

 Tyrosinase is a key enzyme in melanin biosynthesis, and is also involved in the enzymatic browning of plant-derived foods.
Tyrosinase is a key enzyme in melanin biosynthesis, and is also involved in the enzymatic browning of plant-derived foods.
Abstract
Tyrosinase is a key enzyme in melanin biosynthesis, and is also involved in the enzymatic browning of plant-derived foods. Tyrosinase inhibitors are very important in medicine, cosmetics and agriculture. In order to develop more active and safer tyrosinase inhibitors, an efficient approach is to modify natural product scaffolds. In this work, two series of novel tyrosinase inhibitors were designed and synthesized by the esterification of cinnamic acid derivatives with paeonol or thymol. Their inhibitory effects on mushroom tyrosinase were evaluated. Most of these compounds (IC50: 2.0 to 163.8 μM) are found to be better inhibitors than their parent compounds (IC50: 121.4 to 5925.0 μM). Among them, (E)-2-acetyl-5-methoxyphenyl-3-(4-hydroxyphenyl)acrylate (5a), (E)-2-acetyl-5-methoxyphenyl-3-(4-methoxyphenyl)acrylate (5g) and (E)-2-isopropyl-5-methylphenyl-3-(4-hydroxyphenyl)acrylate (6a) showed strong inhibitory activities; the IC50 values were 2.0 μM, 8.3 μM and 10.6 μM, respectively, compared to the positive control, kojic acid (IC50: 32.2 μM). Analysis of the inhibition mechanism of 5a, 5g and 6a demonstrated that their inhibitory effects on tyrosinase are reversible. The inhibition kinetics, analyzed by Lineweaver–Burk plots, revealed that 5a acts as a non-competitive inhibitor while 5g and 6a are mixed-type inhibitors. Furthermore, docking experiments were carried out to study the interactions between 6a and mushroom tyrosinase.
Introduction
Tyrosinase (EC 1.14.18.1) is a multifunctional copper-containing oxidase widely distributed in mammals, plants, microorganisms and insects.1–4 The enzyme is mainly involved in two distinct reactions of melanin synthesis: the hydroxylation of tyrosine by monophenolase and the oxidation of 3,4-dihydroxyphenylalanine (l-dopa) to o-dopaquinone by diphenolase.5 Although melanin production in human skin is a major defense mechanism against UV light, excessive accumulation of epidermal pigmentation can cause various hyperpigmentation disorders, such as melasma, freckles and melanosis.1,6–10 Therefore, tyrosinase inhibitors have become increasingly researched for their application in medicinal and cosmetic products for the prevention or treatment of pigmentation disorders.1,11,12
Apart for the treatment of pigmentation disorders, tyrosinase inhibitors are also applied in insect control and in the food industry. Tyrosinase has several roles in the developmental and defensive functions of insects. It is involved in melanogenesis, wound healing, parasite encapsulation and sclerotization in insects. The development of tyrosinase inhibitors has become an alternative approach for controlling insect pests.2,13,14 In the food industry, tyrosinase is very important in the control of the quality and the economics of fruit and vegetables. Tyrosinase catalyzes the oxidation of phenolic compounds to the corresponding quinones and is responsible for the enzymatic browning of fruit and vegetables. Moreover, the quinone compounds produced in the browning reaction may react with the amino and sulfhydryl groups of proteins irreversibly, which may decrease the digestibility of the protein and the bioavailability of essential amino acids, including lysine and cysteine.2,15 Therefore, tyrosinase inhibitors are also of great interest to the agricultural and food fields.
A large number of naturally occurring and synthetic tyrosinase inhibitors have been reported.1–3,7,10,16–23 However, some well-known inhibitors, such as hydroquinone and kojic acid, are not potent enough to be put into practical use, because of their instability and undesirable side-effects, such as cytotoxicity, skin cancer, dermatitis, and neurodegenerative diseases.19,20,24,25 These problems prompt us to search for safer, more active and stable tyrosinase inhibitors from natural sources.
Many compounds from natural sources have been found to inhibit tyrosinase with moderate activities.11,26 For example, cinnamic acid (Fig. 1) is a natural product obtained from the oil of cinnamon, or from balsams such as styrax. It is reported to have depigmenting ability and effects on the expression of melanin biosynthesis-related enzymes.27,28 Paeonol (Fig. 1), one of the primary bioactive components isolated from the Chinese national flower, the peony, is known as a traditional skin-whitening agent, a potential tyrosinase inhibitor.24 Thymol (Fig. 1) is a naturally occurring monoterpene phenol that is the main constituent of thyme, an herb of the genus Thymus, and of origanum oils. It is reported that thymol can inhibit the oxidation of l-dopa to dopaquinone.29
Fig. 1. Design of novel tyrosinase inhibitors based on natural product scaffolds.

In the search for tyrosinase inhibitors, cinnamic acid and its derivatives have received much attention.27 Our previous studies proved that the substituent in the 4-position of the benzene ring of cinnamic acid is important for the tyrosinase inhibitory activity, while the carboxyl group has no significant effect on the activity. Hence, to develop original, natural and potent tyrosinase inhibitors, cinnamic acid was used as a lead structure. Several cinnamic acid derivatives with different substituents in the 4-position were coupled with paeonol or thymol by esterification, producing two series of cinnamic acid esters (Fig. 1). The compounds synthesized were screened as mushroom tyrosinase inhibitors, and the mechanism of their action was explored. Moreover, computational analyses were performed to highlight their interactions within the catalytic binding site.
Results and discussion
Synthetic procedures
The cinnamic acid esters were prepared according to the general Scheme 1. There are two synthetic routes: 1) using 4-hydroxy cinnamic acid (1a) or 4-hydroxy-3-methoxycinnamic acid (1b) as starting material. Firstly, TBDMSCl was used to protect the hydroxyl group under alkaline conditions. However, since TBDMSCl could react with the hydroxyl and carboxyl groups at the same time, saturated K2CO3 was added to hydrolyze the silyl ester to furnish 2a–b. Secondly, acids 2a–b reacted with SOCl2 to produce the acyl chloride, which was condensed with paeonol or thymol to produce esters 3a–b or 4a–b. Finally, the TBDMS group was removed from 3a–b or 4a–b by 2 M HF solution, giving the final products 5a–b or 6a–b. 2) Using the cinnamic acid derivatives 7c–g as starting material, the esterification of 7c–g with paeonol or thymol in the presence of EDCI and HOBt gives the target products 5c–g or 6c–g directly.
