

 Several synthetic combretastatin A4 (CA-4) derivatives were recently prepared to increase the drug efficacy and stability of the natural product isolated from the South African tree Combretum caffrum.
Several synthetic combretastatin A4 (CA-4) derivatives were recently prepared to increase the drug efficacy and stability of the natural product isolated from the South African tree Combretum caffrum.
Abstract
Several synthetic combretastatin A4 (CA-4) derivatives were recently prepared to increase the drug efficacy and stability of the natural product isolated from the South African tree Combretum caffrum. A group of ten 3-amino-2-azetidinone derivatives, as combretastatin A4 analogues, was selected through docking experiments, synthesized and tested for their anti-proliferative activity against the colon cancer SW48 cell line. These molecules, through the formation of amide bonds in position 3, allow the synthesis of various derivatives that can modulate the activity with great resistance to hydrolytic conditions. The cyclization to obtain the 3-aminoazetidinone ring is highly diastereoselective and provides a trans biologically active isomer under mild reaction conditions with better yields than the 3-hydroxy-2-azetidinone synthesis. All compounds showed IC50 values ranging between 14.0 and 564.2 nM, and the most active compound showed inhibitory activity against tubulin polymerization in vitro, being a potential therapeutic agent against colon cancer.
Introduction
Combretastatins are a group of polyhydroxylated stilbenes isolated from the South African tree Combretum caffrum.1,2 They are natural-occurring well-known microtubule destabilizing agents that inhibit the polymerization of tubulin by binding to its colchicine binding site. Combretastatin A4 (CA-4, Fig. 1), the main component of this family of natural products, is in advanced clinical trials for cancer therapy because it exhibits potent anticancer activity against a panel of human cancer cells including multi-drug resistant ones.3
Fig. 1. The main component of combretastatins: combretastatin A4 and its phosphate derivative.

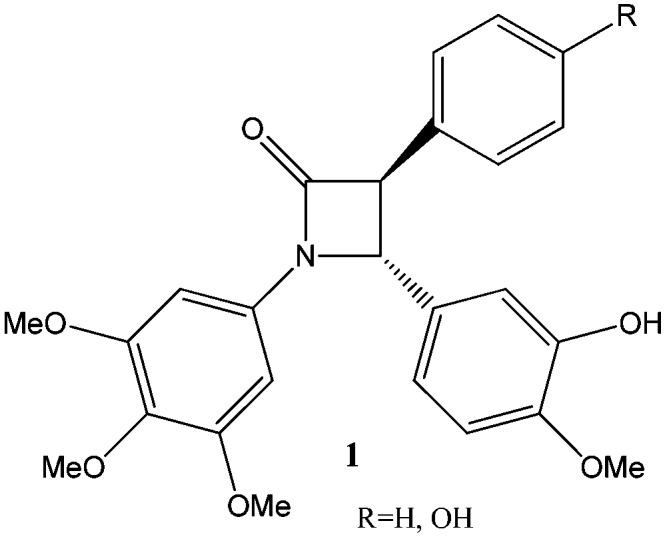
CA-4 has anti-angiogenic effects inducing apoptosis in various human tumor cell lines,4–7 through blocking of the blood flow, vascular rupture and cellular necrosis. Two problems, however, have limited their use as therapeutic agents for a long time: low water solubility and cis/trans CC double bond isomerization, which may occur during storage and administration, causing a dramatic loss of activity. Between the two stereoisomers, the cis form is much more active than the trans one, due to the pharmacophore of the colchicine binding site, which encompasses the trimethoxybenzene ring in cis linked to a methoxy-containing arene.8 The low solubility has been overcome by a water-soluble phosphate prodrug9 (Fig. 1), which is currently under investigation in human clinical trials (phase III) as an anticancer drug.4,9,10 The low cis isomer stability has been approached by designing a variety of conformationally restricted cis-locked analogues. Many structural modifications of combretastatin A4 have been proposed by incorporating the stilbene double bond into a carbocyclic or heterocyclic moiety. Carbocyclic derivatives, containing a phenyl ring, or a triatomic or a pentaatomic ring, have to be cited, as well as nitrogen or oxygen-containing heterocyclics,11 triazoles,12 tetrazoles,13 isooxazoles,14 imidazoles,15 pyrazoles,16 and pyridines.17 Other rings, such as azetidinones, have been inserted between the two phenyl groups of combretastatin A4.18,19 In addition, trans-3,4-disubstituted azetidinones show strong anti-proliferative activity against several cancer cell lines.20,21 Combretastatin-like compounds bearing an azetidinone unit between the A and B rings were presented by N. M. O'Boyle and colleagues.22,23 They firstly synthesized a few derivatives of trans 1,3,4 tri-aryl azetidinone 1 (Fig. 2) which inhibited the polymerization of tubulin with a 6-fold rate of inhibition in comparison with the natural compound CA-4.22,23
Fig. 2. Triaryl derivatives of azetidinone proposed by O'Boyle and coworkers.22.
