

 We present the development of conformationally pre-organised Dyrk1A inhibitors based on the hydroxybenzothiazole urea scaffold.
We present the development of conformationally pre-organised Dyrk1A inhibitors based on the hydroxybenzothiazole urea scaffold.
Abstract
We present the development of conformationally pre-organised Dyrk1A inhibitors based on the hydroxybenzothiazole urea scaffold. The modifications introduced to the discovered hit (AHS-211) proved the crucial role of the urea linker to preserve the bioactive conformation and led to the development of compound b5 as a promising selective Dyrk1A inhibitor.
Introduction
Dyrk1A (the dual-specificity tyrosine phosphorylation-regulated kinase 1A) is a serine/threonine protein kinase that belongs to the DYRK family. The other members in the family are Dyrk1B, Dyrk2, Dyrk3, and Dyrk4. Together with the cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAP kinases), glycogen synthase kinases (GSK) and Ccd2-like kinases (CLKs), they compose CMGC super family of kinases.1,2
DYRK1A gene is allocated in the Down syndrome (DS) critical region of chromosome 21. The cognitive impairments and neurofibrillary degeneration seen in DS as well as in Alzheimer's disease (AD) have been found to be associated with Dyrk1A overexpression.3–5 Additionally, Dyrk1A was reported to be implicated in numerous biological processes such as T cell regulation,6 cell cycle regulation and cell differentiation,6,7 stabilization of cytoskeleton,8 and brain neurodevelopment.9 Drug discovery efforts aim at the development of selective Dyrk1A inhibitors, in particular in light of the putative tumor suppressor activity of the homologous Dyrk2 isoform.10
In this communication we present 6-hydroxybenzothiazol-2-yl urea as a promising new scaffold for Dyrk1A inhibition that allows pre-organisation of the biologically active conformation. We report some modifications for the scaffold exploration as well as scaffold expansion using an in-house discovered hit (AHS-211, Fig. 1) as a starting compound.
Fig. 1. Previously published benzothiazole- and benzothiophene-based Dyrk1A/Clk1 inhibitors (INDY, TG003, A and B) and new benzothiazole urea derivatives (C and AHS-211).

Results and discussion
Compound design
The benzothiazole scaffold was known to be a promising building block for the development of Dyrk1A inhibitors since the discovery of the Dyrk1A/Clk1 inhibitors TG003 and INDY (Fig. 1).11,12 The binding mode of the benzothiazole core to the ATP pocket of Dyr1A was determined by X-ray crystallography for INDY;12 further Dyrk1A co-crystal structures were reported later for acetylated 2-aminobenzothiazole fragments (Fig. 1, structure B).13 Both INDY and our previously reported benzothiophene Clk1/Dyrk1A inhibitor14 (Fig. 1, compound A) contained a hydroxyl group which is important to maintain potency against Dyrk1A. We thought of expanding the hydroxybenzothiazole scaffold to get potent and selective Dyrk1A inhibitors. To this end, we exploited the benzothiazole ring nitrogen to design and synthesize new derivatives which are rigidized via an internal H-bond, avoiding the need to synthesize polycyclic aromatic systems which may have toxicological issues. We envisaged extending the benzothiazole fragment with a urea linker to achieve the desired pseudo 6-membered ring structure between the benzothiazole nitrogen (as HBA) and the distant urea NH (as HBD) as shown in structure CFig. 1. Using ab initio calculations, it was corroborated that the suggested benzothiazole urea scaffold showed the proposed pseudo-ring formation in the most stable conformer (conformer c1, Fig. 2). In contrast, a considerably higher energy content was calculated for the also coplanar rotamer c2 (ΔE = 59 kJ mol–1, Fig. 2), probably attributable to the lack of the energetically favourable H-bond as well as to the unfavourable polar repulsion between the endocyclic nitrogen and the urea oxygen (cf.c2, Fig. 2).
Fig. 2. Lowest energy conformer (c1) and the higher energy linear conformer (c2) of the benzothiazole urea scaffold, exemplified by hit compound AHS-211, as calculated by the B3LYP density functional method. Shown are the fully coplanar conformers with a mesh displaying the electrostatic potential on an isoelectronic density surface (0.025 e a–3). The intramolecular H-bond that stabilizes a pseudo 6-membered ring in c1 is indicated by the overlapping electron density in the center. The calculated energy difference between the conformers is 59 kJ mol–1.

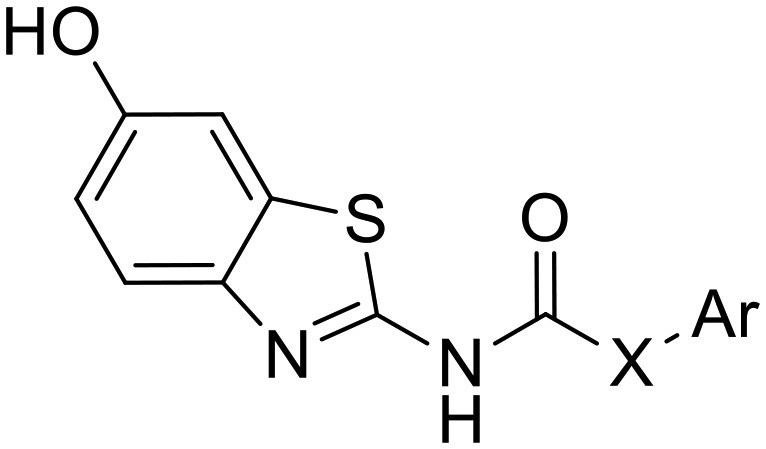
Searching in our in-house library for possible hits that may overlap with the designed target urea derivatives revealed the previously published compound AHS-211 as a good candidate. AHS-211 (compound 23 in ref. 15) was previously published as an inactive compound against 17β-hydroxysteroid dehydrogenase type 1 (17βHSD1).15 Testing AHS-211 against Dyrk1A greatly supported our design concept as it inhibited the recombined enzyme with an IC50 of 0.134 μM. AHS-211 also inhibited Dyrk1B with an IC50 of 0.37 μM, yielding a selectivity factor of 2.7 for Dyrk1A (Table 2). Furthermore, AHS-211 showed 64.7% inhibition when screened against Dyrk2 at 5 μM concentration (Table 2).