Scheme 1. The synthetic route to novel tyrosinase inhibitors. Reagents and conditions: (a) i. TBDMSCl, DIEA, DCM, 0 °C to rt., 20 h; ii. K2CO3, MeOH/THF, rt., 3 h; (b) i. SOCl2, DCM, reflux, 5 h; ii. paeonol or thymol, NEt3, DCM, 0 °C to rt., overnight; (c) 2 M HF solution, THF, reflux., 1 h; (d) paeonol or thymol, EDCI, HOBt, DIEA, DCM, 0 °C to rt., overnight.

Tyrosinase inhibition evaluation
The inhibitory activity of compounds 5a–g and 6a–g on the diphenolase activity of mushroom tyrosinase was investigated using l-dopa as the substrate. The spectrophotometric assay for tyrosinase was performed according to the method reported by Liu et al.30 with some slight modifications. The inhibition percentage was calculated from the equation: inhibition ratio (%) = [(B – S)/B] × 100, where B and S are the absorbances for the blank and samples.
Firstly, the inhibitory activities of all synthesized compounds were screened at a concentration of 100 μM. Among the 14 compounds, 5a, 5c, 5e, 5f, 5g and 6a showed the best activities, with inhibition ratios of more than 40%, while those of the other compounds (5b, 5d, 6b, 6c, 6d, 6e, 6f and 6g) were below 40%. Then, the inhibition rates of compounds 5a, 5c, 5e, 5f, 5g and 6a were tested at more than 8 concentrations. The IC50 values were calculated by Graphpad Prism 7.0 software; the results are summarized in Table 1. For comparison with the activities of the parent structures, the IC50 values of cinnamic acid, 4-hydroxyl cinnamic acid, paeonol and thymol were evaluated under the same conditions. The parent compounds showed moderate inhibitory activities with IC50 values from 121.4 to 5925.0 μM, while the new compounds exhibited good activities with IC50 values from 2.0 to 163.8 μM. Let us consider 5a, prepared by coupling 4-hydroxycinnamic acid with paeonol. The inhibitory activity of 5a is much better than those of its parent structures. The results indicate that the inhibitory activities of the compounds synthesized are significantly improved by combining the two pharmacophores.
Table 1. Tyrosinase inhibitory activities of new compounds.
| Compounds | R1 | R2 | IC50 value (μM) |
| 5a | H | OH | 2.0 |
| 5b | OCH3 | OH | >100 a |
| 5c | H | OCH3 | 77.4 |
| 5d | H | H | >100 a |
| 5e | H | CH3 | 39.0 |
| 5f | H | Cl | 163.8 |
| 5g | H | F | 8.3 |
| 6a | H | OH | 10.6 |
| 6b | OCH3 | OH | >100 a |
| 6c | H | OCH3 | >100 a |
| 6d | H | H | >100 a |
| 6e | H | CH3 | >100 a |
| 6f | H | Cl | >100 a |
| 6g | H | F | >100 a |
| Cinnamic acid | 209.5 | ||
| 4-Hydroxyl cinnamic acid | 4708.5 | ||
| Paeonol | 121.4 | ||
| Thymol | 5925.0 | ||
| Kojic acid | 32.2 |
aThe inhibitory percentage of these compounds at 100 μM is less than 40%.
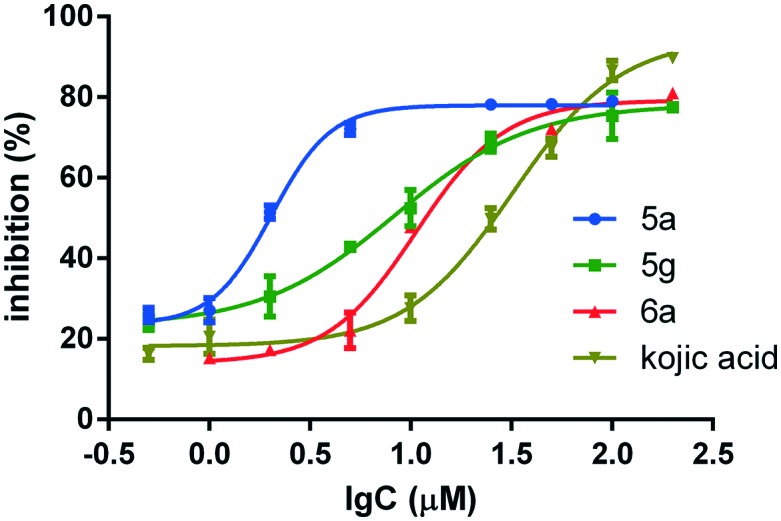
The structural frameworks of series 5a–g and 6a–g are the same, and the main differences between the two series are the substituents on the benzene ring of the phenol moiety. Generally speaking, the paeonol derivatives 5a–g are more active than the thymol derivatives 6a–g; this may be due to the formation of hydrogen bonds between the methoxy and carbonyl groups of 5a–g and tyrosinase. Furthermore, the substituents in the 4-position have large effects on the tyrosinase inhibition activities. The hydroxyl group is much more effective. For example, compounds 5a and 6a exhibit strong inhibition of tyrosinase, with IC50 values of 2.0 and 10.6 μM, respectively (Fig. 2). They are 16 and 3 times more active than kojic acid, which was used as a positive reference. However, the activities fall significantly when methoxyl groups are introduced into the 3-position, as in compounds 5b and 6b. Apart from the hydroxyl group, 4-fluoro also enhances tyrosinase inhibition activity. Compound 5g showed strong inhibition of tyrosinase with an IC50 value of 8.3 μM.
Fig. 2. Tyrosinase inhibition of 5a, 5g, 6a and kojic acid.
