

Abstract
LSD1/KDM1 is a histone demethylase that preferentially removes methyl groups from the mono- and di-methylated lysine 4 in histone H3 (H3K4), key marks for active chromatin for transcriptional activation. LSD1 is essential for pluripotent embryonic stem cells and embryonic teratocarcinoma/carcinoma cells and its expression is often elevated in various cancers. We developed a new LSD1 inhibitor, CBB3001, which potently inhibited LSD1 activity both in vitro and in vivo. CBB3001 also selectively inhibited the growth of human ovarian teratocarcinoma PA-1 and mouse embryonic carcinoma F9 cells, caused the downregulation of pluripotent stem cell proteins SOX2 and OCT4. However, CBB3001 does not have significant inhibition on the growth of human colorectal carcinoma HCT116 cells or mouse fibroblast NIH3T3 cells that do not express these stem cell proteins. Our studies strongly indicate that CBB3001 is a specific LSD1 inhibitor that selectively inhibits teratocarcinoma and embryonic carcinoma cells that express SOX2 and OCT4.
Keywords: LSD1/KDM1 Inhibitor, CBB3001, Histone Demethylase, Methylated H3K4 of Histone H3, Teratocarcinoma, Embryonic Carcinoma, SOX2, OCT4
Graphical Abstract
CBB3001 inhibits the essential function of histone demethylase LSD1 in pluripotent cancer cells that exhibit stem cell properties.
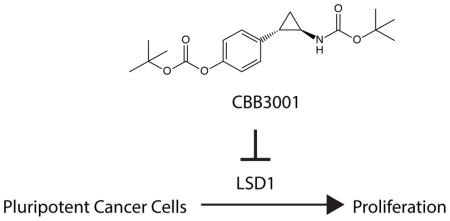
1. Introduction
Lysine-methylation is a major covalent modification of histones that epigenetically regulates gene expression, chromatin structure, and chromatin organization.1, 2 It is well established that the mono-, di- and trimethylations of lysine 4 (K4) in histone H3 (H3K4) are associated with actively transcribed genes with an open chromatin structure.1–3 Histone methylation is dynamically controlled by specific histone methyltransferases and demethylases.3 Specifically, the methylations of H3K4 are primarily catalyzed by histone methyltransferases such as MLL (Mixed Lineage Leukemia) SET-domain methyltransferase complexes which are composed of additional essential components including WDR5 and RBBP5.2, 4 The methylation of H3K4 by WDR5-associated methyltransferases has been shown to be essential for the pluripotency and self-renewal of embryonic stem cells.5 The methylation states of H3K4 are dynamically regulated by a group of histone demethylases including lysine-specific demethylase 1 (LSD1, also called KDM1, AOF2, or BHC110) and JARID1A-1D (also called KDM5A-5D).1, 3, 6 These demethylases specifically remove the methyl group in the mono-, di- and trimethylated H3K4 to regulate gene expression and chromatin structure in various cells.
LSD1 belongs to the flavin adenine dinucleotide (FAD)-dependent amine oxidase family, and specifically catalyzes the demethylation of mono- and di-methylated H3K4 through amine oxidation.1, 6, 7 The unique LSD1-dependent demethylation requires a protonated nitrogen in the methylated histone, precluding it from removing the methyl group from trimethylated H3K4.1, 6, 8 LSD1 is highly conserved in high eukaryotes and it is essential for pluripotency and self-renewal of embryonic stem cells.9 The mouse null mutation of LSD1 genes causes embryonic lethality.9, 10 We have previously shown that the levels of LSD1 are significantly elevated in pluripotent germ cell tumor cells such as teratocarcinoma, embryonic carcinoma, and seminoma cells11 and other cancer cells such as human squamous lung carcinomas.12 Various independent reports also indicate that the expression of LSD1 is often highly elevated in various human cancers.13–22
We have previously developed novel LSD1 inhibitors based on the co-crystal structures of LSD1 protein associated with its H3K4 peptide substrate.11 Our studies showed that these LSD1 inhibitors specifically inhibited the pluripotency and self-renewal of mouse embryonic stem cells and pluripotent teratocarcinoma/embryonic carcinoma cells.11 Our subsequent investigation also revealed that LSD1 inhibition or inactivation is sufficient to block the proliferation of many cancer cells that exhibit stem cell properties, such as expression of SOX2 or other stem cell proteins.12, 23 In these cells, we demonstrated that inhibition or inactivation of LSD1 leads to the downregulation of SOX2, a key pluripotent stem cell transcription factor for embryonic stem cells, embryonic carcinoma and teratocarcinoma cells.11, 12, 23 The SOX2 encoded gene at 3q26.33 is frequently amplified in squamous cell carcinomas of lung, esophagus and oral cavity, small cell lung carcinomas, and glioblatoma multiforme (GBM).24–26 SOX2 is also overexpressed in many other cancers including breast and ovarian carcinomas.27–31 The expression of SOX2 confers the stem cell properties to cancer cells.12, 27, 28 It was also shown that LSD1 is essential for maintaining the oncogenic potential of MLL-AF9 leukemia stem cells and acute myeloid leukemia.32, 33 Thus, LSD1 serves as a critical epigenetic target for various cancer cells with stem cell properties such as expression of SOX2 or other stem cell proteins.12, 27, 28
Here, we report the development of a new LSD1 inhibitor, which is structurally different from our previous LSD1 inhibitors.11, 12 Our studies revealed that this inhibitor potently inhibits LSD1 activity in vitro and in cultured cancer cells. Importantly, this inhibitor selectively impedes the proliferation of teratocarcinoma and embryonic carcinoma cells that express pluripotent stem cell proteins SOX2 and OCT4. However, it has low toxicity towards other cancer cells that do not express these pluripotent stem cell proteins, similar to that of our previously developed LSD1 inhibitors based on the crystal structure of LSD1 protein.11, 12
2. Results and discussion
2.1 Design and organic synthesis of CBB3001
Because the catalytic domain of LSD1 shares significant similarity with other members of the amine oxidase family, most investigation on LSD1 function involves the use of non-selective amine oxidase inhibitors, such as tranylcypromine (trans-2-phenylcycpropylamine, PCPA, Figure 1A), originally developed against two major isoforms of monoamine oxidases, MAO-A and MAO-B, for clinical use as anti-depressants.6, 8, 34–37 Tranylcypromine has been shown to inhibit LSD1 activity with substantially reduced potency as compared to its inhibition of MAOs. It inhibits LSD1 activity through the irreversible modification of the covalently bound FAD at high concentrations (IC50: submillimolar to millimolar), similar to its inhibitory mechanism for MAOs.6, 8, 15, 34, 35, 38, 39 We tried to test a derivative of tranylcypromine, CBB3001, towards LSD1 since the activity of this compound towards LSD1 has never been reported. For the synthesis of CBB3001, we modified the Corey-Chaykovsky chemical synthesis scheme,40–43 as outlined in Figure 1B, to obtain better yield.