Table 2. Inhibition of Dyrk1A, Dyrk1B and Dyrk2 by the phenolic analogues.
| ||||||||
| Cpd# | X | Ar | Dyrk1A |
Dyrk1B |
Selectivity factor c | Dyrk2 | ||
| % inhibition at 0.5 μM a , b | IC50 μM a , b | % inhibition at 0.5 μM a , b | IC50 μM a , b | % inhibition at 5 μM a , d | ||||
| b1 | –NH– |
|
80.8 | 0.163 | 43.6 | 0.710 | 4.3 | 40.4 |
| b2 | –O– |
|
0 | ND | 0 | ND | — | 18.6 |
| b3 | –CH2– |
|
37.8 | ND | 0 | ND | — | 50.1 |
| b4 | –NH– |
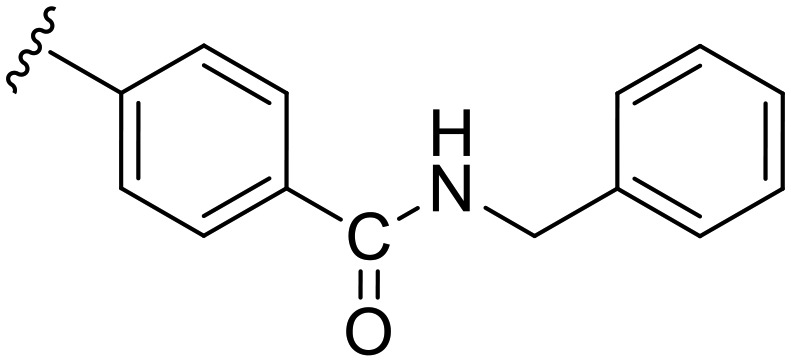
|
89.8 | 0.063 | 69 | 0.130 | 2.1 | 38.1 |
| b5 | –NH– |
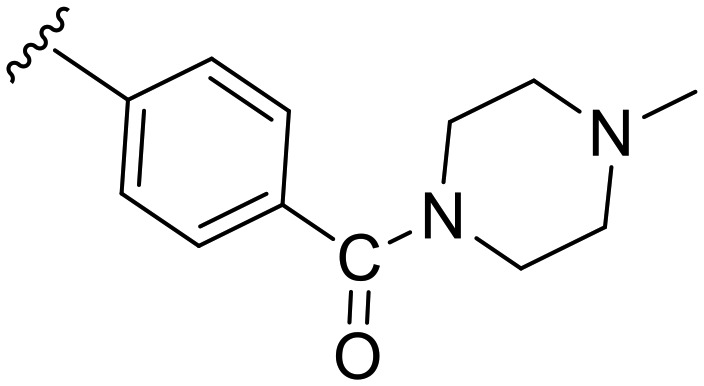
|
99.1 | 0.095 | 21 | 1.513 | 16 | 34.5 |
| AHS-211 | –NH– |
|
74.4 | 0.134 | 54.6 | 0.370 | 2.8 | 64.7 |
aValues are mean values of at least two independent experiments each was done in duplicates; standard deviation <10%.
bThe assay was carried out at ATP conc. of 15 μM.
cSelectivity factor: IC50 (Dyrk1B)/IC50 (Dyrk1A).
dThe assay was carried out at ATP conc. of 100 μM.
Chemistry
The synthesis of the designed urea derivatives was performed mainly through the reaction of the aryl amine derivatives with phenylisocyanates in DMF at room temperature (Scheme 1), or alternatively through the reaction of 2-amino-6-methoxy benzothiazole with phenylchloroformate in the presence of pyridine as a base and dioxane as solvent to give the carbamate compound a4 which is itself was isolated as the oxygen isostere of urea or further reacted in situ with amine derivatives in refluxing dioxane to give the urea compounds (a5 and a6) as shown in Scheme 2. Acylation of 2-amino-6-methoxybenzothiazole was done in acetone in presence of K2CO3 to yield the methylene isostere of urea (Scheme 3). Scheme 4 shows the preparation of the amide derivatives starting from the ester a2 which was hydrolysed to the respective carboxylic acid a8 under alkaline conditions followed by amide coupling with benzyl amine or 1-methylpiperazine using HBTU as coupling agent. Finally, the methyl ether dealkylation was performed using BBr3 in dichloromethane (Scheme 5).
Scheme 1. Synthesis of urea derivatives via isocyanates. Reagents and conditions: (i) DMF, RT, overnight.

Scheme 2. Synthesis of urea derivatives via carbamates. Reagents and conditions: (i) dioxane, pyridine, RT, 1 h (ii) 1.5 equiv. amine derivative, dioxane, overnight, reflux.

Scheme 3. Synthesis of benzothiazoleamide. Reagents and conditions: (i) acetone, Na2CO3, ice cooling then RT, 1 h.

Scheme 4. Synthesis of the benzamide derivatives. Reagents and conditions: (i) aq. KOH, THF, RT, 14 h (ii) 4 equiv. amine derivative, 1.5 equiv. HBTU, TEA, RT, overnight.

Scheme 5. Ether dealkylation. Reagents and conditions: (i) 15 equiv. BBr3, CH2Cl2, –78 °C then room temperature, 20 h.

Biological evaluation
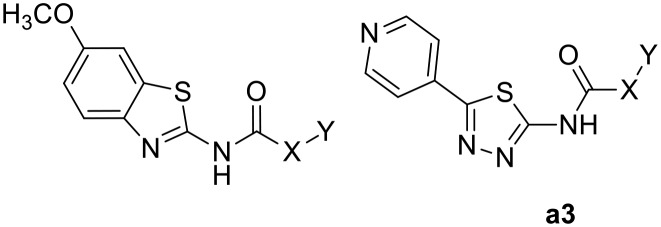
Table 1 shows the screening results of the synthesized methoxy precursors against both Dyrk1A and Dyrk1B while Table 2 shows the screening results for the phenolic analogues against Dyrk1A, Dyrk1B and Dyrk2. As can be seen, the 6-methoxy precursors showed a huge reduction of potency when compared to their 6-hydroxy analogues, indicating that the 6-hydroxyl (born at the benzothiazole) in AHS-211 has a crucial share in binding to Dyrk1A as HBD function; in fact, none of the 6-methoxy derivatives exhibited more than 31% inhibition at 2 μM of the tested compounds (Table 1). Likewise, compound a3, in which the 6-hydroxybenzothiazole moiety was replaced by 5-pyridylthiadiazole, did not show significant activity. In this analogue, the internal H-bond is expected to be conserved and the pyridine nitrogen lies in a similar distance from the urea linker as the 6-methoxy oxygen, thus enabling a similar potential role as HBA; nevertheless, the lack of the HBD impeded the success of this modification.