Mechanism study
The inhibitory mechanism of 5a, 5g and 6a on mushroom tyrosinase was investigated using l-dopa as the substrate. The relationship between enzyme activity and its concentration in the presence of compounds 5a, 5g and 6a was studied. Plots of the remaining enzyme activity versus the enzyme concentration at different inhibitor concentrations give a set of straight lines, which all pass through the origin (Fig. 3). Increasing the inhibitor concentration reduces the slope of the line, indicating that the inhibitor reduces the activity of the enzyme. Thus, the inhibition by compounds 5a, 5g and 6a of the diphenolase activity of tyrosinase is reversible.
Fig. 3. Determination of the mechanism of the inhibition of mushroom tyrosinase by 5a, 5g and 6a.

The kinetic data on the inhibition of l-dopa oxidation by 5a, 5g and 6a were expressed by Lineweaver–Burk double-reciprocal plots. For 5a, the plots of 1/v versus 1/[S] give a group of straight lines with different slopes that intersect at the same point on the x-axis (Fig. 4-A1), suggesting that the Km remains the same, while Vmax decreases with increasing concentrations of 5a. The results indicate that the inhibition type of 5a is non-competitive, which means that the binding site of 5a with tyrosinase is not the active site. 5a reduces the activity of tyrosinase and binds equally well to the enzyme whether or not it is already bound the substrate. For 5g and 6a, all the straight lines intercept in the second quadrant (Fig. 4-B1 and C1), indicating that compounds 5g and 6a are both mixed-type inhibitors. This suggests that they bind not only with free enzyme but also with the enzyme–substrate complex. This complex could prevent the free oxygen molecule taking part in the oxidation of o-diphenols and, thus, cause tyrosinase to lose its catalytic ability. The equilibrium constant for binding of the inhibitor with free enzyme (KI) was obtained from a plot of the slope (Km/Vm) versus the inhibitor concentration (Fig. 4-B2 and C2), and for binding with the enzyme–substrate complex (KIS) from a plot of the vertical intercept (1/Vm) versus the inhibitor concentration (Fig. 4-A3, B3 and C3). The results are shown in Table 2. The KIS values of 5g and 6a are larger than their KI values, indicating that the affinity of the inhibitors for the free enzyme is greater than that for the enzyme–substrate complex.
Fig. 4. Lineweaver–Burk plots (A1, B1 and C1) for mushroom tyrosinase with l-dopa as substrate in the presence of 5a, 5g and 6a. B2 and C2 represent the plot of slope versus the concentration of 5g and 6a for the determination of the inhibition constant KI. A3, B3 and C3 represent the plot of intercept versus the concentration of 5a, 5g and 6a for the determination of the inhibition constant KIS.

Table 2. Type of inhibition mechanism, as well as KI and KIS values of 5a, 5g and 6a.
| Comp. | Inhibition mechanism | K I value (μM) | K IS value (μM) |
| 5a | Non-competitive type | 3.8 | 3.8 |
| 5g | Mixed type | 10.0 | 35.6 |
| 6a | Mixed type | 8.0 | 72.2 |
Molecular docking
As 6a is much more specific for the active site of the enzyme, molecular docking was introduced to predict the binding conformation (Fig. 5). The tyrosinase enzyme structure was prepared by the VMD31 and NAMD232 programs. All the atoms were assigned to the CHARMM force field type. Hydrogens were then optimized. The ligand was prepared with the Avogadro program, and was assigned with Gasteiger charge. The low-energy conformation was obtained by the steepest decent algorithm with the MMFF94 force field. Ledock was used for the docking. In order to validate the docking protocol, the known tyrosinase inhibitors, tropolone and kojic acid (KA) were also docked to the protein. As the result shows (Fig. 5A), the docked conformation of tropolone is well superposed to the conformation in the crystal structure (RMSD < 1.0 Å). In addition, KA was interacted with the two copper ions. An π–π interaction was found between KA and His263; other Van der Waals interactions, with Met280, Ser282, Val283, were also formed (Fig. 5B). These interaction characters are quite similar to that of KA binding to tyrosinase of Bacillus megaterium.33 Then the docking protocol was also subjected to 6a. This reveals that 6a also interacts with copper ions through polar contacts. Hydrophobic interactions with Phe264, Leu275, Pro277, and Val283 are also formed. The ester group interacts with Arg268 through electrostatic attraction (Fig. 5C).
Fig. 5. The binding conformation of tropolone (A), KA (B) or 6a (C) complexed with tyrosinase (PDB ID: ; 2Y9X).34 The conformation was obtained by the Ledock program. The docked tropolone conformation is in green, and the original conformation in crystal structure is in cyan (A). Protein and KA (or 6a) were colored in cyan and green, respectively. The dashed lines show the copper ions interact with ligands. The presentation was prepared with the VMD program.

The kinetics study showed that 5a is a non-competitive inhibitor, while 6a is a mixed-type inhibitor. The docking study of 5a with tyrosinase was also performed to try to explain this difference. The results reveal that the conformation of the cinnamic acid moiety depends on the phenol moiety. The hydroxyl group of 5a does not insert into the active site well. Consequently, 5a does not interact with the active site of tyrosinase directly, which is in agreement with the results of the kinetics study.
Experimental section
General procedure for synthesis of compounds 2a and 2b
tert-Butyldimethylsilyl chloride (9.04 g, 60.0 mmol) was added to a suspension of 1a or 1b (20.0 mmol) in 50 mL DCM at 0 °C, and then DIEA was added to adjust the pH value to 9–10. The mixture was stirred at room temperature for 20 hours. The mixture was washed with 1 M HCl solution, saturated NaHCO3 and brine. The organic layer was dried over MgSO4, and then concentrated under reduced pressure to give a bright yellow oil as an intermediate. This was dissolved in MeOH/THF (6.0 mL, 1/2) in a round-bottomed flask and stirred at room temperature with saturated K2CO3 for 3 hours. The resulting solution was evaporated to dryness. The residue was diluted with DCM, and washed with 1 M HCl solution, saturated NaHCO3 and brine. The organic layer was dried over MgSO4, and concentrated to give a crude product, which was purified by flash chromatography to give 2a or 2b as a white powder (yield: 64% or 72%).