We recently developed a trans-3-hydroxy-2-azetidinone, bearing in position 1 the A ring and in position 4 the B ring of CA-4 (compound 2) (Fig. 3).24 The racemic mixture of this compound showed both high stability and solubility (1700 μM) in aqueous media due to the presence of a second hydroxyl group in position 3. It was synthetized under thermodynamic control of the reaction at 100 °C with a total yield of 33%. Compound 2 induced inhibition of tubulin polymerization and subsequent G2/M arrest in cancer cells.24 At the same time, activation of AMP-activated protein kinase (AMPK) and induction of apoptosis were also reported.24 Moreover, the drug displayed significant nanomolar cytotoxic activity against different cancer cell lines, as well as a tumor growth delay in a colorectal cancer mouse xenograft model, confirming its potential therapeutic action against colon cancer.25 In the present paper, we report the synthesis of compound 3 (Fig. 3), in which the hydroxyl group of compound 2 was substituted by an amino group, as well as a series of its derivatives. Remarkably, the synthesis of compound 3 was easier than that of compound 2 because in the Staudinger reaction, the trans closure of the azetidinone ring took place at room temperature with an overall yield of 50%, with only a few traces of the cis isomer.
Fig. 3. Azetidinone derivatives proposed by Tripodi and coworkers.24.

In order to select derivatives of compound 3 which could have pronounced activity, we performed a preliminary docking study before biological tests. The presence of the amino group permits the easy preparation of a series of derivatives by transforming NH2 to its corresponding amido group. The variability in size and functions of the introduced substituents can be the source of valuable insights into substrate binding site interactions.
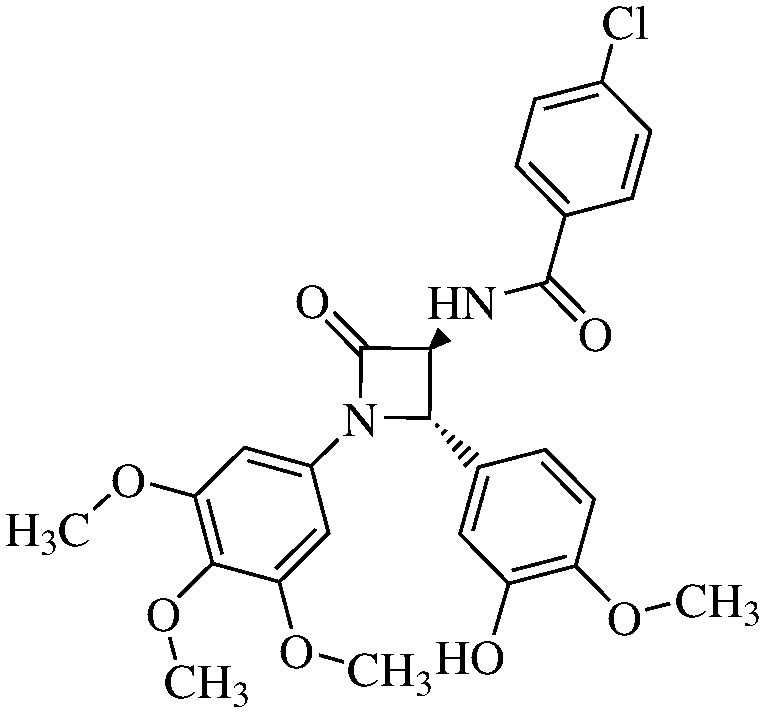
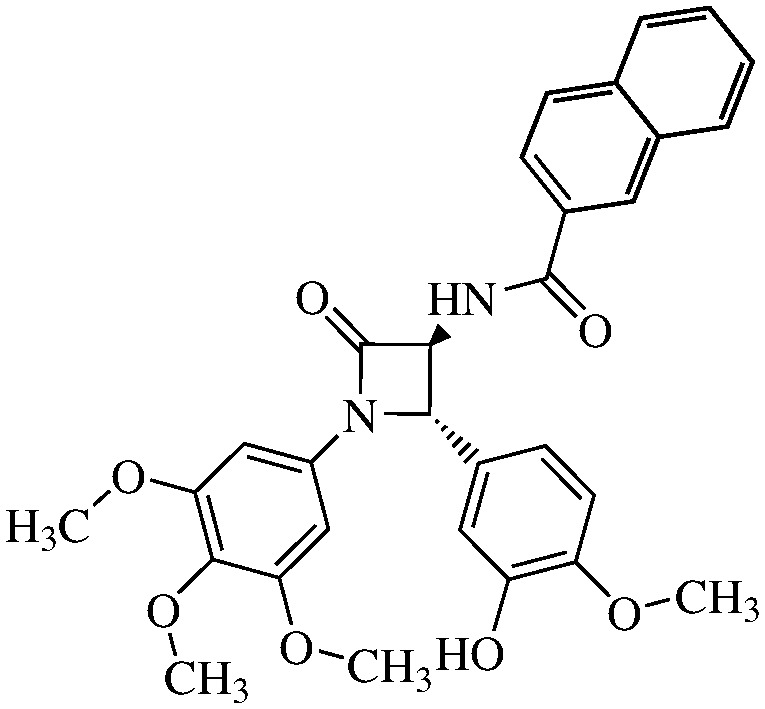