Table 1. Inhibition of Dyrk1A and Dyrk1B by the methoxy precursors and compound a3.
| ||||
| Cpd# | X | Y | Dyrk1A | Dyrk1B |
| % inhibition at 0.5 μM a | % inhibition at 0.5 μM a | |||
| a1 | –NH– |
|
8 | 0 |
| a3 | –NH– |
|
19.6 | 12 |
| a4 | –O– |
|
13.3 | 0 |
| a5 | –NH– |
|
13.8 | 0 |
| a6 | –NH– |
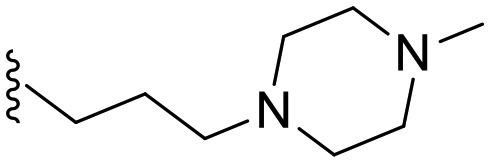
|
20.3 | 0 |
| a7 | –CH2– |
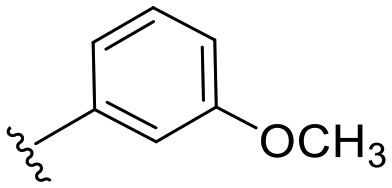
|
21.2 | 25 |
| a8 | –NH– |
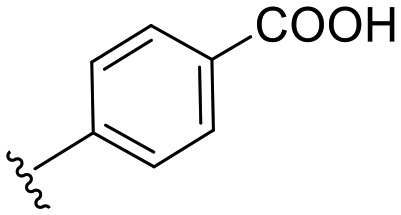
|
25.5 | 0 |
| a9 | –NH– |
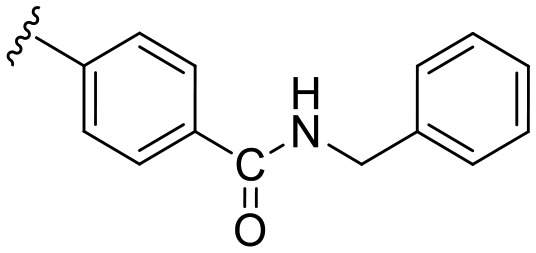
|
30.5 | 0 |
| a10 | –NH– |
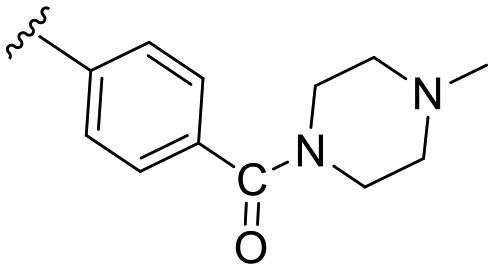
|
28.1 | 0 |
aValues are mean values of at least two independent experiments each was done in duplicates; standard deviation <10%. The ATP concentration in the assay was 15 μM.
In contrast, the deletion of the 3-hydroxyl (at the phenyl ring) in AHS-211 almost did not influence the activity, as observed with compound b1 (IC50 = 0.163 μM against Dyrk1A). Of note, the latter change reduced the potency against Dyrk1B, increasing the selectivity factor to 4.3 toward Dyrk1B, and also reduced the off-target activity against Dyrk2 when compared with the hit AHS-211 (Table 2).
Compounds b2–b3 were prepared to prove the importance of the urea linker in maintaining the biologically active conformer; in these compounds, the –NH– assumed to act as HBD and help the pseudo ring formation was replaced by the isosteric –O– and –CH2–, respectively. Compound b2 gave no inhibition for Dyrk1A and Dyrk1B at 0.5 μM while its urea analogue b1 showed more than 80% inhibition at the same concentration (Table 2). Since the carbamate-derivative b2 was least likely to adopt a coplanar conformation similar to c1 (Fig. 2) because of the electronic repulsion between the oxygen and the ring nitrogen, the complete loss of inhibitory activity supported our hypothesis that the biological activity was associated with a c1-like molecule shape. In further accordance, the more neutral electrostatic potential of the methylene bridge in b3 led to a partial recovery of the inhibitory potency, but failed to reach the potency of AHS-211 due to the lack of the conformation-stabilizing H-bond. Of interest, compound b3 showed its highest inhibitory potency against Dyrk2 (IC50 ≈ 5 μM, [ATP] = 100 μM), probably due to the additional presence of the m-hydroxy function at the phenyl, which also enhanced the Dyrk2 inhibitory activity of AHS-211 (Table 2).
Having proven the crucial role of the urea motif, we envisaged some substantial modifications of compound b1 to improve potency and selectivity for Dyrk1A; further attempts aimed at optimization of the drug-like properties, such as water solubility and log P. To achieve this, two main types of modifications were planned: the first one was to replace the phenyl ring in b1 with an alkyl chain linked to a basic alicyclic amine like morpholine and N-methyl piperazine. This is represented by the 6-methoxy precursors a5 and a6, respectively. Unfortunately, the unsuccessful O-demethylation of both compounds hampered testing the effect of such a modification. However, the inhibition noted for these two precursors at 0.5 μM did not indicate a potential advantage over the other modifications presented in Table 1. The second approach was to extend compound b1 through amide derivatives arising from the phenyl ring. Two amide derivatives offering diverse possible interactions with the binding site were prepared: the benzyl amide b4 and the methyl piperazine amide b5. Although the extension with the 4-benzyl amide in compound b4 enhanced the potency toward Dyrk1A (IC50 = 0.063 μM) by more than 2-fold compared with b1 and AHS-211, it did not show any improvement in selectivity over Dyrk1B (Table 2). On the other hand, compound b5 was almost similar to AHS-211 in potency against Dyrk1A, yet, it showed more than 15 fold selectivity for Dyrk1A over Dyrk1B. Importantly, both b4 and b5 displayed superior selectivity for Dyrk1A over Dyrk2 when compared with AHS-211.