Figure 1.
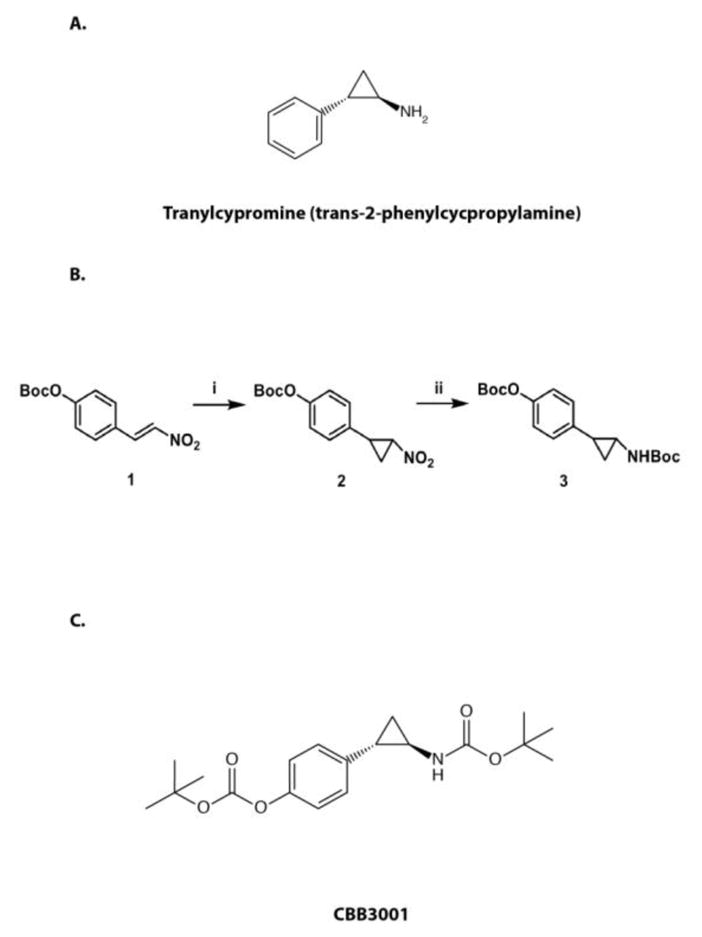
The synthesis scheme of CBB3001.
A. The structure of tranylcypromine (trans-2-phenylcycpropylamine).
B. Scheme for chemical synthesis of CBB3001: i) a) Boc2O, triethylamine, DMAP, DCM; b) Trimethylsulfoxonium iodide, NaH, DMSO. ii) a) Zn, HCl (aq), i-PrOH; b) Boc2O, TEA, dichloromethane (DCM). Compound 3 is CBB3001.
C. The structure of CBB3001.
2.1.1 Preparation of tert-butyl 4-(2-nitrocyclopropyl)phenyl carbonate (2)
To obtain compound 2 (Figure 1B), the mixture of (E)-4-(2-Nitrovinyl)phenol, Boc2O, DMAP, and triethylamine in dichloromethane was allowed to react at room temperature overnight. Water was then added and the organic layer was separated, washed, dried over Na2SO4, filtered and evaporated to dryness. The residue was dissolved in DMSO. Trimethylsulfoxonium iodide was added to a suspension of 60% NaH in DMSO and then (E)-tert-butyl 4-(2-nitrovinyl)phenyl carbonate DMSO solution was added dropwise to react for another 2 hrs. The reaction was quenched with NH4Cl and diluted with ethyl acetate. The organic layer was separated, washed, dried over Na2SO4, filtered and evaporated to dryness. The resulting mixture was purified via column chromatography using ethyl acetate and hexanes as eluents to afford compound 2 with 32% yield.
2.1.2 Preparation of tert-butyl 2-(4-(tert-butoxycarbonyloxy)phenyl)cyclopropylcarbamate (3, CBB3001)
To obtain compound 3 or CBB3001 (Figure 1B), compound 2 in a mixture of i-PrOH and 1N HCl was added Zn powder to react at room temperature for 1 hr. The reaction was quenched with NaHCO3 and diluted with ethyl acetate. The organic layer was separated, washed, dried, filtered and evaporated to dryness. The residue was dissolved in dichloromethane and mixed with triethylamine and Boc2O and the mixture was stirred overnight. Water was subsequently added and the organic layer was separated, washed with 1N HCl and then brine, dried, filtered and evaporated to dryness. The crude product was purified via column chromatography using ethyl acetate and hexanes as eluents to afford compound 3, which is designated as CBB3001 (Figure 1B and 1C), with 23% yield.
2.2. Biochemical evaluation: CBB3001 potently inhibits LSD1 demethylase activity in vitro
To test whether CBB3001 has any inhibitory effect on the catalytic demethylase activity of LSD1 protein, we constructed an in-frame fusion protein between the amino-terminus of human LSD1 protein and the carboxy-terminus of glutathione-S-transferase (GST).11 This fusion GST-LSD1 protein was expressed in bacterial BL21 strain that is deficient in protease activity. The expressed GST-LSD1 protein was subsequently purified by the glutathione affinity chromatography (Figure 2A). Since the LSD1 demethylase activity specifically removes the methyl group from the mono- and di-methylated H3K4 in a stepwise manner, we used a histone H3 peptide that contains a di-methylated lysine 4 (H3K4me2) as the substrate for assaying the catalytic activity of LSD1 in vitro.11 Our studies showed that the purified GST-LSD1 protein exhibited a potent demethylase activity by removing the methyl group from the H3K4me2 peptide to produce the mono- and non-methylated H3K4 peptide products, which can be analyzed by mass spectrometry (Figure 2B).11 Using the demethylation assay, we found that inclusion of various concentrations of CBB3001 in the demethylation reaction leads to the potent inhibition of LSD1 demethylase activity (Figure 2B). Quantification of inhibitory profile revealed that the IC50 of CBB3001 is about 21.25 μM toward the demethylase activities of LSD1 under our reaction conditions (Figure 2C), indicating that this compound has potent inhibitory effects towards the purified LSD1 demethylase activity in vitro.
Figure 2.

Analysis of CBB3001 and tranylcypromine (PCPA) on LSD1-dependent demethylation in vitro.