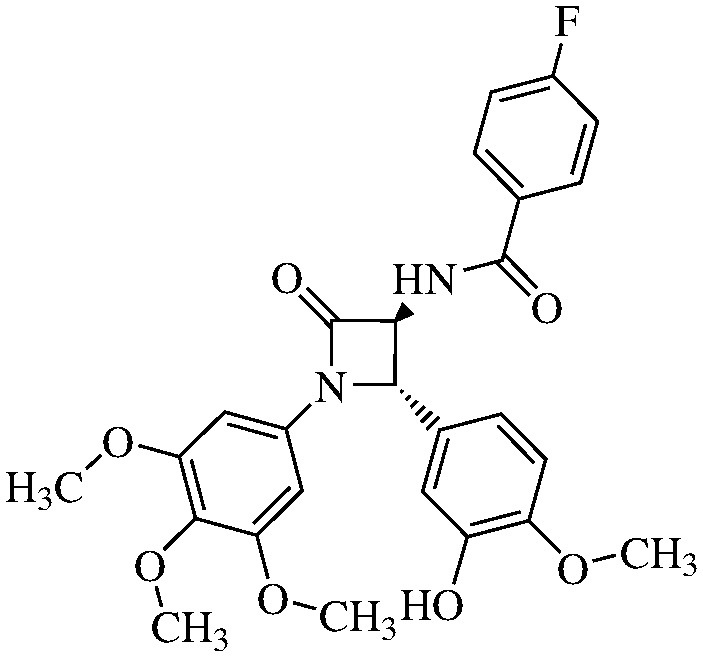
We hypothesized the preparation of 10 derivatives (compounds 11a–j) (Table 1), where the R residue derived from the acidic part of the amido group varies from acetic to naphthoic acids. We included short chain, aromatic, heteroaromatic, cyclic and polycyclic derivatives with diverse functional groups.
Table 1. Structures and IC50 values of the indicated compounds in SW48 cell lines measured by the MTT assay after 72 h of treatment (0.1% DMSO was used as a vehicle control).
| Entry | Compound | IC50 (nM) | Entry | Compound | IC50 (nM) |
| 11a |
|
270.7 ± 76.6 | 11f |
|
220.2 ± 76.5 |
| 11b |
|
384.2 ± 117.3 | 11g |
|
49.4 ± 1.1 |
| 11c |
|
108.4 ± 11.6 | 11h |
|
244.2 ± 64.2 |
| 11d |
|
564.2 ± 61.7 | 11i |
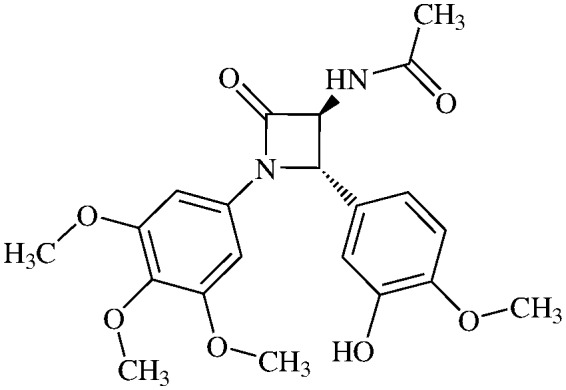
|
14.0 ± 2.1 |
| 11e |
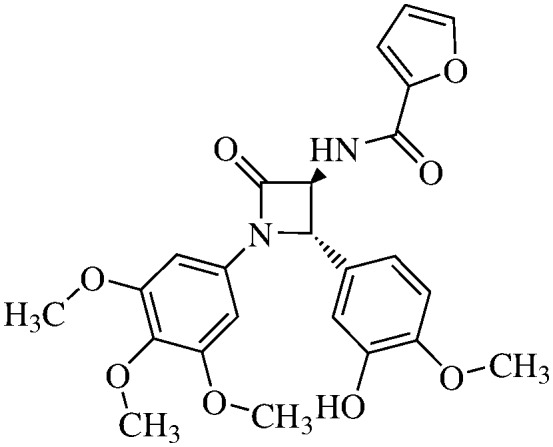
|
18.6 ± 1.7 | 11j |
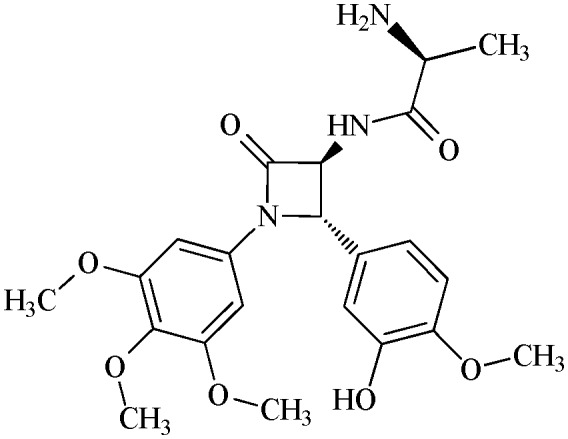
|
90.6 ± 7.3 |
First, we examined the docking of compounds 2a and 2b showing anticancer activity in previous studies (Fig. 4).24 Molecular docking was performed generating 100 binding poses for each compound. Considering the similarity of the energies obtained from the molecular docking of these compounds (Tables S1 and S2†), we performed a cluster analysis on the 100 conformations obtained from each run using an RMSD threshold of 2 Å. We considered the most populated cluster of each compound as the best docked conformation. Compound 2a (enantiomer SS) showed the known interaction of the trimethoxyphenyl group that came into contact with Cysβ241 and Valβ238. In addition, it interacted with Aspβ251 through hydrogen bonds. Compound 2b (enantiomer RR) maintained the interaction of the trimethoxyphenyl group with Cysβ241 and Valβ238. In contrast, it did not interact with Aspβ251, suggesting that the two enantiomers did not share the same docking conformation.