As compound b5 was more selective over Dyrk1B, it was selected for an extended selectivity profiling against all kinases that were frequently reported as being co-inhibited by previously published Dyrk1A inhibitor classes (Table 3).16–18 This profiling revealed that our combination of modifications led to a significant increase in selectivity compared with the previously published benzothiazole-based Dyrk1A inhibitors such as INDY, which strongly inhibited, among other kinases, Dyrk2, PIM1 and CK1δ.12
Table 3. Selectivity profile of b5vs. the related kinases a .
| Kinase | % inhibition at 5 μM | Kinase | % inhibition at 5 μM |
| CDK5/p25 | 2 ± 1 | Haspin | 1 ± 1 |
| CLK1 | 72 ± 0.5 | PIM1 | 29 ± 2.5 |
| CK1δ | 9 ± 2 | SRPK1 | 5 ± 0.5 |
| HIPK1 (Myak) | 50 ± 0.5 | MLCK (MLCK2) | 45 ± 3 |
| NTRK2 (TRKB) | 13 ± 1 | STK17A (DRAK1) | 39 ± 0 |
| Dyrk1A | 85 ± 0.5 |
aValues are mean values of at least two duplicates; testing was done at an ATP concentration of 100 μM.
Additionally, b5 displayed good selectivity for Dyrk1A over Clk1, one of the most common off targets of many reported Dyrk1A inhibitors, with a selectivity factor >7 (IC50 of b5 against Clk1 = 730 nM, [ATP] = 15 μM).
Because of its marked selectivity and good potency, we aimed at analyzing the metabolic stability of b5 against human hepatic CYP enzymes. Compared to testosterone, which was included as a reference known to be rapidly metabolized, b5 showed a promising stability (half-life >60 min, Table 4). It is worth mentioning that at 60 min incubation time, 88% of the initial compound concentration could be still detected.
Table 4. Metabolic stability of compounds 4b and 5b against human CYP enzymes.
aIncubation with pooled human liver S9 fraction at 37 °C; samples taken at 0, 15, 30, 60 min, determination of the parent compound by MS.
bReference compound with low metabolic stability.
These data were encouraging, suggesting that b5 might also be applicable in in vivo models. As it was expected, compound b4 exhibited lower, yet, appreciable stability toward hepatic CYP enzymes with a metabolic half-life of 33 min which is mostly attributed to the presence of the unsubstituted benzyl group (Table 4).
Next we aimed at identifying the putative binding mode of b5 to Dyrk1A in order to explain its high potency and selectivity. To this end, we employed molecular docking to Dyrk1A coordinates derived from a co-crystal structure with a 5-hydroxy benzothiazole fragment (PDB code: ; 5A3X). Thus, we anticipated that any realistic docking pose should exhibit a significant overlap with the original co-crystallized fragment; this was the case with the docking pose showing the highest score (depicted in Fig. 3). Another criterion for plausibility was that b5 was binding in the H-bond-stabilized low energy conformation, which was in full agreement with the observed structure–activity relationships and the ab initio calculations. The docking model predicts several classical and non-classical H-bonds contributing to the binding affinity inside the ATP pocket (Fig. 3A). Of note, because of the shifted position, the 6-hydroxy function was only acting as an HBD, thus explaining the loss of potency seen with the methoxy precursors.
Fig. 3. Predicted binding mode of b5 in the ATP binding pocket of Dyrk1A. Compound b5 was docked to the Dyrk1A coordinates derived from the co-crystal structure with 2-acetamido-5-hydroxy benzothiazole (PDB code: ; 5A3X) using MOE. The pose with the highest score is depicted, showing binding of b5 (yellow) in the lowest energy conformation of the scaffold (cf.c1, Fig. 2). A. Inside the pocket, the ligand is anchored via two crucial H-bonds involving the backbone carbonyl of the hinge region residue Leu241, and the conserved Lys188 as a donor. Several CH–π and an NH–π interaction (with Phe238) additionally enhance the affinity. At the border of the pocket, the protonated piperazine amine is predicted to form a salt bridge with Asp287. H-bonds and ionic bonds are indicated by blue lines and H–π bonds by brown lines. B. Top view on the ATP binding pocket visualized by a transparent surface. The ligand is bound as in A; through the central pseudo 6-membered ring, ideal shape complementarity to the pocket is achieved.

Noteworthy, Rothweiler et al. had identified an alternative binding mode, inverted by 180°, with a 5-methoxybenzothiazole fragment in the ATP binding pocket of Dyrk1A.13 Using the corresponding crystal structure coordinates (PDB: ; 5A4E) we also evaluated the possibility of an inverted binding mode for our best compound b5. Indeed, we found that theoretically, binding of b5 might also occur in a flipped orientation, with the hydroxyl then forming an H-bond with Asp307 (Fig. S1, ESI†). However, we considered this inverted pose to be less likely, because it did not well explain the drop of potency observed with the methoxy precursor a10 (28% inhibition at 0.5 μM, Table 1). With a10, the H-bond interaction in the hypothetical flipped mode simply switched from Asp307 to Lys188, now with the methoxy oxygen as acceptor (not shown). Thus, the binding pose shown in Fig. S1,† although it cannot be totally ruled out, would predict a comparable potency for b5 and a10, in contrast to our experimental results.
An important structural feature enhancing the selectivity over Dyrk1B was the amide-linked N-methyl piperazine extension, which was predicted to engage in an ionic interaction with Asp287 in our model. Although this residue is conserved in Dyrk1B, it could be speculated that its position is slightly shifted in the Dyrk1B 3D structure, thus causing a less efficient ionic interaction. Such a shift in the spatial position of the corresponding Asp257 can be observed in the X-ray structures of the homologous Dyrk2 (data not shown), however, it cannot be verified with Dyrk1B due to the lack of a published 3D structure.
Conclusion
Our design concept of attaching a urea linker to the benzothiazole core, followed by a scaffold expansion, led to a new class of potent and selective Dyrk1A inhibitors. Of note, the best compound, b5, did not appreciably inhibit the homologous Dyrk2 isoform and even showed a remarkable 15 fold selectivity over the most closely related isoform Dyrk1B. In addition, the activity toward the atypical kinase haspin that was strongly inhibited by most of the previously reported Dyrk1A inhibitors was completely abolished with b5. Together with the favorable balance of lipophilic/hydrophilic functions, further testing of b5 as a potential agent for the treatment of Dyrk1A-related neuropathological disorders can be envisaged.