A. Purified recombinant GST-LSD1 protein used for the in vitro demethylation assay.
B. CBB3001 or tranylcypromine inhibits LSD1 demethylase activity in vitro. Recombinant LSD1 was incubated at 30 °C for 1 hour with the di-methylated H3K4 peptide and various concentrations of CBB3001. The demethylated products, mono-methylated (H3K4Me1) and non-methylated (H3K4me0), were analyzed by mass-spectrometry (MS). From top to bottom panels: 1) 1 μg di-methylated H3K4 peptide without GST-LSD1; 2) 1 μg dimethylated peptide + 2 μg GST-LSD1; 3) 1 μg di-methylated H3K4 peptide + 2 μg GST-LSD1 + 30 μM CBB3001; 4 and 5) 1 μg di-methylated H3K4 peptide + 2 μg GST-LSD1 + 50 μM or 500 μM tranylcypromine in 20 μl reaction as indicated.
C. The MS peak areas of control and various CBB3001 treated samples were integrated as described in Experimental and used to calculate IC50 of CBB3001 at 0, 1, 2, 5, 10, 15, 20, 30, 40, 50, 100 μM, respectively.
In contrast, our analyses revealed that tranylcypromine (PCPA) at 500 μM caused significant inhibition of LSD1 demethylase activity, while 50 μM tranylcypromine did not have substantially inhibitory effects (Figure 2B). Our results indicate that CBB3001 is significantly more potent than tranylcypromine for LSD1 inhibition.
2.3. Biological evaluation: CBB3001 is active in cancer cells to inhibit LSD1 activity in vivo
LSD1 was identified as a histone demethylase that removes the methyl group from mono- and di-methylated H3K4. Inhibition or reduced expression of LSD1 in vivo causes the accumulation of the mono- and di-methylated forms of H3K4 but not trimethylated H3K4.6, 11, 12, 15, 38 To determine whether CBB3001 inhibits LSD1 in vivo, we treated a human colorectal carcinoma cells, HCT116, with various concentrations of CBB3001 (Figure 3A and 3B). We found that treatment of this compound at 20 μM was sufficient to induce the increased levels of the mono- and di-methylated H3K4 in histone H3, while there was no change on the level of trimethylated H3K4 (Figure 3C). We also treated a human ovarian teratocarcinoma cell, PA-1, originally derived from a metastatic ovary of a 12 year old Caucasian female, with various concentrations of CBB3001 (Figure 3A and 3B). Similar to the effects observed on HCT116 cells, treatment of PA-1 cells with this LSD1 inhibitor also increased the levels of the mono- and di-methylated H3K4 in histone H3 but not the level of trimethylated H3K4 (Figure 3C). These studies indicate that CBB3001 indeed inhibits the histone demethylase activity of LSD1 in various cancer cells.
Figure 3.

In vivo analysis of CBB3001 on LSD1 demethylation of methylated H3K4 in histone H3.
A. In vivo effects of CBB3001 on cancer cells. Actively growing HCT116 and PA-1 cells were treated with various concentrations of CBB3001 for 16 hours and examined. Cells were examined and cells images were acquired with 10×10 lens of Nikon ECLIPSE Ti-S microscope equipped with NIS-Elements BR 3.1 software. Triplicated cells (technical repeats) were used for examination and one set of representative treated cells was shown.
B. Triplicated treated cells (technical repeats) from Figure 3A were harvested by trypsin digestion, diluted, and blindly spotted onto a hemacytometer. Cells in four corners of the hemacytometer were counted to obtain average cells per dish. The differences between control and CBB3001 cells in triplicated samples were plotted. Experiments were repeated for three independent times with the same conclusion and one example is shown. Statistically significant differences were determined using a two-tailed equal-variance independent t-test. Different data sets were considered to be statistically significant when the P-value was <0.05 (*), 0.01 (**) or 0.001 (***).
C. Total histones were extracted from control and CBB3001 treated cells and the levels of mono-, di-, and trimethylated H3K4 and histone H3 were monitored by Western blotting with specific antibodies indicated on the left, respectively, and quantified.
2.4. CBB3001 selectively inhibits pluripotent PA-1 teratocarcinoma cells but does not have general cytotoxicity towards HCT116 cells
Although CBB3001 caused the increased levels of mono- and di-methylated H3K4 in both HCT116 and PA-1 cells, we found that CBB3001 did not appear to affect the growth of HCT116 cells, even at high concentrations of CBB3001 (>40 μM, Figure 3A and 3B). In contrast, we observed that CBB3001 potently inhibited the growth of PA-1 cells between 5–20 μM after 12–16 hours of treatment (Figure 3A and 3B).
We and others have previously shown that the activity of LSD1 is essential for the self-renewal of pluripotent mouse embryonic stem cells and embryonic teratocarcinoma/carcinoma cells but not for non-pluripotent cancer cells such as HeLa and 293 cells or mouse fibroblast NIH3T3 cells, due to the specific requirement of LSD1 for the expression of pluripotent stem cell proteins SOX2 and OOC4 in mouse embryonic stem cells and embryonic teratocarcinoma/carcinoma cells.11, 12, 23, 44–46 The relative refractory effects of CBB3001 on HCT116 cells suggest that CBB3001 does not have general cytotoxicity towards cancer cells such as HCT116 cells that do not express these stem cell proteins. Thus, the selective inhibition of PA-1 teratocarcinoma cells by CBB3001 indicates that this compound is highly specific for LSD1 inhibition in teratocarcinoma cells that express SOX2 and OCT4.
2.5. CBB3001 selectively inhibits the expression of pluripotent stem cell proteins SOX2 and OCT4 to block the proliferation of pluripotent PA-1 teratocarcinoma cells
LSD1 has been shown to be specifically required for the self-renewal of pluripotent mouse embryonic stem cells and embryonic teratocarcinoma/carcinoma cells.11, 12, 23 Inactivation of LSD1 leads to the downregulation of pluripotent stem cell proteins such as SOX2 or OCT4, which are essential for the pluripotency and self-renewal of pluripotent stem cells.11, 12, 23 We found that treatment of CBB3001 indeed induced the downregulation of SOX2 and OCT4 proteins in PA-1 cells (Figure 4A and 4B). To further confirm the effect of CBB3001 in PA-1 cells, we also used siRNA-mediated gene silencing to knockdown the expression of LSD1 in PA-1 cells by specific siRNAs. We found that knockdown of LSD1 indeed caused the increased levels of mono-and di-methylated H3K4, downregulation of SOX2 and OCT4 proteins, and the growth inhibition of 1 cells (Figure 4C and 4D). These studies confirm that CBB3001 is a specific inhibitor of LSD1 both in vitro and in vivo.
Figure 4.

CBB3001 downregulates SOX2 and OCT4 expression in PA-1 cells.
A. PA-1 cells were treated at indicated concentrations of CBB3001 for 16 hours and cells were examined and cells images were acquired as in Figure 3A.