Fig. 4. SS an RR enantiomers of compound 2, previously tested as anticancer agents.

Then, molecular docking studies were carried out on compounds 11a–j in order to assess their ability to bind to the colchicine site of β tubulin.
RR enantiomers showed low binding capability, displaying a large number of clusters of different conformations. Compound 11i resulted to be the most interesting compound among the RR enantiomers, displaying a very populated cluster containing 85% of the conformations. Details regarding binding energies and clustering are reported in Table S1.†
Much more interesting results were obtained from the molecular docking of SS enantiomers. In this case, most of the docked compounds showed a more highly populated cluster with respect to RR compounds and binding energies around –5 kcal mol–1 (Table S2†). Compounds 11d, 11f and 11g were not suitable to bind within the colchicine site, due to the steric hindrance of the R substituents of the amide group. This was supported by highly diversified docked conformations. Compounds 11a, 11b, 11c, 11e, 11h and 11j shared the same orientation within the binding site (Fig. 5). Like combretastatin compounds,8 the trimethoxyphenyl group came into contact with Cysβ241 and Valβ238. Moreover, the amidic group in position 3 interacted with Metβ259 through a hydrogen bond (Fig. 6). The most interesting compound, however, was 11i, showing a single cluster of 100 conformations.
Fig. 5. Superimposition of docking poses of compounds 11a (blue), 11b (red), 11c (orange), 11e (yellow), 11h (green), and 11j (purple).

Fig. 6. Interactions of the compound 11a. In the illustration, tubulin α (orange) and β (cyan) subunits are represented. Trimethoxyphenyl group came into contact with residues Cysβ241 and Valβ238, as well as HB with residue Metβ259. This interaction pattern was common to compounds 11a, 11b, 11c, 11e, 11h, and 11j.

This compound showed a different docking pose with respect to the aforementioned ones. It interacted with Asnα101 and Aspβ251 through hydrogen bonds, while the orientation of the trimethoxyphenyl group was towards residues Cysβ241 and Valβ238 as in the previous cases (Fig. 7). Moreover, compound 11i showed a very similar docking pose to compounds already studied by our group, namely trans 3-hydroxy-1,4-diaryl-2-azetidinone (2b) (Fig. 8).
Fig. 7. Interactions of compound 11i. In the illustration, tubulin α (orange) and β (cyan) subunits were represented. Trimethoxyphenyl group is in contact with residues Cysβ241 and Valβ238. Moreover, two hydrogen bonds were observed between the amide group in position 3 and Asnα101 as well as between the ketone oxygen of the scaffold and Aspβ251. This interaction pattern was common to compounds 11i and 2b.

Fig. 8. Superimposition of 11i (blue) and 2a (red) docking poses.

In keeping with these results, the syntheses of all the derivatives were performed.
Chemistry
In this work, we focused our attention on the preparation of a small library of 3-amido-1,4-diaryl-2-azetidinones 11a–j (Table 1), with the goal of improving the biological activity and elucidating the biological effects by the introduction of specific ligands useful to selectively target tumor cells.
3,4-trans-3-Amino-4-(3-hydroxy-4-methoxyphenyl)-1-(3,4,5-trimethoxyphenyl)-azetidin-2-one 9 was synthesized as previously reported by the authors,24via the Staudinger approach between a properly selected imine 6 and phthalylglycyl chloride as the ketene precursor, followed by hydrolysis with hydrazine dihydrochloride at 60 °C (Scheme 1). The stereochemistry of the products obtained from the Staudinger cycloaddition was affected by the experimental conditions (such as solvent, temperature and order of addition of the reagents) and by the type of the ketene precursor: acetoxyacetyl chloride or phthalylglycyl chloride, used to introduce an OH group or a NH2 group, respectively, at the position 3 of the azetidin-2-one. In the latter case, the best results were obtained by dropwise addition of a 4-fold excess of acid chloride 7 to a methylene chloride solution of imine 6 and dry triethylamine at 0 °C, followed by stirring at room temperature for twenty-four hours. Hydrolysis of the Staudinger adduct with hydrazine afforded only trans-diastereoisomer 9 in 61% total yield from the imine. A different approach,26 based on the use of in situ generated N-chloramines as imine precursors, was tested, without improving the yields of the reaction.