General procedure for synthesis of compounds 3a–b and 4a–b
SOCl2 (800 μL, 11.0 mmol) was added to a solution of 2a or 2b (1.0 mmol) in DCM (5 mL), and the mixture was refluxed for 5 hours. Then the solution was heated to 90 °C for evaporation. After cooling to room temperature, the residue was dissolved in dry DCM (5 mL). Paeonol or thymol (1.0 mmol) was added at 0 °C, and then NEt3 (1 mL, 7.2 mmol). The mixture was stirred at room temperature overnight. The reaction mixture was washed with 0.1 M HCl solution, saturated NaHCO3 and brine. The organic layer was dried over MgSO4, and concentrated to give a crude product, which was purified by chromatography to give 3a–b or 4a–b as a light yellow oil (yield: 50–64%).
General procedure for synthesis of compounds 5a–b and 6a–b
2 M HF solution (4.0 mL) was added to a solution of 3a–b or 4a–b (0.19 mmol) in THF (4.0 mL), and the mixture was refluxed for 1 hour. The reaction mixture was washed with saturated NaHCO3, 0.1 M HCl solution, H2O and brine. The organic layer was dried over MgSO4, and concentrated to give a crude product, which was purified by chromatography to give 5a–b or 6a–b as a pale yellow powder (yield: 70–82%).
General procedure for synthesis of compounds 5c–g and 6c–g
0 °C, to a solution of 7c–g (2.0 mmol) in dry DCM (5 mL) was added paeonol or thymol (2.2 mmol), EDCI (575 mg, 3.00 mmol), HOBt (405 mg, 3.00 mmol), and then DIEA was added to adjust the pH value of the solution to 9–10. The mixture was stirred at room temperature overnight, then washed with 0.1 M HCl, saturated NaHCO3 and brine. The organic layer was dried over MgSO4, and concentrated to give a crude product, which was purified by chromatography to give 5c–g or 6c–g as a grey powder (yield: 72–85%).
(E)-3-(4-(tert-Butyldimethylsilyloxy)phenyl)acrylic acid (2a)
1H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 15.9 Hz, 1H), 7.45 (d, J = 8.6 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 6.31 (d, J = 15.9 Hz, 1H), 0.99 (s, 9H), 0.23 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 172.66, 158.45, 146.94, 130.20, 127.54, 120.74, 115.02, 25.77, 18.40, –4.23; MS (ESI): m/z 277.20 [M – H]–, 555.53 [2 M – H]–.
(E)-3-(4-(tert-Butyldimethylsilyloxy)-3-methoxyphenyl)acrylic acid (2b)
1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 15.8 Hz, 1H), 7.09–6.99 (m, 2H), 6.86 (d, J = 8.4 Hz, 1H), 6.31 (d, J = 15.9 Hz, 1H), 3.85 (s, 3H), 1.00 (s, 9H), 0.18 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 172.54, 151.42, 148.19, 147.29, 128.10, 122.87, 121.29, 115.02, 111.24, 55.62, 25.80, 18.63, –4.44; MS (ESI): m/z 307.48 [M – H]–.
(E)-2-Acetyl-5-methoxyphenyl-3-(4-(tert-butyldimethylsilyloxy)-phenyl)acrylate (3a)
1H NMR (400 MHz, CDCl3) δ 7.91–7.78 (m, 2H), 7.50 (d, J = 8.5 Hz, 2H), 6.92–6.80 (m, 3H), 6.68 (d, J = 2.1 Hz, 1H), 6.54 (d, J = 15.9 Hz, 1H), 3.86 (s, 3H), 2.52 (s, 3H), 1.00 (s, 9H), 0.24 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 196.03, 165.57, 163.83, 158.59, 151.74, 147.29, 132.34, 130.30, 127.55, 124.00, 120.77, 114.58, 112.03, 109.25, 55.86, 29.83, 25.77, 18.41, –4.21; MS (APCI): m/z 449.57 (M + Na)+.
(E)-2-Acetyl-5-methoxyphenyl-3-(4-(tert-butyldimethylsilyloxy)-3-methoxyphenyl)acrylate (3b)
1H NMR (300 MHz, CDCl3) δ 7.89–7.79 (m, 2H), 7.12–7.07 (m, 2H), 6.90–6.81 (m, 2H), 6.67 (d, J = 2.5 Hz, 1H), 6.54 (d, J = 15.9 Hz, 1H), 3.88–3.83 (m, 6H), 2.52 (s, 3H), 1.00 (s, 9H), 0.18 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 196.07, 165.58, 163.82, 151.70, 151.40, 148.26, 147.63, 132.38, 128.07, 123.86, 123.01, 121.29, 114.57, 111.98, 111.18, 109.27, 55.86, 55.61, 29.80, 25.79, 18.64, –4.44; MS (APCI): m/z 479.57 (M + Na)+.
(E)-2-Isopropyl-5-methylphenyl-3-(4-(tert-butyldimethylsilyloxy)-phenyl)acrylate (4a)
1H NMR (300 MHz, CDCl3) δ 7.83 (d, J = 15.9 Hz, 1H), 7.50 (d, J = 8.6 Hz, 2H), 7.22 (d, J = 7.9 Hz, 1H), 7.04 (d, J = 7.9 Hz, 1H), 6.92–6.84 (m, 3H), 6.53 (d, J = 15.9 Hz, 1H), 3.03 (m, 1H), 2.33 (s, 3H), 1.21 (d, J = 6.9 Hz, 6H), 1.00 (s, 9H), 0.24 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 166.16, 158.36, 148.16, 146.30, 137.37, 136.68, 130.11, 127.65, 127.19, 126.56, 123.00, 120.76, 115.01, 27.30, 25.77, 23.23, 21.02, 18.41, –4.21; MS (APCI): m/z 411.34 (M + H)+.