B. The treated cells were lysed with 0.5% SDS-containing lysis buffer and the levels of SOX2, OCT4, LSD1, mono-, di-, and trimethylated H3K4 and histone H3 were monitored by Western blotting with specific antibodies indicated on the left, respectively, and quantified.
C. PA-1 cells were transfected with 50 nM control (luciferase, luc) and LSD1 siRNAs for 48 hours and cells were examined and cells images were acquired as in Figure 4A.
D. The levels of SOX2, OCT4, LSD1, mono-, di-, and trimethylated H3K4 and histone H3 were monitored by Western blotting with specific antibodies indicated on the left, respectively, and quantified.
2.6. CBB3001 also selectively blocks the growth of mouse embryonic carcinoma F9 cells but not mouse fibroblast NIH3T3 cells that do not express pluripotent stem cell proteins SOX2 and OCT4
Since PA-1 is a teratocarcinoma cell that express SOX2 and OCT4, we also tested whether other cancer cells such as mouse embryonic carcinoma F9 cells are sensitive to CBB3001.11, 12, 23 Our studies showed that treatment of F9 cells with CBB3001 led to the growth inhibition and downregulation of SOX2 and OCT4 (Figure 5A-5C). In contrast, CBB3001 did not have significant effects on the growth of mouse fibroblast cell NIH3T3 that does not express SOX2 or OCT4 (Figure 5A-5C).11 These studies are consistent with our previous and current observations that PA-1 and F9 cells are selectively sensitive to CBB3001 because LSD1 is specifically required for the expression of pluripotent stem cell proteins SOX2 and OCT4 in these cells.11, 12, 23
Figure 5.
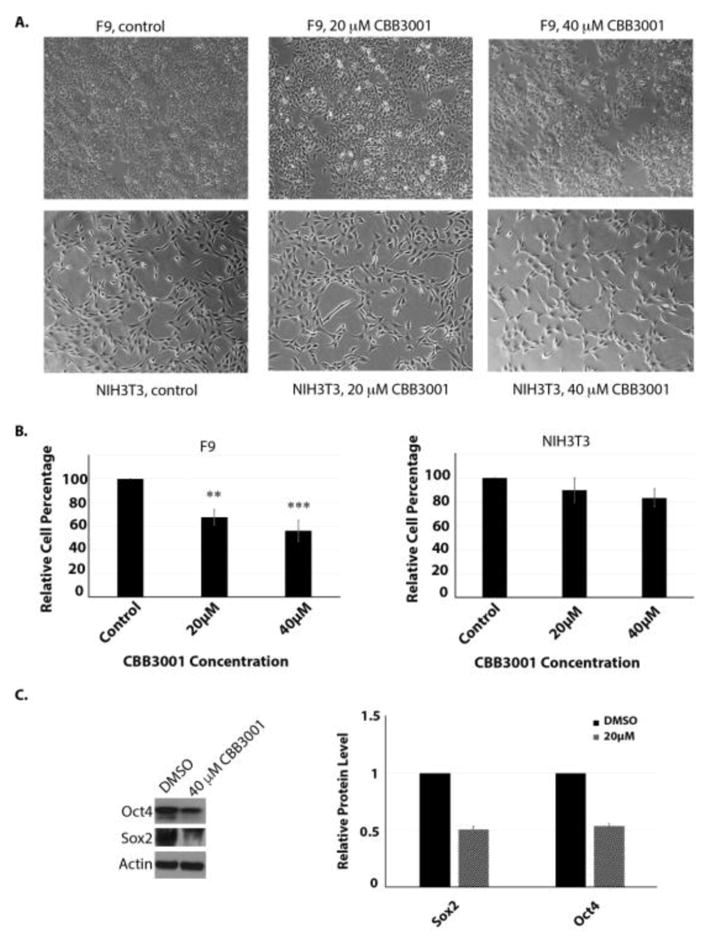
CBB3001 Selectively inhibits the growth of F9 cells.
A. Actively growing F9 and NIH3T3 cells were treated with various concentrations of CBB3001 for 30 hours and examined. Cells were examined and cells images were acquired as in Figure 4A. Triplicated cells (technical repeats) were used for examination and one set of representative treated cells was shown.
B. Triplicated treated cells (technical repeats) from Figure 5A were harvested by trypsin digestion, diluted, and blindly spotted onto a hemacytometer. Cells in four corners of the hemacytometer were counted to obtain average cells per dish. The differences between control and CBB3001 cells in triplicated samples were plotted. Statistically significant differences were determined using a two-tailed equal-variance independent t-test. Different data sets were considered to be statistically significant when the P-value was <0.05 (*), 0.01 (**) or 0.001 (***).
C. The treated F9 cells were lysed with 0.5% SDS-containing lysis buffer and the levels of SOX2, OCT4, and actin (loading control) were monitored by Western blotting with specific antibodies indicated on the left, respectively, and quantified.
2.7. Tranylcypromine (PCPA) non-specifically inhibits the growth of all tested cells at high concentrations
To compare the selectivity and potency of CBB3001, we also examined the effects of tranylcypromine in our cells. Our studies revealed that tranylcypromine exhibited general cytotoxicity towards the growth of PA-1, F9, HCT116, and NIH3T3 cells in a dose-dependent manner, especially at high concentrations (> 200 μM, Figure 6 and 7). Our studies are consistent with previous reports that tranylcypromine is a non-specific inhibitor that is toxic to the growth of many cells.8, 11, 47
Figure 6.

Tranylcypromine inhibits the growth of both HCT116 and PA-1 cells.
A. In vivo effects of tranylcypromine on HCT116 and PA-1 cells. Actively growing HCT116 and PA-1 cells were treated with various concentrations of tranylcypromine (PCPA) for 16 hours and examined. Cells were examined and cells images were acquired as in Figure 3A. Triplicated cells (technical repeats) were used for examination and one set of representative treated cells was shown.
B. Triplicated treated cells (technical repeats) from Figure 6A were harvested by trypsin digestion, diluted, and blindly spotted onto a hemacytometer as in Figure 3B. The differences between control and tranylcypromine-treated cells in triplicated samples were plotted. Experiments were repeated for three independent times with the same conclusion and one example is shown. Statistically significant differences were determined using a two-tailed equal-variance independent t-test. Different data sets were considered to be statistically significant when the P-value was <0.05 (*), 0.01 (**) or 0.001 (***).
Figure 7.

Tranylcypromine inhibits the growth of both F9 and NIH3T3 cells.
A. In vivo effects of tranylcypromine on F9 and NIH3T3 cells. Actively growing F9 and NIH3T3 cells were treated with various concentrations of tranylcypromine (PCPA) for 16 hours and examined. Cells were examined and cells images were acquired as in Figure 3A. Triplicated cells (technical repeats) were used for examination and one set of representative treated cells was shown.