Scheme 1. Reagents and conditions: a) anhydrous EtOH; b) Et3N/CH2Cl2; c) NH2NH2·2HCl, Et3N, MeOH, 60 °C; d) N,N′-disuccinimidyl carbonate, Et3N, CH3CN; e) NH2NH2·2HCl, Et3N, MeOH, r.t.

With respect to previously reported 3-hydroxy-1,4-diaryl-2-azetidinones, 3-amino-1,4-diaryl-2-azetidinones could a priori display different and hopefully improved activity, and the nitrogen atom at position 3 can be derivatized to give stable amides, easily prepared by reaction with a properly selected carboxylic acid, RCOOH. In order to obtain a library of compounds able to interact with the receptors in a variety of ways, R has been varied in terms of polarity, size, and ability to act as a hydrogen bond acceptor or donor and to give different types of interactions, namely dipole–dipole, β-stacking and hydrophobic interactions.
Amides 11a–i were obtained by a five-step synthesis. As already described,24 imine 6 was obtained by reaction of compounds 4 and 5 in anhydrous ethyl alcohol: the solid compound 6 was subjected to Staudinger cyclization with phthalyl glycine chloride and was transformed into β-lactam 8, which was easily hydrolyzed to compound 9. 3,4-trans-3-Amino-4-(3-hydroxy-4-methoxyphenyl)-1-(3,4,5-trimethoxyphenyl)-azetidin-2-one 9 was first added to an acetonitrile solution of the selected carboxylic acid, previously activated by reaction with N,N′-disuccinimidyl carbonate (overnight at room temperature). A mixture of the N,O-diacyl derivative 10 and amide 11 was obtained. Pure samples of 11a–j were obtained by flash-chromatography, whereas, the presence of N,O-diacyl derivatives 10a–j was recognized in the MS spectra of the mixtures (10a–j and 11a–j) which were directly treated with hydrazine hydrochloride at room temperature to hydrolyse the ester moiety. The target amides 11a–j were thus obtained with total yields ranging from 60% to 81% depending on the R substituents of the amide group. The synthesis of the amide 11j required one additional step as tert-butoxycarbonyl alanine was used as an acylating reagent: the removal of the protective tert-butoxycarbonyl moiety was easily accomplished by treatment with trifluoroacetic acid in methylene chloride.
Biological results
Compounds 11a–j were tested against the SW48 human colon cancer cell line by testing their antiproliferative activity. They retained their nanomolar cytotoxic activity, and among them 11i, with a small alkyl group (methyl), showed the highest activity, comparable to the activity of natural CA-4 and of the previously reported 3-hydroxy azetidinone 2 (Table 1, Fig. S1†). The activity was decreased by the introduction of a primary amino group on the small alkyl chain in 11j and, even more, by the substitution of the methyl group with the more sterically demanding cyclohexyl group in 11h. Heteroaromatic rings, as in 11e and 11g, were approximately comparable to that in 11i. In the presence of aromatic rings, as in 11a–11d, the activity was further reduced. The results of the activity tests were consistent with the assessments from the docking study, where the compounds containing bulky aromatic residues did not converge to a single structure.
CA-4 is known to act by inhibiting tubulin polymerization.1 Thus, we evaluated the effect of compound 11i, which showed the highest activity against SW48 cells, on the in vitro polymerization of purified tubulin, and CA-4 was used as a positive control (Table 2, Fig. S2†). In the assembly assay, 11i showed an IC50 value of 5.05 μM, higher than that of CA-4, but indicative of the fact that one of the molecular targets of this active compound was tubulin, as reported for other azetidinone compounds.24
Table 2. In vitro inhibition of tubulin polymerization.
| Compound | IC50 μM tubulin polymerization |
| 11i | 5.05 ± 1.2 |
| CA-4 | 1.32 ± 0.2 |
Conclusions
Insertions of azetidinone rings into the combretastatin skeleton have been valid modifications because they prevented the isomerization of the cis double bond, while preserving the activity of some derivatives comparable to that of the natural compound. They made the natural compound more stable and the synthesis provided good yields. In particular, the 3-aminoazetidinones, compared to the 3-hydroxyazetidinones, which bear a hydroxyl group in position 3 of the β-lactam ring, were synthesized with higher yields since the Staudinger reaction almost exclusively provided the 3,4-trans isomer. Furthermore, the derivatization of the amino group afforded amide derivatives which were stable under hydrolytic conditions. Remarkably, these new derivatives showed nanomolar anti-proliferative activity against SW48 cells, probably acting by inhibiting tubulin polymerization, opening the route to a new class of potential therapeutic agents against colon cancer.