Experimental section
Chemistry
Solvents and reagents were obtained from commercial suppliers and used as received. A Bruker DRX 500 spectrometer was used to obtain 1H NMR and 13C NMR spectra, in some cases Bruker Fourier 300 was used. The chemical shifts are referenced to the residual protonated solvent signals. At least 95% purity in all the tested compounds was obtained by means of HPLC coupled with mass spectrometry. Mass spectra (HPLC-ESI-MS) were obtained using a TSQ quantum (Thermo Electron Corporation) instrument prepared with a triple quadrupole mass detector (Thermo Finnigan) and an ESI source. All samples were inserted using an autosampler (Surveyor, Thermo Finnigan) by an injection volume of 10 μL. The MS detection was determined using a source CID of 10 V and carried out at a spray voltage of 4.2 kV, a nitrogen sheath gas pressure of 4.0 × 105 Pa, a capillary temperature of 400 °C, a capillary voltage of 35 V and an auxiliary gas pressure of 1.0 × 105 Pa. The stationary phase used was a RP C18 NUCLEODUR 100-3 (125 × 3 mm) column (Macherey-Nagel). The solvent system consisted of water containing 0.1% TFA (A) and 0.1% TFA in acetonitrile (B). HPLC-method: flow rate 400 μL min–1. The percentage of B started at an initial of 5%, was increased up to 100% during 16 min, kept at 100% for 2 min, and flushed back to 5% in 2 min. High resolution precise mass spectra were recorded on Thermo Fisher Scientific (TF, Dreieich, Germany) Q Exactive Focus system equipped with heated electrospray ionization (HESI)-II source and Xcalibur software (version 4.0.27.19). Before analysis external mass calibration was done according to the manufacturer's recommendations. The samples were dissolved and diluted in methanol in a concentration of 3 μM and directly injected into the Q Exactive Focus using the integrated syringe pump. All the data analyses were done in positive ion mode using voltage scans and the data collected in continuous mode. Melting points were determined using a Mettler FP1 melting point apparatus and are uncorrected.
General procedure for synthesis of urea derivatives
Method A
A solution of the aryl amine (2 mmol) and the respective isocyanate derivative (2.4 mmol) in DMF (10 mL) was left to stir at room temperature for 3 h. The product was precipitated by adding a 50 mL of ice/water mixture then purified with column chromatography.
Method B
To a solution of 6-methoxy-2-aminobenzothiazole (0.36 gm, 2 mmol) in dioxane (6 mL) containing pyridine (179 μL, 2.22 mmol) phenyl chloroformate (0.25 mL, 2 mmol) was added dropwise at room temperature. After 10 min, the carbamate derivative a4 precipitated, then the corresponding amine derivative (2 mmol) was added and the reaction mixture was heated to 100 °C for 5 h. After cooling to room temperature, the reaction mixture was diluted with ethyl acetate and washed with water. The combined organic layers were dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified with column chromatography to give the desired compounds. Compound a4 was isolated by filtration in case of the carbamate is needed as a product to be tested or for further demethylation.
Phenyl (6-methoxybenzo[d]thiazol-2-yl)carbamate (a4)
The carbamate derivative was isolated by filtration as mentioned above. White solid; yield: 88%; mp >300 °C; 1H NMR (500 MHz, DMSO) δ 9.34 (s, 1H), 7.63 (d, J = 8.8 Hz, 1H), 7.58 (d, J = 2.6 Hz, 1H), 7.49–7.43 (m, 2H), 7.40 (d, J = 8.8 Hz, 1H), 7.35–7.25 (m, 2H), 7.02 (td, J = 8.7, 2.6 Hz, 1H), 3.80 (s, 3H); MS (ESI): m/z = 301.12 (M + H)+.
1-(6-Methoxybenzo[d]thiazol-2-yl)-3-phenylurea (a1)
The title compound was prepared according to the general procedure for synthesis of urea derivatives (method A) by the reaction of 2-amino-6-methoxybenzothiazole and phenylisocynate. White solid; yield: 78%; mp 201–202 °C; 1H NMR (500 MHz, DMSO) δ 10.64 (s, 1H), 9.12 (s, 1H), 7.55 (t, J = 9.2 Hz, 1H), 7.54–7.44 (m, 3H), 7.39–7.25 (m, 2H), 7.08–7.01 (m, 1H), 6.98 (dd, J = 8.8, 2.6 Hz, 1H), 3.80 (s, 3H); 13C NMR (126 MHz, DMSO) δ 157.32, 155.67, 151.73, 142.83138.47, 132.48, 128.91, 122.88, 120.15, 118.75, 114.36, 104.92, 55.58; MS (ESI): m/z = 300.14 (M + H)+.
Ethyl 4-(3-(6-methoxybenzo[d]thiazol-2-yl)ureido)benzoate (a2)
The title compound was prepared according to the general procedure for synthesis of urea derivatives (method A) by the reaction of 2-amino-6-methoxybenzothiazole and 4-(ethoxycarbonyl)phenylisocyanate. White solid; yield: 73%; mp 290–291 °C; 1H NMR (500 MHz, DMSO) δ 10.73 (s, 1H), 9.51 (s, 1H), 8.02–7.87 (m, 2H), 7.76–7.62 (m, 2H), 7.66 (d, J = 8.5 Hz, 2H), 6.99 (dd, J = 8.8, 2.6 Hz, 1H), 4.29 (q, J = 7.1 Hz, 2H), 3.80 (s, 3H), 1.31 (t, J = 7.1 Hz, 3H); MS (ESI): m/z = 372.02 (M + H)+.
1-Phenyl-3-(5-(pyridin-4-yl)-1,3,4-thiadiazol-2-yl)urea (a3)
The title compound was prepared according to the general procedure for synthesis of urea derivatives (method A) by the reaction of 5-(pyridin-4-yl)-1,3,4-thiadiazol-2-amine and phenylisocynate. White solid; yield: 88%; mp 153–154.5 °C; 1H NMR (500 MHz, DMSO) δ 11.34 (s, 1H), 9.11 (s, 1H), 8.72 (dd, J = 4.4, 1.7 Hz, 2H), 7.88 (dd, J = 4.5, 1.7 Hz, 2H), 7.51 (d, J = 7.6 Hz, 2H), 7.42–7.25 (m, 2H), 7.16–6.98 (m, 1H); 13C NMR (126 MHz, DMSO) δ 161.25, 158.83, 151.37, 150.68, 138.21, 137.19, 128.94, 123.19, 120.53, 118.90; MS (ESI): m/z = 297.80 (M + H)+.