B. Triplicated treated cells (technical repeats) from Figure 7A were harvested by trypsin digestion, diluted, and blindly spotted onto a hemacytometer as in Figure 3B. The differences between control and tranylcypromine-treated cells in triplicated samples were plotted. Experiments were repeated for three independent times with the same conclusion and one example is shown. Statistically significant differences were determined using a two-tailed equal-variance independent t-test. Different data sets were considered to be statistically significant when the P-value was <0.05 (*), 0.01 (**) or 0.001 (***).
2.8. CBB3001 is more selective and potent than several recently reported LSD1 inhibitors
Recently, in addition to tranylcypromine, several other compounds such as ORY-1001 (ORY or RG-6016) and GSK2879552 (GSK) were reported to inhibit the demethylase activity of LSD1 and to block the growth of many cancer cells with high potency (reported IC50 of ORY=20 nM and IC50 of GSK=240 nM for inhibiting the growth of many cancer cells).48, 49 To compare the selectivity and potency of CBB3001, we also tested the effects of GSK2879552 and ORY-1001 on our cells, in parallel to CBB3001 (Figure 8 and 9). Our studies indicate that under our assay conditions (single dose and 16–30 hours), there were no significant inhibitory effects of both GSK2879552 and ORY-1001 at their reported concentrations or much higher concentrations (GSK2879552 at 5 μM and ORY-1001 at 2 μM) in PA-1, HCT116, F9 and NIH3T3 cells (Figure 8 and 9), although a low level of cytotoxicity of GSK2879552 (< 10–20%) towards all these cell lines was sometimes observed. Thus, our studies indicate that CBB3001 exhibits selective and potent inhibitory effects on teratocarcinoma and embryonic carcinoma cells that are consistent with the requirement of LSD1 for the expression of pluripotent SOX2 and OCT4 these cells.
Figure 8.

Effects of ORY-1001, GSK2879552, and CBB3001 on HCT116 and PA-1 cells.
A. In vivo effects of tranylcypromine on HCT116 and PA-1 cells. Actively growing HCT116 and PA-1 cells were treated with various concentrations of CBB3001, GSK2879552 (GSK), or ORY-1001 (ORY) for 16 hours and examined. Cells were examined and cells images were acquired as in Figure 3A. Triplicated cells (technical repeats) were used for examination and one set of representative treated cells was shown.
B. Triplicated treated cells (technical repeats) from Figure 8A were harvested by trypsin digestion, diluted, and blindly spotted onto a hemacytometer as in Figure 3B. Statistically significant differences were determined using a two-tailed equal-variance independent t-test. Different data sets were considered to be statistically significant when the P-value was <0.05 (*), 0.01 (**) or 0.001 (***).
Figure 9.

Effects of ORY-1001, GSK2879552, and CBB3001 on F9 and NIH3T3 cells.
A. In vivo effects of tranylcypromine on F9 and NIH3T3 cells. Actively growing F9 and NIH3T3 cells were treated with various concentrations of CBB3001, GSK2879552 and ORY-1001 for 16 hours and examined. Cells were examined and cells images were acquired as in Figure 3A. Triplicated cells (technical repeats) were used for examination and one set of representative treated cells was shown.
B. Triplicated treated cells (technical repeats) from Figure 9A were harvested by trypsin digestion, diluted, and blindly spotted onto a hemacytometer as in Figure 3B. The differences between control and tranylcypromine-treated cells in triplicated samples were plotted. Experiments were repeated for three independent times with the same conclusion and one example is shown. Statistically significant differences were determined using a two-tailed equal-variance independent t-test. Different data sets were considered to be statistically significant when the P-value was <0.05 (*), 0.01 (**) or 0.001 (***).
2.8. The growth inhibitory effects of CBB3001 on PA-1 cells are not reversible
Since previous studies indicated that LSD1 maintains the self-renewal and pluripotency of mouse embryonic stem cells and other embryonic carcinoma/teratocarcinoma cells by preventing them from differentiation,11, 12, 23 we tested whether the growth inhibitory effects of CBB3001 are reversible. We found that after treatment of PA-1 cells with CBB3001 for 24 hours, the growth inhibitory effects of CBB3001 were not reversible if the compound was removed from the culture medium for another 24 hours (Figure 10A and 10B). It is possible that by downregulating pluripotent stem cell proteins such as SOX2 or OCT4 (Figure 4) in these cells by CBB3001, the cells are induced to undergo differentiation and stop growth, as previously reported in LSD1-inactivated embryonic stem cells or embryonic carcinoma/teratocarcinoma cells.
Figure 10.

The growth inhibitory effects of CBB3001 is not reversible in PA-1 cells.
A. Actively growing PA-1 cells were treated with various concentrations of CBB3001 for 24 hours and the chemicals were removed. The cells were extensively washed and re-cultured in fresh cell culture medium without CBB3001 for 24 additional hours. Cells were examined and cells images were acquired as I Figure 3A. Triplicated cells (technical repeats) were used for examination and one set of representative treated cells was shown.
B. Triplicated treated cells (technical repeats) from Figure 10A were harvested by trypsin digestion, diluted, and blindly spotted onto a hemacytometer. Cells in four corners of the hemacytometer were counted to obtain average cells per dish. The differences between control and CBB3001 cells in triplicated samples were plotted. Experiments were repeated for three independent times with the same conclusion and one example is shown.
C. PA-1 cells were transfected with 50 nM control (luciferase, Luc) or SOX2 siRNAs for 48 hours. Left and middle panels: Cells were examined and cells images were acquired as in Figure 4A. Right panel: The levels of SOX2 and CUL1 (loading control) were examined in transfected cells by specific antibodies.
2.9. Dowregulation of SOX2 is sufficient to block the growth of PA-1 cells
PA-1 is a teratocarcinoma cell line that expresses pluripotent stem cell proteins such as SOX2 and OCT4.12, 50 As an example to demonstrate the importance of these pluripotent stem cell proteins for the growth of PA-1 cells, we also used siRNA-mediated gene silencing to knockdown the expression of SOX2 (Figure 10C). Our examination revealed that downregulation ofby specific siRNAs is sufficient to block the growth of PA-1 cells, indicating that the downregulation of pluripotent stem cell proteins such as SOX2 by CBB3001 serves as a mechanism for the selectivity of CBB3001 towards PA-1 cells.