Experimental section
Docking studies
The tubulin X-ray structure in a complex with combretastatin was obtained from the protein data bank (PDB ID 5LYJ 8). The pocket analysis and the docking calculations were performed only on one of the dimers in the asymmetric unit, labelled chains A and B. Combretastatin was removed from the structure before docking our compounds. Docking was carried out with Autodock 4.2,27 employing a Lamarckian genetic algorithm.28 100 independent runs per molecule were performed. In each run, a population of 50 individuals evolved along 27 000 generations and a maximum number of 25 million energy evaluations were performed. The best fit (lowest docked energy) solutions of the 100 independent runs were stored for subsequent analysis. The visual inspection of docked structures was carried out using VMD.29
Chemistry
All commercially available reagents were purchased from Sigma-Aldrich and used without further purification. Preparative separations were usually performed by flash-column chromatography on silica gel (Merck grade 9385). Thin-layer chromatography was conducted on silica gel (Merck, 10 × 5 cm, silica gel 60 F254): zones were detected visually by ultraviolet irradiation (254 nm) or by exposition to iodine vapors. 1H NMR and 13C NMR spectra were recorded at 300 MHz and 400 MHz on Bruker instruments (AC 300 UltrashieldTM 400), using deuterochloroform solutions, unless otherwise specified, and chemical shifts were represented as δ-values relative to the internal standard TMS. ESI-mass spectra were recorded on a Bruker Esquire 3000 Plus. The synthesized compounds subjected to biological tests have a purity of ≥95% determined by HPLC (Phenomenex, Nucleosil 250 × 3.2 mm column, 5 μm, C18; mobile phase 0.05 M phosphate buffer pH 7/acetonitrile, 7 : 3; flow rate 1.5 mL min–1), and they have been filtered in sterile atmosphere with 0.20 μm filters.
3-Hydroxy-4-methoxybenzylidene-(3,4,5-trimethoxyphenyl)amine (6)
Under an inert atmosphere, compound 4 (1.9, 10.1 mmol) and compound 5 (1.5 g, 10.1 mmol) were dissolved together at room temperature with stirring, in 10 mL ethanol previously dried on molecular sieves. The reaction was followed by TLC (pre-treatment of the plate with CH2Cl2 : Et3N 95 : 5, then eluted with AcOEt : Esano 1 : 1). After 2 h, NMR analysis showed the formation of imine 6 which was observable also by the presence of a precipitate. The mixture was filtered to collect the solid product. The filter was washed twice with dichloromethane to dissolve the reaction product. The solvent was removed and compound 6 was collected, pure enough for the next step (yields = 93%). 1H-NMR (CDCl3): δ 8.41 (1H, s), 7.63 (1H, s), 7.46 (1H, d, J = 7.2 Hz), 6.96 (1H, d, J = 7.2 Hz), 6.53 (2H, s), 5.77 (1H, s), 3.99 (3H, s), 3.92 (6H, s), 3.88 (3H, s). EI-MS (m/z): 318 (M+).
(±)-3,4-trans-3-Amino-4-(3-hydroxy-4-methoxyphenyl)-1-(3,4,5-trimethoxyphenyl)-azetidin-2-one ((±)-trans-9)
A solution of phthalylglycyl chloride (5.38 g, 24 mmol), in anhydrous methylene chloride (7 mL), was added dropwise at 0 °C under nitrogen to a stirred solution of imine 6 (1.9 g, 6 mmol) and TEA (8.3 mL, 60 mmol) in anhydrous methylene chloride (7 mL). The solution was maintained at 0 °C for 1 h and then allowed to reach room temperature and stirred for 24 h. HCl (1 N) was added (40 mL), and the two phases were stirred for 40 min. The aqueous phase was separated and extracted twice with dichloromethane. Organic phases were washed with a saturated solution of NaHCO3, dried with sodium sulfate, filtered, and concentrated under reduced pressure, affording a brown oil which was purified by flash column chromatography (silica gel; eluent gradient of n-hexane/ethyl acetate from 7/3 to 4/6). The isomer (±)-trans-8 only was isolated with a 65% yield. The isomer (±)-cis-8 was formed only in traces from the reaction. (±)-trans-8: 1H-NMR (CDCl3) δ 8–7.6 (8H, m), 7.29 (1H, dd, J1 = 8.4 Hz, J2 = 2.2 Hz), 7.19 (1H, d, J = 2.2 Hz), 7.00 (1H, d, J = 8.4 Hz), 6.57 (2H, s), 5.28 (2H, s), 4.71 (2H, s), 3.85 (3H, s), 3.76 (3H, s), 3.70 (6H, s). 13C NMR (CDCl3): δ 167.46, 166.96, 165.41, 162.03, 140.09, 134.79, 134.53, 133.40, 132.15, 131.86, 128.37, 126.40, 125.08, 124.27, 123.87, 121.47, 113.64, 95.50, 67.78, 61.07, 56.24, 38.89. ESI-MS: m/z 691 (M+).