1-(6-Methoxybenzo[d]thiazol-2-yl)-3-(2-morpholinoethyl)urea (a5)
The title compound was prepared according to the general procedure for synthesis of urea derivatives (method B) using 4-(2-aminoethyl)morpholine. Beige solid; yield: 68%; mp 179–181 °C; 1H NMR (500 MHz, DMSO) δ 10.64 (s, 1H), 7.52–7.48 (m, 1H), 7.47 (d, J = 2.6 Hz, 1H), 6.94 (dd, J = 8.8, 2.6 Hz, 1H), 6.77 (s, 1H), 3.78 (s, 3H), 3.64–3.54 (m, 4H), 3.28 (dd, J = 11.8, 6.1 Hz, 2H), 2.46–2.34 (m, 6H); 13C NMR (126 MHz, DMSO) δ 157.87, 155.46, 153.77, 143.22, 132.56, 120.12, 114.07, 104.80, 66.17, 57.27, 55.55, 53.11, 36.15; MS (ESI): m/z = 337.04 (M + H)+.
1-(6-Methoxybenzo[d]thiazol-2-yl)-3-(3-(4-methylpiperazin-1-yl)propyl)urea (a6)
The title compound was prepared according to the general procedure for synthesis of urea derivatives (method B) using 1-(3-aminopropyl)-4-methylpiperazine. White solid; yield: 61%; mp 192–194 °C; 1H NMR (500 MHz, DMSO) δ 10.76 (s, 1H), 7.47 (dd, J = 15.2, 5.7 Hz, 2H), 7.12 (s, 1H), 6.94 (dd, J = 8.8, 2.6 Hz, 1H), 3.78 (s, 3H), 3.42–3.31 (m, 4H), 3.17 (dd, J = 12.6, 6.5 Hz, 2H), 2.48–2.39 (m, 4H), 2.34 (dd, J = 17.2, 10.0 Hz, 2H), 2.25 (s, 3H), 1.66–1.56 (m, 2H); 13C NMR (126 MHz, DMSO) δ 157.78, 155.41, 150.70, 143.22, 132.59, 120.05, 114.01, 104.78, 62.59, 55.55, 54.89, 54.10, 52.01, 44.99, 26.44; MS (ESI): m/z = 363.99 (M + H)+.
N-(6-Methoxy-benzothiazol-2-yl)-2-(3-methoxy-phenyl)-acetamide (a7)
3-Methoxyphenylacetyl chloride (0.18 g, 1 mmol) was added gradually to a stirred solution of 6-methoxy-2-aminobenzothiazole (0.16 g, 1 mmol) and Na2CO3 (0.16 g, 1.5 mmol) in acetone (20 mL) under ice cooling. The mixture was stirred at room temperature under a nitrogen atmosphere for 2 h then added to 100 mL of water/ice mixture, the solid obtained was separated by filtration followed by column chromatogrpahy purification to give the title compound as a white solid; yield: 82%; mp 151–152 °C; 1H NMR (500 MHz, DMSO) δ 12.44 (s, 1H), 7.63 (d, J = 8.8 Hz, 1H), 7.55 (d, J = 2.5 Hz, 1H), 7.25 (t, J = 7.9 Hz, 1H), 7.02 (dd, J = 8.8, 2.6 Hz, 1H), 6.92 (dd, J = 9.4, 4.9 Hz, 2H), 6.84 (dd, J = 7.9, 2.2 Hz, 1H), 3.79 (s, 3H), 3.77 (s, 2H), 3.75 (s, 3H); 13C NMR (126 MHz, DMSO) δ 169.72, 159.24, 156.11, 155.83, 142.57, 136.13, 132.71, 129.42, 121.47, 121.10, 115.11, 114.89, 112.23, 104.68, 55.57, 54.97, 41.83; MS (ESI): m/z = 329.19 (M + H)+.
4-(3-(6-Methoxybenzo[d]thiazol-2-yl)ureido)benzoic acid (a8)
The compound was prepared by alkaline ester hydrolysis of compound a2 where 2 M aqueous potassium hydroxide solution (10 mL) was added to a stirred solution of a2 (1 mmol) in THF (50 ml). After stirring at room temperature for 14 h the solvent was evaporated under reduced pressure. The residue was diluted with water (20 mL) and the solution was acidified using 2 M HCl. The product was extracted with ethylacetate (3 × 15 ml). The organic layers were combined and dried over MgSO4 followed by evaporation of solvent under reduced pressure to give the crude product that was used directly for the next steps without further purification. White solid; yield: 93%; mp 284–286 °C; 1H NMR (300 MHz, DMSO) δ 11.10 (s, 1H), 10.41 (s, 1H), 7.93 (t, J = 6.0 Hz, 2H), 7.67 (d, J = 8.8 Hz, 2H), 7.59 (d, J = 8.8 Hz, 1H), 7.52 (t, J = 4.0 Hz, 1H), 6.99 (dd, J = 8.8, 2.6 Hz, 1H), 3.81 (s, 3H); MS (ESI): m/z = 343.95 (M + H)+.
General procedure for amide synthesis
The appropriate amine (4 equiv.) was added dropwise to the solution of the benzoic acid derivative (a8) (0.34 g, 1 mmol), HBTU (0.57 g, 1.5 mmol) and TEA (0.5 mL) in DCM (20 mL). The reaction mixture was stirred at room temperature overnight. The solvent was evaporated under reduced pressure. The residue was partitioned between 50 mL of ethyl acetate and 20 mL of water, and the aqueous layer was extracted with 20 mL of ethyl acetate (over 3 times). The combined organic extracts were dried over anhydrous MgSO4, the solvent was removed under reduced pressure, and the product was purified by column chromatography to give the descried amide derivative.
N-Benzyl-4-(3-(6-methoxybenzo[d]thiazol-2-yl)ureido)benzamide (a9)
The title compound was prepared according to the general procedure for amide synthesis using benzylamine. White solid; yield: 64%; mp 284–286 °C; 1H NMR (300 MHz, DMSO) δ 10.76 (s, 1H), 9.39 (s, 1H), 8.94 (t, J = 5.9 Hz, 1H), 7.90 (d, J = 8.7 Hz, 2H), 7.67–7.50 (m, 4H), 7.36–7.30 (m, 4H), 7.25 (dq, J = 6.0, 4.1 Hz, 1H), 7.00 (dd, J = 8.8, 2.6 Hz, 1H), 4.49 (d, J = 6.0 Hz, 2H), 3.81 (s, 3H); MS (ESI): m/z = 433.04 (M + H)+.