(E)-2-Isopropyl-5-methylphenyl-3-(4-(tert-butyldimethylsilyloxy)-3-methoxyphenyl)acrylate (4b)
1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 15.9 Hz, 1H), 7.22 (d, J = 7.9 Hz, 1H), 7.14–7.06 (m, 2H), 7.04 (d, J = 7.8 Hz, 1H), 6.92–6.84 (m, 2H), 6.52 (d, J = 15.9 Hz, 1H), 3.87 (s, 3H), 3.09–2.99 (m, 1H), 2.33 (s, 3H), 1.21 (d, J = 6.9 Hz, 6H), 1.01 (s, 9H), 0.19 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 166.11, 151.47, 148.21, 148.12, 146.65, 137.41, 136.69, 128.26, 127.20, 126.59, 123.02, 122.76, 121.34, 115.13, 111.18, 55.65, 27.32, 25.81, 23.24, 21.01, 18.65, –4.43; MS (APCI): m/z 441.27 [M + H]+.
(E)-2-Acetyl-5-methoxyphenyl-3-(4-hydroxyphenyl)acrylate (5a)
1H NMR (400 MHz, CDCl3) δ 7.90–7.77 (m, 2H), 7.45 (d, J = 14.9 Hz, 2H), 6.91–6.81 (m, 3H), 6.69 (d, J = 2.5 Hz, 1H), 6.50 (d, J = 15.9 Hz, 1H), 5.93 (brs, 1H), 3.87 (s, 3H), 2.54 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 197.37, 165.82, 164.10, 158.94, 151.78, 147.42, 132.58, 130.56, 126.60, 123.59, 116.22, 113.95, 112.00, 109.48, 55.91, 29.49; MS (ESI): m/z 311.32 [M – H]–.
(E)-2-Acetyl-5-methoxyphenyl-3-(4-hydroxy-3-methoxyphenyl)-acrylate (5b)
1H NMR (400 MHz, CDCl3) δ 7.86 (d, J = 8.8 Hz, 1H), 7.82 (d, J = 15.9 Hz, 1H), 7.14 (dd, J = 8.2, 1.8 Hz, 1H), 7.10 (d, J = 1.8 Hz, 1H), 6.94 (d, J = 8.1 Hz, 1H), 6.84 (dd, J = 8.8, 2.5 Hz, 1H), 6.68 (d, J = 2.5 Hz, 1H), 6.52 (d, J = 15.9 Hz, 1H), 6.02 (s, 1H), 3.94 (s, 3H), 3.86 (s, 3H), 2.52 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.18, 165.60, 163.84, 151.69, 148.66, 147.61, 146.99, 132.39, 126.80, 123.85, 114.96, 114.24, 111.97, 109.75, 109.31, 56.13, 55.86, 29.73; MS (ESI): m/z 341.05 [M – H]–.
(E)-2-Acetyl-5-methoxyphenyl-3-(4-methoxyphenyl)acrylate (5c)
1H NMR (400 MHz, CDCl3) δ 7.87 (s, 1H), 7.84 (d, J = 6.7 Hz, 1H), 7.55 (d, J = 8.7 Hz, 2H), 6.93 (d, J = 8.7 Hz, 2H), 6.84 (dd, J = 8.8, 2.4 Hz, 1H), 6.68 (d, J = 2.5 Hz, 1H), 6.54 (d, J = 15.9 Hz, 1H), 3.85 (s, 6H), 2.52 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 195.97, 165.53, 163.79, 162.00, 151.70, 147.16, 132.29, 130.31, 126.94, 123.94, 114.56, 114.34, 111.96, 109.24, 55.81, 55.52, 29.76; MS (APCI): m/z 349.39 (M + Na)+.
2-Acetyl-5-methoxyphenyl-cinnamate (5d)
1H NMR (400 MHz, CDCl3) δ 7.95–7.82 (m, 2H), 7.65–7.55 (m, 2H), 7.48–7.37 (m, 3H), 6.85 (dd, J = 8.8, 2.5 Hz, 1H), 6.75–6.63 (m, 2H), 3.85 (s, 3H), 2.52 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 195.84, 165.20, 163.78, 151.51, 147.35, 134.15, 132.35, 130.91, 129.05, 128.50, 123.71, 117.01, 111.93, 109.25, 55.79, 29.58; MS (APCI): m/z 319.39 [M + Na]+.
(E)-2-Acetyl-5-methoxyphenyl-3-p-tolylacrylate (5e)
1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 8.4 Hz, 1H), 7.86 (s, 1H), 7.50 (d, J = 8.0 Hz, 2H), 7.22 (d, J = 7.9 Hz, 2H), 6.84 (dd, J = 8.8, 2.4 Hz, 1H), 6.69 (d, J = 2.5 Hz, 1H), 6.64 (d, J = 16.0 Hz, 1H), 3.85 (s, 3H), 2.52 (s, 3H), 2.39 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 195.90, 165.38, 163.76, 151.59, 147.44, 141.48, 132.31, 131.43, 129.80, 128.53, 123.80, 115.83, 111.94, 109.21, 55.78, 29.67, 21.60; MS (APCI): m/z 333.48 [M + Na]+.
(E)-2-Acetyl-5-methoxyphenyl-3-(4-chlorophenyl)acrylate (5f)
1H NMR (400 MHz, CDCl3) δ 7.91–7.78 (m, 2H), 7.52 (d, J = 8.5 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H), 6.85 (dd, J = 8.8, 2.4 Hz, 1H), 6.71–6.61 (m, 2H), 3.85 (s, 3H), 2.51 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 195.88, 165.04, 163.84, 151.44, 145.78, 136.85, 132.71, 132.48, 129.67, 129.37, 123.56, 117.68, 111.93, 109.34, 55.83, 29.45; MS (APCI): m/z 353.38 (M + Na)+.