Hydrazine dihydrochloride (752.5 mg, 6.223 mmol) was added, at 0 °C and under nitrogen, to a stirred suspension of (±)-trans-8 (870 mg, 1.245 mmol) in methanol (10 mL). TEA (3.5 mL, 24.89 mmol) was then added dropwise. The mixture was allowed to reach room temperature and then warmed at 50 °C for 5 h. The solvent was removed at reduced pressure, and the residue was treated with 1 N HCl (40 mL) and extracted with dichloromethane (3 × 10 mL). The aqueous phase was made alkaline by 3 N NaOH and extracted with dichloromethane (3 × 10 mL). The organic phase was dried (Na2SO4), and the solvent was removed at reduced pressure. A solid was obtained, which was purified by flash chromatography (silica gel; eluent n-hexane/ethyl acetate, 2/8) to afford the stereoisomer (±)-trans-9 (0.437 g, 1.17 mmol, 94% yield) as a yellow solid. 1H-NMR (CDCl3): δ 6.92 (1H, s), 6.89 (2H, s), 6.54 (2H, s), 4.54 (1H, d, J = 2.2 Hz), 4.05 (1H, d, J = 2.2 Hz), 3.88 (3H, s), 3.76 (3H, s), 3.71 (6H, s). 13C NMR (CDCl3): δ 168.05, 153.65, 147.11, 146.51, 134.80, 133.80, 130.14, 118.14, 117.99, 112.33, 111.22, 95.38, 69.70, 66.70, 61.09, 56.22. ESI-MS: m/z 374 (M+).
General procedure for the preparation of compounds 11a–h and 11j
To a stirred solution of the selected carboxylic acid RCOOH (0.88 mmol) in dry acetonitrile (6 mL), N,N′-disuccinimidyl carbonate DSC (0.88 mmol) and dry triethylamine (1.603 mmol) were added at 0 °C, under a nitrogen atmosphere. The stirring was continued overnight and 3,4-trans-3-amino-4-(3-hydroxy-4-methoxyphenyl)-1-(3,4,5-trimethoxyphenyl)-azetidin-2-one 9 (0.4 mmol) was added. The reaction was monitored by TLC (silica gel, eluted with methylene chloride–methanol), treated with a saturated solution of NaHCO3, and extracted with methylene chloride. The combined organic extracts were dried (Na2SO4) and the solvent was removed under reduced pressure. A mixture of 11a–i and 10a–i in varying amounts was obtained (the latter generally detected in the MS and/or NMR spectrum of the crude material) and directly hydrolyzed to 11a–i.
When Boc-N-methyl-l-alanine was used as an acyl donor, a mixture of Boc-protected N-acylated and N,O-di-acylated derivatives was obtained, directly hydrolyzed to the protected Boc-N-acyl derivative and deprotected with trifluoroacetic acid to afford 11j.
Hydrolysis of the ester group was achieved by addition of hydrazine dichloride (0.174 mmol) and dry triethyl amine (0.34 mmol) to a stirred solution of the mixture of 10a–j and 11a–j (0.087 mmol) in methanol (4 mL), under a nitrogen atmosphere. The reaction was monitored by TLC analysis (silica-gel). The reaction was then treated with KHSO4 solution (5%) and extracted with methylene chloride. The combined organic extracts were dried and the solvent was removed under reduced pressure. The crude material was flash chromatographed on silica gel, eluting with a gradient of methylene chloride–methanol affording pure 11a–j.
N,O-(Diacetyl)-3,4-trans-3-amino-1-(3,4,5-trimethoxyphenyl)-4-(3-hydroxy-4-methoxyphenyl)-azetidin-2-one (10i)
The title compound was prepared as well by addition of acetic anhydride (2 mL) to 3,4-trans-3-amino-4-(3-hydroxy-4-methoxyphenyl)-1-(3,4,5-trimethoxyphenyl)-azetidin-2-one 1. Removal of acetic anhydride under reduced pressure afforded the diacyl derivative in quantitative yields.
10i 1H NMR (300 MHz, CDCl3): δ ppm 7.19 (dd, 1H, J = 8.5, 1.7 Hz), 7.05 (d, 1H, J = 1.7 Hz), 6.95 (d, 1H, J = 8.5 Hz), 6.80 (d, 1H, J = 7.5 Hz), 6.48 (s, 2H), 4.94 (d, 1H, J = 1.7 Hz), 4.53 (dd, 1H, J = 7.4, 1.7 Hz), 3.81 (s, 3H), 3.75 (s, 3H), 3.69 (s, 6H), 2.28 (s, 3H), 2.06 (s, 3H). 13C (300 MHz, CDCl3): 171.45, 169.45, 164.88, 154.08, 152.16, 140.84, 135.12, 134.10, 129.46, 125.23, 121.82, 113.64, 95.73, 66.55, 63.27, 61.57, 56.67, 23.35, 21.25. MS (ESI): m/z 481 (M + Na).