1-(6-Methoxybenzo[d]thiazol-2-yl)-3-(4-(4-methylpiperazine-1-carbonyl)phenyl)urea (a10)
The title compound was prepared according to the general procedure for amide synthesis using N-methylpiperazine. White solid; yield: 72%; mp 284–286 °C; 1H NMR (500 MHz, DMSO) δ 10.83 (s, 1H), 9.41 (s, 1H), 7.57 (ddd, J = 11.9, 6.6, 3.6 Hz, 3H), 7.52 (d, J = 2.6 Hz, 1H), 7.41–7.33 (m, 2H), 6.99 (dd, J = 8.8, 2.6 Hz, 1H), 3.79 (s, 3H), 3.48 (s, 4H), 2.35 (s, 4H), 2.22 (s, 3H); 13C NMR (126 MHz, DMSO) δ 168.74, 157.74, 155.71, 152.27, 141.75, 139.85, 132.25, 129.78, 128.19, 119.84, 118.11, 114.41, 104.98, 55.58, 54.41, 45.70, 45.43; MS (ESI): m/z = 426.03 (M + H)+.
General procedure for ether dealkylation
1 M BBr3 solution in CH2Cl2 (15 equiv.) was added dropwise via syringe under nitrogen to a stirred solution of the methyl ether derivative (1 equiv.) in CH2Cl2 at –78 °C. Then the reaction was maintained at –78 °C for 1 hour, after that allowed to reach room temperature and stirred for an additional 20 h. The mixture was cooled to 0 °C and H2O was carefully added (15–25 mL). The product was then repeatedly extracted with EtOAc and the organic layers were dried over anhydrous Na2SO4. Upon solvent removal the residue was purified by column chromatography.
1-(6-Hydroxybenzo[d]thiazol-2-yl)-3-phenylurea (b1)
The title compound was prepared by demethylation of compound a1 according to the general procedure for ether dealkylation. Off-white solid; yield: 83%; mp 260–262 °C; 1H NMR (500 MHz, CDCl3) δ 10.57 (s, 1H), 9.44 (s, 1H), 9.12 (s, 1H), 7.54–7.40 (m, 3H), 7.33 (t, J = 7.9 Hz, 2H), 7.23 (d, J = 2.2 Hz, 1H), 7.03 (dd, J = 17.6, 10.3 Hz, 1H), 6.84 (dd, J = 8.7, 2.4 Hz, 1H); MS (ESI): m/z = 286.08 (M + H)+; HRMS m/z = 286.0627 (calculated = 286.0645).
Phenyl (6-hydroxybenzo[d]thiazol-2-yl)carbamate (b2)
The title compound was prepared by demethylation of compound a4 according to the general procedure for ether dealkylation. White solid; yield: 68%, mp 119–120 °C; 1H NMR (300 MHz, DMSO) δ 12.36 (s, 1H), 9.52 (s, 1H), 7.53 (d, J = 8.7 Hz, 1H), 7.45 (dt, J = 10.6, 2.2 Hz, 2H), 7.35–7.24 (m, 4H), 6.88 (dd, J = 8.7, 2.5 Hz, 1H); MS (ESI): m/z = 286.98 (M + H)+; HRMS m/z = 287.0466 (calculated = 287.0485).
N-(6-Hydroxybenzo[d]thiazol-2-yl)-2-(3-hydroxyphenyl)acetamide (b3)
The title compound was prepared by demethylation of compound a7 according to the general procedure for ether dealkylation. Beige powder; yield: 70%; mp 227–228 °C; 1H NMR (500 MHz, DMSO) δ 12.36 (s, 1H), 9.52 (s, 1H), 9.37 (s, 1H), 7.54 (dd, J = 8.5, 5.9 Hz, 1H), 7.26 (q, J = 2.5 Hz, 1H), 7.09 (dt, J = 25.2, 12.8 Hz, 1H), 6.92–6.81 (m, 1H), 6.81–6.71 (m, 2H), 6.65 (ddd, J = 8.1, 2.3, 1.1 Hz, 1H), 3.68 (s, 2H); 13C NMR (126 MHz, DMSO) δ 169.68, 157.32, 154.93, 154.15, 141.53, 136.04, 132.70, 129.34, 121.07, 119.80, 116.03, 115.19, 113.81, 106.45, 41.85; MS (ESI): m/z = 301.15 (M + H)+; HRMS m/z = 301.0622 (calculated = 301.0641).
N-Benzyl-4-(3-(6-hydroxybenzo[d]thiazol-2-yl)ureido)benzamide (b4)
The title compound was prepared by demethylation of compound a9 according to the general procedure for ether dealkylation. Light brown solid; yield: 73%; mp 200–201 °C; 1H NMR (500 MHz, DMSO) δ 10.75 (s, 1H), 9.47 (s, 1H), 9.42 (s, 1H), 8.95 (t, J = 6.0 Hz, 1H), 7.89 (d, J = 8.8 Hz, 2H), 7.60 (d, J = 8.6 Hz, 2H), 7.53–7.40 (m, 1H), 7.36–7.28 (m, 4H), 7.28–7.19 (m, 2H), 6.85 (dd, J = 8.7, 2.5 Hz, 1H), 4.48 (d, J = 6.0 Hz, 2H);13C NMR (126 MHz, DMSO) δ 165.60, 154.88, 154.21, 147.81, 140.37, 139.82, 131.12, 128.30, 128.23, 127.17, 126.66, 122.55, 117.72, 115.32, 114.81, 112.70, 106.70, 42.53; MS (ESI): m/z = 418.97 (M + H)+; HRMS m/z = 419.1149 (calculated = 419.1172).
1-(6-Hydroxybenzo[d]thiazol-2-yl)-3-(4-(4-methylpiperazine-1-carbonyl)phenyl)urea (b5)
The title compound was prepared by demethylation of compound a10 according to the general procedure for ether dealkylation. White solid; yield: 48%; mp 153–155 °C; 1H NMR (300 MHz, DMSO) δ 10.80 (s, 1H), 9.46 (s, 1H), 9.39 (s, 1H), 7.57 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 8.6 Hz, 1H), 7.37 (d, J = 8.5 Hz, 2H), 7.23 (d, J = 2.3 Hz, 1H), 6.84 (dd, J = 8.6, 2.4 Hz, 1H), 3.51 (s, 4H), 2.36 (s, 4H), 2.22 (s, 3H); MS (ESI): m/z = 412.03 (M + H)+; HRMS m/z = 412.1415 (calculated = 412.1438).