3. Conclusion
In this report, we report a new chemical compound, CBB3001, that specifically inhibits the activity of histone demethylase LSD1. Unlike tranylcypromine that inhibits LSD1 at high concentrations34, 35, 51, this new LSD1 inhibitor specifically and potently inhibits the demethylase activity of LSD1 in vitro, with IC50 about 21 μM (Figure 1 and 2). While tranylcypromine exhibits general cytotoxicity towards all our cell lines (Figure 6 and 7), we found CBB3001 does not have general cytotoxicity towards cancer cells such as HCT116 or NIH3T3 cells (Figure 3–5). However, as expected, CBB3001 displays strong growth inhibitory effects on the growth of pluripotent teratocarcinoma PA-1 or embryonic carcinoma F9 cells that express SOX2 and OCT4 (Figure 3 and 4). Our further studies revealed that CBB3001 potently inhibited the expression of pluripotent stem cell proteins SOX2 or OCT4 in PA-1 and F9 cells (Figure 3–5).11, 12, 50 Since HCT116 or NIH3T3 cells do not express SOX2 and/or OCT4 or other stem cell proteins such as NANOG, CBB3001 does not have significant growth inhibitory effects towards HCT116 and NIH3T3 cells (Figure 3 and 5). Our studies revealed that CBB3001 represents another class of chemical compounds that exhibits potent and specific inhibitory effects toward LSD1. Our studies further indicate that LSD1 serves as a critical epigenetic target in cancer cells that express pluripotent stem cell proteins.
4. Experimental
4.1. Organic synthesis
Preparation of tert-butyl 4-(2-nitrocyclopropyl)phenyl carbonate (2)
We modified a chemical scheme to synthesize CBB3001.40–43 (E)-4-(2-Nitrovinyl)phenol (193 mg), Boc2O (280 mg), DMAP (15 mg) and triethylamine (244 μL) in 5 mL DCM was stirred at room temperature overnight. Then water was added into the flask and the organic layer was separated, washed with brine, dried over Na2SO4, filtered and evaporated to dryness. The residue was dissolved in 1 mL DMSO. To a suspension of 60% NaH (61 mg) in 7 mL DMSO was added trimethylsulfoxonium iodide (335 mg) [1]. The mixture was stirred at room temperature for 30 min. Then (E)-tert-butyl 4-(2-nitrovinyl)phenyl carbonate DMSO solution was added dropwise into the flask. The reaction was stirred at room temperature for another 2 hrs. The reaction was quenched with NH4Cl (aq) and diluted with ethyl acetate. The organic layer was separated and washed with brine, dried over Na2SO4, filtered and evaporated to dryness. The resulting mixture was purified via column chromatography using EA and hexanes as eluents to afford 106 mg compound 2. Yield 32%. 1H NMR (400 MHz, CDCl3) δ 7.11 (s, 4H), 4.48 – 4.26 (m, 1H), 3.21 – 3.04 (m, 1H), 2.32 – 2.11 (m, 1H), 1.66 – 1.59 (m, 1H), 1.53 (s, 9H); GC-MS m/z 179.0 (M-Boc).
Preparation of tert-butyl 2-(4-(tert-butoxycarbonyloxy)phenyl)cyclopropylcarbamate (CBB3001)
Compound 2 (106 mg) was stirred in a mixture of 15 mL i-PrOH and 3.8 mL 1N HCl (aq). Then Zn powder (247 mg) was added into the flask. The mixture was stirred at room temperature for 1 hr. The reaction was quenched with NaHCO3 (aq) and diluted with ethyl acetate. The organic layer was separated and washed with brine, dried over Na2SO4, filtered and evaporated to dryness. The residue was dissolved in 15 mL DCM and mixed with triethylamine (210 μL) and Boc2O (250 mg). The mixture was stirred at room temperature overnight. Subsequently, 10 mL water was added into the flask and the organic layer was separated, washed with 1N HCl (aq) x1, brine x 1, dried over Na2SO4, filtered and evaporated to dryness. The crude product was purified via column chromatography using EA and hexanes as eluents to afford 31 mg compound 3. Yield 23%.1H NMR (400 MHz, CDCl3) δ 7.14 (d, J = 8.4 Hz, 2H), 7.05 (d, J = 8.5 Hz, 2H), 4.85 (br s, 1H), 2.87 – 2.57 (m, 1H), 2.20 – 1.93 (m, 1H), 1.55 (s, 9H), 1.45 (s, 9H), 1.22 – 1.06 (m, 2H); LR-MS m/z 250.2 (M-Boc + H+).
4.3. Cells, tissue culture, and growth inhibitory assays
HCT116, PA-1, F9 and NIH3T3 cells were purchased from ATCC and cultured in RPMI-1640, DMEM, or MEM medium supplemented with 10% fetal bovine serum and 1% antibiotics (Invitrogen). The cells have been recently authenticated and tested based on the pseudo diploid genome for HCT116 cells and protein markers such as SOX2 or OCT4 in PA-1 and F9 cells and p53 and p21 in HCT116 and NIH3T3 cells. We used previous described procedures for growth inhibitory assays.11, 12 Active growing PA-1, F9, HCT116 or NIH3T3 cells (30–50% confluent) were treated with dimethylsulfoxide (DMSO, vehicle control) or with various concentrations of CBB3001 for 12–24 hours. Cells were examined and cell images were acquired with 10X10 lens of Nikon ECLIPSE Ti-S microscope equipped with NIS-Elements BR 3.1 software. Triplicated cells (technical repeats) were harvested by trypsin digestion, diluted, and blindly spotted onto a hemacytometer. Cells in four corners of the hemacytometer were counted and to obtain average cells per dish. The differences between control and CBB3001 treated cells were compared and plotted. Experiments were repeated for three independent times with the same conclusion and one example is shown.
4.4. In vitro demethylation assays
Human LSD1 cDNA were cloned into pGEXKG as a GST-LSD1 fusion protein. The plasmid was transformed into E coli BL21 strain that is deficient for protease activity to minimize the proteolytic degradation of the fusion protein. The GST-LSD1 was expressed and purified from bacteria using the glutathione Sepharose beads (GE Healthcare). The recombinant LSD1 protein was tested for demethylation assays using the di-methylated H3K4 peptide as previously reported.11 Methylated H3K4 peptides were incubated with recombinant LSD1 protein at 30 °C for 1 hour according to previously described methods.6 A typical 20 μl reaction contained 2 μg GST-LSD1, 1 μg methylated histone H3K4 peptides, and various concentrations of CBB3001 or tranylcypromine. The reaction products were analyzed by mass-spectrometry. The conversion efficiency of the di-methylated (Me2) H3K4 peptide by the demethylases to mono- (Me1) and non-methylated (Me0) H3K4 is calculated using the integrated product peak areas in an Orbitrap XL mass-spectrometry (Thermo Scientific) system using the following formula:11
The in vitro IC50 of CBB3001 for LSD1 inhibition was calculated using the conversion efficiency and the GraphPad Prism5 software as previously described.11
4.5. Chemical, antibodies, and Western blotting
ORY-1001 (RG-6016), tranylcypromine, and GSK2879552 were purchased from APExBio. Anti-LSD1 (A300-215A) and anti-SOX2 (A301-741 A) antibodies were from Bethyl Laboratories; Anti-mono-methylated (ab8895), di-methylated, and trimethylated (ab8580) K4 of histone H3 (ab32356), and histone H3 (ab1791) antibodies were obtained from Abcam. Log-phase growing cancer cells were treated with CBB3001 for indicated time and the cells were then directly lysed in a buffer containing 0.5% SDS. Proteins in the lysates were equalized and analyzed by Western blotting using antibodies against SOX2, OCT4, LSD1, H3K4Me1, H3K4Me2, H3K4me3 and histone H3, and other proteins as described previously.11
4.6. Small RNA Interference
Cells were transfected with 50 nM siRNAs using the DharmaFECT reagent 1 (Dharmacon) for 30–36 hours for mRNA analysis using quantitative real-time RT-PCR or for 48–60 hours for Western blotting as described previously11. The siRNA sequences are: human LSD1: GGAAGAAGAUAGUGAAAAC and human Sox2: CGCUCAUGAAGAAGGAUAA.