(E)-2-Acetyl-5-methoxyphenyl-3-(4-methoxyphenyl)acrylate (5g)
1H NMR (400 MHz, CDCl3) δ 7.88 (s, 1H), 7.85 (d, J = 7.5 Hz, 1H), 7.64–7.53 (m, 2H), 7.11 (t, J = 8.6 Hz, 2H), 6.85 (dd, J = 8.8, 2.5 Hz, 1H), 6.68 (d, J = 2.5 Hz, 1H), 6.61 (d, J = 16.0 Hz, 1H), 3.86 (s, 3H), 2.52 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 195.97, 165.59, 165.20, 163.85, 163.08, 151.50, 146.02, 132.48, 130.54, 130.51, 130.47, 130.45, 123.62, 116.82, 116.80, 116.40, 116.18, 111.96, 109.33, 55.86, 29.53; MS (APCI): m/z 337.41 [M + Na]+.
(E)-2-Isopropyl-5-methylphenyl-3-(4-hydroxyphenyl)acrylate (6a)
1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 15.9 Hz, 1H), 7.49 (d, J = 8.6 Hz, 2H), 7.23 (d, J = 7.9 Hz, 1H), 7.05 (d, J = 7.8 Hz, 1H), 6.89 (s, 1H), 6.84 (d, J = 8.6 Hz, 2H), 6.52 (d, J = 15.9 Hz, 1H), 5.99 (brs, 1H), 3.10–2.98 (m, 1H), 2.33 (s, 3H), 1.21 (d, J = 6.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 166.62, 158.47, 148.13, 146.59, 137.38, 136.75, 130.42, 127.26, 126.94, 126.62, 122.96, 116.14, 114.50, 27.32, 23.21, 20.97; MS (ESI): m/z 295.54 [M – H]–.
(E)-2-Isopropyl-5-methylphenyl-3-(4-hydroxy-3-methoxyphenyl)-acrylate (6b)
1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 15.9 Hz, 1H), 7.23 (d, J = 7.9 Hz, 1H), 7.15 (d, J = 8.2 Hz, 1H), 7.11 (s, 1H), 7.05 (d, J = 7.8 Hz, 1H), 6.96 (d, J = 8.2 Hz, 1H), 6.89 (s, 1H), 6.52 (d, J = 15.9 Hz, 1H), 5.96 (s, 1H), 3.96 (s, 3H), 3.10–3.00 (m, 1H), 2.34 (s, 3H), 1.22 (d, J = 6.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 166.12, 148.50, 148.21, 147.02, 146.63, 137.39, 136.68, 127.19, 127.09, 126.58, 123.58, 123.01, 114.99, 114.80, 109.72, 56.15, 27.32, 23.22, 20.98; MS (ESI): m/z 325.60 [M – H]–.
(E)-2-Isopropyl-5-methylphenyl-3-(4-methoxyphenyl)acrylate (6c)
1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 15.9 Hz, 1H), 7.56 (d, J = 8.8 Hz, 2H), 7.22 (d, J = 7.9 Hz, 1H), 7.04 (d, J = 7.9 Hz, 1H), 6.94 (d, J = 8.7 Hz, 2H), 6.89 (s, 1H), 6.53 (d, J = 15.9 Hz, 1H), 3.86 (s, 3H), 3.09–2.99 (m, 1H), 2.33 (s, 3H), 1.21 (d, J = 6.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 166.20, 161.85, 148.20, 146.26, 137.40, 136.68, 130.18, 127.19, 127.12, 126.57, 123.01, 114.84, 114.58, 55.57, 27.30, 23.22, 21.00; MS (APCI): m/z 311.35 [M + H]+.
2-Isopropyl-5-methylphenyl-cinnamate (6d)
1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 16.0 Hz, 1H), 7.67–7.56 (m, 2H), 7.50–7.38 (m, 3H), 7.24 (d, J = 7.9 Hz, 1H), 7.06 (d, J = 7.9 Hz, 1H), 6.91 (s, 1H), 6.69 (d, J = 16.0 Hz, 1H), 3.12–2.99 (m, 1H), 2.35 (s, 3H), 1.23 (d, J = 6.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 165.83, 148.09, 146.57, 137.32, 136.71, 134.34, 130.80, 129.12, 128.44, 127.27, 126.60, 122.93, 117.37, 27.31, 23.21, 20.99; MS (APCI): m/z 281.21 [M + H]+.
(E)-2-Isopropyl-5-methylphenyl-3-p-tolylacrylate (6e)
1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 16.0 Hz, 1H), 7.49 (d, J = 8.0 Hz, 2H), 7.28–7.18 (m, 3H), 7.04 (d, J = 7.9 Hz, 1H), 6.89 (s, 1H), 6.62 (d, J = 16.0 Hz, 1H), 3.09–2.99 (m, 1H), 2.39 (s, 3H), 2.33 (s, 3H), 1.21 (d, J = 6.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 166.03, 148.14, 146.59, 141.32, 137.35, 136.68, 131.63, 129.85, 128.45, 127.22, 126.57, 122.97, 116.30, 27.30, 23.21, 21.66, 20.99; MS (APCI): m/z 295.26 [M + H]+.
(E)-2-Isopropyl-5-methylphenyl-3-(4-chlorophenyl)acrylate (6f)
1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 16.0 Hz, 1H), 7.54 (d, J = 8.5 Hz, 2H), 7.41 (d, J = 8.5 Hz, 2H), 7.24 (d, J = 7.9 Hz, 1H), 7.06 (d, J = 7.9 Hz, 1H), 6.89 (s, 1H), 6.65 (d, J = 16.0 Hz, 1H), 3.09–2.99 (m, 1H), 2.34 (s, 3H), 1.22 (d, J = 6.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 165.60, 148.02, 145.09, 137.28, 136.76, 132.83, 129.60, 129.43, 127.36, 126.64, 122.86, 118.03, 27.30, 23.21, 20.99; MS (APCI): m/z 315.18 [M + H]+.
(E)-2-Isopropyl-5-methylphenyl-3-(4-fluorophenyl)acrylate (6g)
1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 16.0 Hz, 1H), 7.65–7.55 (m, 2H), 7.23 (d, J = 7.9 Hz, 1H), 7.12 (t, J = 8.6 Hz, 2H), 7.05 (d, J = 7.9 Hz, 1H), 6.89 (s, 1H), 6.59 (d, J = 16.0 Hz, 1H), 3.10–2.98 (m, 1H), 2.34 (s, 3H), 1.21 (d, J = 6.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 165.72, 165.53, 163.02, 148.06, 145.24, 137.30, 136.75, 130.63, 130.60, 130.41, 130.32, 127.32, 126.62, 122.90, 117.21, 117.18, 116.42, 116.20, 27.31, 23.21, 20.99; MS (APCI): m/z 299.29 [M + H]+.