Biological assays
Protein kinase assays were done exactly as described before,14 except that the ATP concentration were varied as indicated in the legends. Metabolic stability assay is described in the ESI.†
Molecular modeling
Ab initio calculations of the equilibrium conformer of AHS-211 and energy levels were carried out as previously described.19 Molecular docking simulations of the b5 binding to human Dyrk1A (extracted from PDB entries ; 5A3X or ; 5A4E, respectively) were performed as previously described.20
Conflicts of interest
The authors declare no competing interest.
Supplementary Material
Acknowledgments
The support by the Deutsche Forschungsgemeinschaft (DFG) (EN381/2-3) to ME is greatly acknowledged. We thank Mariam G. Tahoun (Helmholtz Institute for Pharmaceutical Research Saarland) for carrying out the metabolic stability studies.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00142a
References
- Becker W., Sippl W. Rev. Geophys. 2011;278:246–256. doi: 10.1111/j.1742-4658.2010.07956.x. [DOI] [PubMed] [Google Scholar]
- Becker W., Weber Y., Wetzel K., Eirmbter K., Tejedor F. J., Joost H.-G. J. Biol. Chem. 1998;273:25893–25902. doi: 10.1074/jbc.273.40.25893. [DOI] [PubMed] [Google Scholar]
- Wegiel J., Dowjat K., Kaczmarski W., Kuchna I., Nowicki K., Frackowiak J., Mazur Kolecka B., Wegiel J., Silverman W. P., Reisberg B., deLeon M., Wisniewski T., Gong C.-X., Liu F., Adayev T., Chen-Hwang M.-C., Hwang Y.-W. Acta Neuropathol. 2008;116:391–407. doi: 10.1007/s00401-008-0419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Liang Z., Wegiel J., Hwang Y.-W., Iqbal K., Grundke-Iqbal I., Ramakrishna N., Gong C.-X. FASEB J. 2008;22:3224–3233. doi: 10.1096/fj.07-104539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R., Kamino K., Yamamoto M., Nuripa A., Kida T., Kazui H., Hashimoto R., Tanaka T., Kudo T., Yamagata H., Tabara Y., Miki T., Akatsu H., Kosaka K., Funakoshi E., Nishitomi K., Sakaguchi G., Kato A., Hattori H., Uema T., Takeda M. Hum. Mol. Genet. 2007;16:15–23. doi: 10.1093/hmg/ddl437. [DOI] [PubMed] [Google Scholar]
- Khor B., Conway K. L., Xavier R. J., Gagnon J. D., Aldrich L. N., Sundberg T. B., Schirmer M., Shamji A. F., Schreiber S. L., Goel G., Roche M. I., Medoff B. D., Tran K., Tan P. H., Shaw S. Y., Paterson A. M., Sharpe A. H., Mordecai S., Dombkowski D., Bhan A. K., Roychoudhuri R., Restifo N. P., O'Shea J. J. eLife. 2015;4:e05920. [Google Scholar]
- Soppa U., Schumacher J., Pasqualon T., Becker W., Florencio O. V., Tejedor F. J. Cell Cycle. 2014;13:2084–2100. doi: 10.4161/cc.29104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori-McKenney K. M., Li T., Meltzer S., Jan L. Y., McKenney R. J., Vale R. D., Huang H. H., Wiita A. P., Jan Y. N. Neuron. 2016;90:551–563. doi: 10.1016/j.neuron.2016.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejedor F. J., Hammerle B. Rev. Geophys. 2011;278:223–235. doi: 10.1111/j.1742-4658.2010.07954.x. [DOI] [PubMed] [Google Scholar]
- Becker W. Cell Cycle. 2012;11:3389–3394. doi: 10.4161/cc.21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraki M., Ohkawara B., Hosoya T., Onogi H., Koizumi J., Koizumi T., Sumi K., Yomoda J.-I., Murray M. V., Kimura H., Furuichi K., Shibuya H., Krainer A. R., Suzuki M., Hagiwara M. J. Biol. Chem. 2004;279:24246–24254. doi: 10.1074/jbc.M314298200. [DOI] [PubMed] [Google Scholar]
- Ogawa Y., Nonaka Y., Goto T., Ohnishi E., Hiramatsu T., Kii I., Yoshida M., Ikura T., Onogi H., Shibuya H., Hosoya T., Ito N., Hagiwara M. Nat. Commun. 2010;1:86. doi: 10.1038/ncomms1090. [DOI] [PubMed] [Google Scholar]
- Rothweiler U., Stensen W., Brandsdal B. R. O., Isaksson J., Leeson F. A., Engh R. A., Svendsen J. S. M. E. J. Med. Chem. 2016;59:9814–9824. doi: 10.1021/acs.jmedchem.6b01086. [DOI] [PubMed] [Google Scholar]
- Schmitt C., Miralinaghi P., Mariano M., Hartmann R. W., Engel M. ACS Med. Chem. Lett. 2014;5:963–967. doi: 10.1021/ml500059y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spadaro A., Negri M., Marchais-Oberwinkler S., Bey E., Frotscher M. PLoS One. 2012;7:e29252. doi: 10.1371/journal.pone.0029252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahtouh T., Elkins J. M., Filippakopoulos P., Soundararajan M., Burgy G., Durieu E., Cochet C., Schmid R. S., Lo D. C., Delhommel F., Oberholzer A. E., Pearl L. H., Carreaux F., Bazureau J.-P., Knapp S., Meijer L. J. Med. Chem. 2012;55:9312–9330. doi: 10.1021/jm301034u. [DOI] [PubMed] [Google Scholar]
- Bain J., McLauchlan H., Elliott M., Cohen P. Biochem. J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuny G. D., Robin M., Ulyanova N. P., Patnaik D., Pique V., Casano G., Liu J.-F., Lin X., Xian J., Glicksman M. A., Stein R. L., Higgins J. M. G. Bioorg. Med. Chem. Lett. 2010;20:3491–3494. [PMC free article] [PubMed] [Google Scholar]
- Miralinaghi P., Schmitt C., Hartmann R. W., Frotscher M., Engel M. ChemMedChem. 2014;9:2294–2308. doi: 10.1002/cmdc.201402050. [DOI] [PubMed] [Google Scholar]
- ElHady A. K., Abdel-Halim M., Abadi A. H., Engel M. J. Med. Chem. 2017;60:5377–5391. doi: 10.1021/acs.jmedchem.6b01915. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.