4.7. Statistical Information
Experiments were usually performed with at least three independent repeats (biological replicates) to ensure the results. For cell number assays, triplicated cell samples were used to determine and compare the statistically significant differences between means of control (DMSO) and CBB3001 treated cells using two-tailed paired t-test. Quantitative data are expressed by bar graph and standard deviations (S.D.) are expressed as mean and error bars. Different data sets were considered to be statistically significant when the P-value was <0.05 (*), 0.01 (**) or 0.001 (***).52
Acknowledgments
This work was supported by grants from National Institutes of Health (R15GM116087 to HZ) and from the Cancer Research Gift Fund to the College of Sciences at University of Nevada, Las Vegas (HZ).
Footnotes
Competing Financial Interests
The authors declare there are no competing financial interests in this research work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Shi Y. Nat Rev Genet. 2007;8:829. doi: 10.1038/nrg2218. [DOI] [PubMed] [Google Scholar]
- 2.Klose RJ, Zhang Y. Nat Rev Mol Cell Biol. 2007;8:307. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- 3.Agger K, Christensen J, Cloos PA, Helin K. Curr Opin Genet Dev. 2008;18:159. doi: 10.1016/j.gde.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, Roeder RG, Brivanlou AH, Allis CD. Cell. 2005;121:859. doi: 10.1016/j.cell.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 5.Ang YS, Tsai SY, Lee DF, Monk J, Su J, Ratnakumar K, Ding J, Ge Y, Darr H, Chang B, Wang J, Rendl M, Bernstein E, Schaniel C, Lemischka IR. Cell. 2011;145:183. doi: 10.1016/j.cell.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA. Cell. 2004;119:941. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 7.Forneris F, Binda C, Battaglioli E, Mattevi A. Trends Biochem Sci. 2008;33:181. doi: 10.1016/j.tibs.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Culhane JC, Cole PA. Curr Opin Chem Biol. 2007;11:561. doi: 10.1016/j.cbpa.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, Gaudet F, Li E, Chen T. Nat Genet. 2009;41:125. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- 10.Shilatifard A. Curr Opin Cell Biol. 2008;20:341. doi: 10.1016/j.ceb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Lu F, Ren Q, Sun H, Xu Z, Lan R, Liu Y, Ward D, Quan J, Ye T, Zhang H. Cancer Research. 2011;71:7238. doi: 10.1158/0008-5472.CAN-11-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X, Lu F, Wang J, Yin F, Xu Z, Qi D, Wu X, Cao Y, Liang W, Liu Y, Sun H, Ye T, Zhang H. Cell reports. 2013;5:445. doi: 10.1016/j.celrep.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Z, Li S, Song W, Li X, Li Q, Zhang Z, Han Y, Zhang X, Miao S, Du R, Wang L. PLoS One. 2013;8:e70077. doi: 10.1371/journal.pone.0070077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao ZK, Dong P, Gu J, Chen L, Zhuang M, Lu WJ, Wang DR, Liu YB. Tumour Biol. 2013;34:173. doi: 10.1007/s13277-012-0525-x. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, Liang J, Sun L, Yang X, Shi L, Li R, Li Y, Zhang Y, Li Q, Yi X, Shang Y. Cell. 2009;138:660. doi: 10.1016/j.cell.2009.05.050. [DOI] [PubMed] [Google Scholar]
- 16.Serce N, Gnatzy A, Steiner S, Lorenzen H, Kirfel J, Buettner R. BMC clinical pathology. 2012;12:13. doi: 10.1186/1472-6890-12-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagasawa S, Sedukhina AS, Nakagawa Y, Maeda I, Kubota M, Ohnuma S, Tsugawa K, Ohta T, Roche-Molina M, Bernal JA, Narvaez AJ, Jeyasekharan AD, Sato K. PLoS One. 2015;10:e0118002. doi: 10.1371/journal.pone.0118002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X, Song Y. PLoS One. 2012;7:e35065. doi: 10.1371/journal.pone.0035065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao ZK, Yu HF, Wang DR, Dong P, Chen L, Wu WG, Ding WJ, Liu YB. World J Gastroenterol. 2012;18:6651. doi: 10.3748/wjg.v18.i45.6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jie D, Zhongmin Z, Guoqing L, Sheng L, Yi Z, Jing W, Liang Z. Dig Dis Sci. 2013;58:1581. doi: 10.1007/s10620-012-2552-2. [DOI] [PubMed] [Google Scholar]
- 21.Yu Y, Wang B, Zhang K, Lei Z, Guo Y, Xiao H, Wang J, Fan L, Lan C, Wei Y, Ma Q, Lin L, Mao C, Yang X, Chen X, Li Y, Bai Y, Chen D. Biochem Biophys Res Commun. 2013;437:192. doi: 10.1016/j.bbrc.2013.05.123. [DOI] [PubMed] [Google Scholar]
- 22.Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R, Kirfel J. Carcinogenesis. 2010;31:512. doi: 10.1093/carcin/bgp324. [DOI] [PubMed] [Google Scholar]