Tyrosinase activity assay
The compounds synthesized were tested for diphenolase inhibitory activity of tyrosinase using l-dopa as substrate. Mushroom tyrosinase (Sigma-aldrich, 2687 U mg–1) was used for the bioassay. All the compounds were dissolved in DMSO. The final concentration of DMSO in the test solution was 2.0%. Firstly, 813 μL of phosphate buffer (pH 6.86), 7 μL of mushroom tyrosinase (final concentration: 33.3 U mL–1) and 20 μL of the inhibitor solution were combined in a centrifuge tube. After pre-incubation for 10 min at room temperature, 160 μL of l-dopa (final concentration: 0.4 mM) was added to the mixture, and the mixture was examined immediately. The enzyme reaction was monitored for 1 min by measuring the change in absorbance at 475 nm corresponding to the formation of dopachrome. Inhibition of the tyrosinase reaction was calculated as follows: inhibition ratio (%) = [(B – S)/B] × 100, where B and S are the absorbances of the blank and samples.
The inhibition mechanism of 5a, 5g and 6a was determined by the following method: a series of diluted inhibitor solutions was prepared (5a: 0, 1, 2, and 5 μM, 5g: 0, 2, 8, and 20 μM, 6a: 0, 5, 10 and 20 μM). The l-dopa concentration (0.4 mM) was kept constant. The inhibition rates were measured by the above method with different concentrations of tyrosinase (0.00, 8.3, 16.7, and 33.3 U mL–1).
The inhibition type of the enzyme was assayed by Lineweaver–Burk plots. A series of diluted inhibitor solutions was prepared (5a: 0, 1, 2, and 5 μM, 5g: 0, 2, 8, and 20 μM, 6a: 0, 5, 10 and 20 μM). The tyrosinase concentration (33.3 U mL–1) was kept constant. The inhibition rates were measured by the above method with different concentrations of l-dopa (0.2, 0.4, 0.8, and 1.6 mM).
Molecular docking
The fungal tyrosinase structure (PDBID: 2Y9X) was downloaded from the RCSB PDB database. All the ligand and water atoms were removed. The copper ions in the active site were kept. Hydrogen atoms were added by the VMD31 Autopsf plugin. Then all the hydrogens were optimized by the NAMD232 minimization procedure, and other heavy atoms were kept fixed. The final conformation and psf topology file were extracted for docking. The ligand structure was prepared by the Avogadro program (; http://avogadro.cc/) using a steepest descent algorithm with the MMFF94 force field. Gasteiger charges were calculated for each atom. In addition, to validate the docking, other well known ligands, tropolone and kojic acid, were also docked to this tyrosinase. Docking was performed by the Ledock program (http://; www.lephar.com), integrated with a simulated annealing search and genetic algorithm. The docking score was estimated from van der Waals interactions, weighted hydrogen bonds and electrostatic interactions. 20 search runs were performed. Finally, the generated poses were clustered again (RMSD < 2.0 Å). The cluster with lowest energy was extracted for presentation. The figure was prepared by the VMD program.
Conclusions
Two series of novel tyrosinase inhibitors based on natural product scaffolds have been prepared and evaluated. The tyrosinase inhibitory activities were significantly improved by the combination of the two natural product scaffolds. Most of the compounds synthesized showed better activities than their parent compounds. Among them, 5a, 5g and 6a exhibited activities in the low-micromolar range (IC50 = 2.0 μM, 8.3 μM and 10.6 μM, respectively), better than kojic acid (IC50 = 32.2 μM), used as reference. The presence of the hydroxyl group at the 4-position of the cinnamic acid moiety improved the activity. Study of the inhibition mechanism showed that 5a, 5g and 6a are all reversible inhibitors. The mode of inhibition of the enzyme, determined by analysis of Lineweaver–Burk plots, indicated that 5a is a non-competitive type inhibitor, while 5g and 6a are mixed-type inhibitors. The molecular docking of 6a and tyrosinase indicates that chelating copper at the active site is an important mechanism. These results suggest that the newly synthesized compounds could be potential agents for the prevention and treatment of pigmentation and the inhibition of melanogenesis, or may serve as structural templates for designing and developing novel tyrosinase inhibitors. These findings encourage us to continue our efforts towards the optimization of the pharmacological profile of these cinnamic acid ester derivatives.
Conflicts of interest
The authors declare no competing interest.
