Stamatoyannopoulos JA. 2015. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat Genet 47: 1393–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakovic B, Habibi E, Wang SY, Arts RJW, Davar R, Megchelenbrink W, Kim B, Kuznetsova T, Kox M, Zwaag J, et al. 2016. β-Glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell 167: 1354–1368.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet C, Miller P, Hemmings H, Walaas SI, Greengard P. 1984. DARPP-32, a dopamine-and adenosine 3′:5′-monophosphate-regulated phosphoprotein enriched in dopamine-innervated brain regions. III. Immunocytochemical localization. J Neurosci 4: 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu K, Zaba LC, Giresi PG, Li R, Longmire M, Kim YH, Greenleaf WJ, Chang HY. 2015. Individuality and variation of personal regulomes in primary human T cells. Cell Syst 1: 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport JL, Giedd JN, Gogtay N. 2012. Neurodevelopmental model of schizophrenia: update 2012. Mol Psychiatry 17: 1228–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussos P, Mitchell AC, Voloudakis G, Fullard JF, Pothula VM, Tsang J, Stahl EA, Georgakopoulos A, Ruderfer DM, Charney A, et al. 2014. A role for noncoding variation in schizophrenia. Cell Rep 9: 1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. 2014. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511: 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood RI, Hashimoto T, O'Donnell CW, Lewis S, Barkal AA, van Hoff JP, Karun V, Jaakkola T, Gifford DK. 2014. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat Biotechnol 32: 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, Giusti-Rodriguez P, Hodge RD, Miller JA, Muñoz-Manchado AB, et al. 2018. Genetic identification of brain cell types underlying schizophrenia. Nat Genet 50: 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa AMM, Zhu Y, Raghanti MA, Kitchen RR, Onorati M, Tebbenkamp ATN, Stutz B, Meyer KA, Li M, Kawasawa YI, et al. 2017. Molecular and cellular reorganization of neural circuits in the human lineage. Science 358: 1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K. 2014. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158: 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A, Marques S, Munguba H, He L, Betsholtz C, et al. 2015. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347: 1138–1142. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based Analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N. 2014. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34: 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.