- 23.Yin F, Lan R, Zhang X, Zhu L, Chen F, Xu Z, Liu Y, Ye T, Sun H, Lu F, Zhang H. Mol Cell Biol. 2014;34:158. doi: 10.1128/MCB.00631-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, Ramos AH, Woo MS, Weir BA, Getz G, Beroukhim R, O’Kelly M, Dutt A, Rozenblatt-Rosen O, Dziunycz P, Komisarof J, Chirieac LR, Lafargue CJ, Scheble V, Wilbertz T, Ma C, Rao S, Nakagawa H, Stairs DB, Lin L, Giordano TJ, Wagner P, Minna JD, Gazdar AF, Zhu CQ, Brose MS, Cecconello I, UR, Marie SK, Dahl O, Shivdasani RA, Tsao MS, Rubin MA, Wong KK, Regev A, Hahn WC, Beer DG, Rustgi AK, Meyerson M. Nat Genet. 2009;41:1238. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hussenet T, Dali S, Exinger J, Monga B, Jost B, Dembele D, Martinet N, Thibault C, Huelsken J, Brambilla E, du Manoir S. PLoS One. 2010;5:e8960. doi: 10.1371/journal.pone.0008960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maier S, Wilbertz T, Braun M, Scheble V, Reischl M, Mikut R, Menon R, Nikolov P, Petersen K, Beschorner C, Moch H, Kakies C, Protzel C, Bauer J, Soltermann A, Fend F, Staebler A, Lengerke C, Perner S. Hum Pathol. 2011;42:1078. doi: 10.1016/j.humpath.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 27.Tam WL, Ng HH. Cancer Cell. 2014;26:3. doi: 10.1016/j.ccr.2014.06.024. [DOI] [PubMed] [Google Scholar]
- 28.Weina K, Utikal J. Clinical and translational medicine. 2014;3:19. doi: 10.1186/2001-1326-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leis O, Eguiara A, Lopez-Arribillaga E, Alberdi MJ, Hernandez-Garcia S, Elorriaga K, Pandiella A, Rezola R, Martin AG. Oncogene. 2012;31:1354. doi: 10.1038/onc.2011.338. [DOI] [PubMed] [Google Scholar]
- 30.Matsuoka J, Yashiro M, Sakurai K, Kubo N, Tanaka H, Muguruma K, Sawada T, Ohira M, Hirakawa K. The Journal of surgical research. 2012;174:130. doi: 10.1016/j.jss.2010.11.903. [DOI] [PubMed] [Google Scholar]
- 31.Sholl LM, Barletta JA, Yeap BY, Chirieac LR, Hornick JL. Am J Surg Pathol. 2010;34:1193. doi: 10.1097/PAS.0b013e3181e5e024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, Ciceri F, Blaser JG, Greystoke BF, Jordan AM, Miller CJ, Ogilvie DJ, Somervaille TC. Cancer Cell. 2012;21:473. doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 33.Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, Klein HU, Popescu AC, Burnett A, Mills K, Casero RA, Jr, Marton L, Woster P, Minden MD, Dugas M, Wang JC, Dick JE, Muller-Tidow C, Petrie K, Zelent A. Nat Med. 2012;18:605. doi: 10.1038/nm.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang M, Culhane JC, Szewczuk LM, Gocke CB, Brautigam CA, Tomchick DR, Machius M, Cole PA, Yu H. Nat Struct Mol Biol. 2007;14:535. doi: 10.1038/nsmb1255. [DOI] [PubMed] [Google Scholar]
- 35.Szewczuk LM, Culhane JC, Yang M, Majumdar A, Yu H, Cole PA. Biochemistry. 2007;46:6892. doi: 10.1021/bi700414b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benelkebir H, Hodgkinson C, Duriez PJ, Hayden AL, Bulleid RA, Crabb SJ, Packham G, Ganesan A. Bioorganic & medicinal chemistry. 2011;19:3709. doi: 10.1016/j.bmc.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 37.Gooden DM, Schmidt DM, Pollock JA, Kabadi AM, McCafferty DG. Bioorg Med Chem Lett. 2008;18:3047. doi: 10.1016/j.bmcl.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, Ora I, Pajtler K, Klein-Hitpass L, Kuhfittig-Kulle S, Metzger E, Schule R, Eggert A, Buettner R, Kirfel J. Cancer Res. 2009;69:2065. doi: 10.1158/0008-5472.CAN-08-1735. [DOI] [PubMed] [Google Scholar]
- 39.Stavropoulos P, Hoelz A. Expert Opin Ther Targets. 2007;11:809. doi: 10.1517/14728222.11.6.809. [DOI] [PubMed] [Google Scholar]
- 40.Neelamegam R, Ricq EL, Malvaez M, Patnaik D, Norton S, Carlin SM, Hill IT, Wood MA, Haggarty SJ, Hooker JM. ACS chemical neuroscience. 2012;3:120. doi: 10.1021/cn200104y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corey EJ, Chaykovsky J Am Chem Soc. 1962;84:3782. [Google Scholar]
- 42.Corey EJ, Chaykovsky J Am Chem Soc. 1965;87:1353. [Google Scholar]
- 43.Ciaccio JAaA, CE Synthetic communications. 2006;36:1333. [Google Scholar]
- 44.Adamo A, Barrero MJ, Izpisua Belmonte JC. Cell Cycle. 2011;10:3215. doi: 10.4161/cc.10.19.17052. [DOI] [PubMed] [Google Scholar]
- 45.Adamo A, Sese B, Boue S, Castano J, Paramonov I, Barrero MJ, Izpisua Belmonte JC. Nat Cell Biol. 2011;13:652. doi: 10.1038/ncb2246. [DOI] [PubMed] [Google Scholar]
- 46.Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, Young RA. Nature. 2012;482:221. doi: 10.1038/nature10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Culhane JC, Wang D, Yen PM, Cole PA. J Am Chem Soc. 2010;132:3164. doi: 10.1021/ja909996p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, Schneck JL, Carson JD, Liu Y, Butticello M, Bonnette WG, Gorman SA, Degenhardt Y, Bai Y, McCabe MT, Pappalardi MB, Kasparec J, Tian X, McNulty KC, Rouse M, McDevitt P, Ho T, Crouthamel M, Hart TK, Concha NO, McHugh CF, Miller WH, Dhanak D, Tummino PJ, Carpenter CL, Johnson NW, Hann CL, Kruger RG. Cancer Cell. 2015;28:57. doi: 10.1016/j.ccell.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 49.Maes T, Tirapu I, Mascaró C, Ortega A, Estiarte A, Valls N, Castro-Palomino J, Arjol CB, Kurz G, Genomics O, de Llobregat C. 2013 ASCO Annual Meeting. 2013:e13543. Abstract#. [Google Scholar]
- 50.Seo EJ, Kim DK, Jang IH, Choi EJ, Shin SH, Lee SI, Kwon SM, Kim KH, Suh DS, Kim JH. Oncotarget. 2016;7:55624. doi: 10.18632/oncotarget.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Culhane JC, Szewczuk LM, Liu X, Da G, Marmorstein R, Cole PA. J Am Chem Soc. 2006;128:4536. doi: 10.1021/ja0602748. [DOI] [PubMed] [Google Scholar]
- 52.Fay DS, Gerow K. WormBook: the online review of C. elegans biology. 2013. p. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]