Supplementary Material
Acknowledgments
This work was supported by the Natural Science Foundation of Guangdong Province (No. 2016A030310441), the Science Foundation for Young Teachers of Wuyi University (No. 2015zk03 and 2016td01), the Department of Education of Guangdong Province (No. 2016KCXTD005) and the Natural Science Foundation of Jiangsu Province (BK20170312). The authors thank Dr. John S. Lomas for his help in correcting the English of this article.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00099a
References
- Chang T. S. Int. J. Mol. Sci. 2009;10:2440–2475. doi: 10.3390/ijms10062440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. J., Uyama H. Cell. Mol. Life Sci. 2005;62:1707–1723. doi: 10.1007/s00018-005-5054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo S. Y., Sharma V. K., Sharma N. J. Agric. Food Chem. 2003;51:2837–2853. doi: 10.1021/jf020826f. [DOI] [PubMed] [Google Scholar]
- Durán N., Rosa M. A., D'Annibale A., Gianfreda L. Enzyme Microb. Technol. 2002;31:907–931. [Google Scholar]
- Sánchez-Ferrer Á., Neptuno Rodríguez-López J., García-Cánovas F., García-Carmona F. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1995;1247:1–11. doi: 10.1016/0167-4838(94)00204-t. [DOI] [PubMed] [Google Scholar]
- Amer M., Metwalli M. Int. J. Dermatol. 1998;37:449–450. doi: 10.1046/j.1365-4362.1998.00499.x. [DOI] [PubMed] [Google Scholar]
- Oyama T., Yoshimori A., Takahashi S., Yamamoto T., Sato A., Kamiya T., Abe H., Abe T., Tanuma S.-I. Bioorg. Med. Chem. Lett. 2017;27:2868–2872. doi: 10.1016/j.bmcl.2017.04.074. [DOI] [PubMed] [Google Scholar]
- Rossi A. M., Perez M. I. Facial. Plast. Surg. Clin. North Am. 2011;19:313–324. doi: 10.1016/j.fsc.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Pillaiyar T., Manickam M., Jung S. H. Cell. Signalling. 2017;40:99–115. doi: 10.1016/j.cellsig.2017.09.004. [DOI] [PubMed] [Google Scholar]
- Lee S. Y., Baek N., Nam T. G. J. Enzyme Inhib. Med. Chem. 2016;31:1–13. doi: 10.3109/14756366.2015.1004058. [DOI] [PubMed] [Google Scholar]
- Parvez S., Kang M., Chung H. S., Bae H. Phytother. Res. 2007;21:805–816. doi: 10.1002/ptr.2184. [DOI] [PubMed] [Google Scholar]
- Taofiq O., González-Paramás A. M., Martins A., Barreiro M. F., Ferreira I. C. F. R. Ind. Crops Prod. 2016;90:38–48. [Google Scholar]
- Yu L. J. Agric. Food Chem. 2003;51:2344–2347. doi: 10.1021/jf0208379. [DOI] [PubMed] [Google Scholar]
- Sugumaran M. FEBS Lett. 1991;293:4–10. [Google Scholar]
- Friedman M. J. Agric. Food Chem. 1996;44:631–653. [Google Scholar]
- Ha Y. M., Kim J. A., Park Y. J., Park D., Choi Y. J., Kim J. M., Chung K. W., Han Y. K., Park J. Y., Lee J. Y., Moon H. R., Chung H. Y. MedChemComm. 2011;2:542–549. [Google Scholar]
- Jiang Y., Du Z., Xue G., Chen Q., Lu Y., Zheng X., Conney A. H., Zhang K. Molecules. 2013;18:3948–3961. doi: 10.3390/molecules18043948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. R., Lee H. J., Choi Y. J., Park Y. J., Woo Y., Kim S. J., Park M. H., Lee H. W., Chun P., Chung H. Y., Moon H. R. MedChemComm. 2014;5:1410–1417. [Google Scholar]
- Pintus F., Matos M. J., Vilar S., Hripcsak G., Varela C., Uriarte E., Santana L., Borges F., Medda R., Di Petrillo A., Era B., Fais A. Bioorg. Med. Chem. 2017;25:1687–1695. doi: 10.1016/j.bmc.2017.01.037. [DOI] [PubMed] [Google Scholar]
- Matos M. J., Varela C., Vilar S., Hripcsak G., Borges F., Santana L., Uriarte E., Fais A., Di Petrillo A., Pintus F., Era B. RSC Adv. 2015;5:94227–94235. [Google Scholar]
- Hwang K. S., Yang J. Y., Lee J., Lee Y. R., Kim S. S., Kim G. R., Chae J. S., Ahn J. H., Shin D. S., Choi T. Y., Bae M. A. J. Dermatol. Sci. 2017;89:165–171. doi: 10.1016/j.jdermsci.2017.11.004. [DOI] [PubMed] [Google Scholar]
- Hu X., Yu M. H., Yan G. R., Wang H. Y., Hou A. J., Lei C. J. Asian Nat. Prod. Res. 2017 doi: 10.1080/10286020.2017.1350653. [DOI] [PubMed] [Google Scholar]
- Rezaei M., Mohammadi H. T., Mahdavi A., Shourian M., Ghafouri H. Int. J. Biol. Macromol. 2017;108:205–213. doi: 10.1016/j.ijbiomac.2017.11.147. [DOI] [PubMed] [Google Scholar]
- Zhu T. H., Cao S. W., Yu Y. Y. Int. J. Biol. Macromol. 2013;62:589–595. doi: 10.1016/j.ijbiomac.2013.09.056. [DOI] [PubMed] [Google Scholar]
- Zhao D. Y., Zhang M. X., Dong X. W., Hu Y. Z., Dai X. Y., Wei X., Hider R. C., Zhang J. C., Zhou T. Bioorg. Med. Chem. Lett. 2016;26:3103–3108. doi: 10.1016/j.bmcl.2016.05.006. [DOI] [PubMed] [Google Scholar]
- Yang Y. F., Lai X. Y., Lai G. Y., Jiang Z. D., Ni H., Chen F. J. Funct. Foods. 2016;27:140–149. [Google Scholar]
- Cui Y., Hu Y. H., Yu F., Zheng J., Chen L. S., Chen Q. X., Wang Q. Int. J. Biol. Macromol. 2017;95:1289–1297. doi: 10.1016/j.ijbiomac.2016.11.027. [DOI] [PubMed] [Google Scholar]
- Taofiq O., González-Paramás A., Barreiro M., Ferreira I. Molecules. 2017;22:1–24. doi: 10.3390/molecules22020281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satooka H., Kubo I. J. Agric. Food Chem. 2011;59:8908–8914. doi: 10.1021/jf2014149. [DOI] [PubMed] [Google Scholar]
- Liu J., Wu F., Chen L., Zhao L., Zhao Z., Wang M., Lei S. Food Chem. 2012;135:2872–2878. doi: 10.1016/j.foodchem.2012.07.055. [DOI] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. J. Mol. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deri B., Kanteev M., Goldfeder M., Lecina D., Guallar V., Adir N., Fishman A. Sci. Rep. 2016;6:34993. doi: 10.1038/srep34993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismaya W. T., Rozeboom H. J., Weijn A., Mes J. J., Fusetti F., Wichers H. J., Dijkstra B. W. Biochemistry. 2011;50:5477–5486. doi: 10.1021/bi200395t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.