Spectroscopic data of compounds 11a–j
N-(4-Chlorobenzoil)-3,4-trans-3-amino-1-(3,4,5-trimethoxyphenyl)-4-(3-hydroxy-4-methoxyphenyl)-azetidin-2-one
11a (65% yield). 1H NMR (400 MHz, CDCl3): δ ppm 7.79 (d, 2H, J = 7.5 Hz), 7.43 (d, 2H, J = 7.5 Hz), 7.23 (d, 1H, J = 6.5 Hz, NH), 6.98 (d, 1H, J = 2.5 Hz), 6.94 (dd, 1H, J = 7.5, 2.5 Hz), 6.88 (d, 1H, J = 7.5 Hz), 6.55 (s, 2H), 5.00 (d, 1H, J = 2.0 Hz), 4.81 (dd, 1H, J = 7.5 Hz, 2.0 Hz), 3.92 (s, 3H), 3.77 (s, 3H), 3.72 (s, 6H). 13C (400 MHz, CDCl3): 168.13, 165.29, 154.31, 147.43, 146.57, 136.15, 135.06, 130.76, 130.86, 129.90, 129.15, 118.92, 116.33, 116.14, 112.57, 111.63, 95.72, 66.83, 63.93, 61.51, 56.25, 56.13, 51.44. MS (ESI): m/z 535–537 (M + Na).
10a. MS (ESI): m/z 673–675 (M + Na).
N-(4-Fluorobenzoyl)-3,4-trans-3-amino-1-(3,4,5-trimethoxyphenyl)-4-(3-hydroxy-4-methoxyphenyl)-azetidin-2-one
11b (66% yield). 1H NMR (400 MHz, CDCl3): δ ppm 7.90 (dd, 2H, J = 8.5, JH–F = 5.5 Hz), 7.82 (d, 1H, J = 7.4 Hz, NH), 7.08 (dd, 2H, J = 8.5, JH–F = 8.4 Hz), 6.95 (d, 1 H, J = 1.5 Hz), 6.90 (dd, 1H, J = 8.1, 1.5 Hz), 6.84 (d, 1H, J = 8.1 Hz), 6.52 (s, 2H), 5.01 (s, 1H), 4.85 (d, 1 H, J = 7.4 Hz), 3.89 (s, 3H), 3.74 (s, 3H), 3.68 (s, 6H).13C (400 MHz, CDCl3): 168.05, 166.71 (JC–F = 253 Hz), 165.44, 154.95, 148.63, 147.87, 135.97, 134.87, 131.31 (JC–F = 9.1 Hz), 130.77, 130.44, 120.70, 116.98 (JC–F = 21.9 Hz), 113.55, 112.41, 95.82, 66.73, 63.73, 61.58, 56.65, 56.64, 51.45. MS (ESI): m/z 519 (M + Na). 10b. MS (ESI): m/z 641 (M + Na).
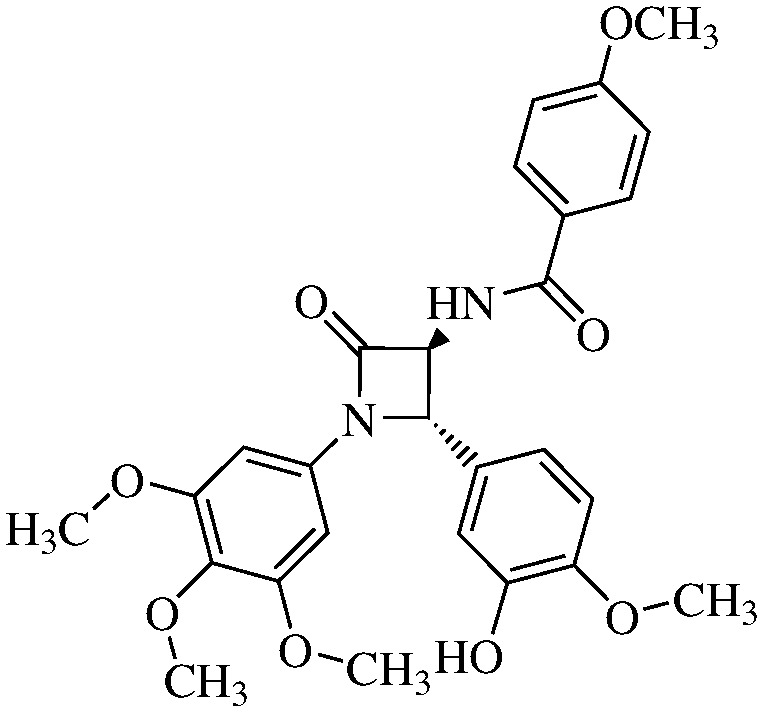
N-(4-Methoxybenzoyl)-3,4-trans-3-amino-1-(3,4,5-trimethoxyphenyl)-4-(3-hydroxy-4-methoxyphenyl)-azetidin-2-one
11c (64% yield). 1H NMR (400 MHz, CDCl3): δ ppm 7.82 (d, 2H, J = 7.6 Hz), 6.98 (s, 1H), 6.93 (dd, 1H, J1 = 7.5 Hz), 6.86 (d, 1H, J = 7.5 Hz), 6.56 (s, 2H), 5.25 (s, 1H), 4.79 (d, 1H, J = 6.4 Hz), 5.85 (d, 1H, J = 6.4 Hz, NH), 3.89 (s, 3H), 3.87 (s, 3H), 3.77 (s, 3H), 3.70 (s, 6H). 13C (300 MHz, CDCl3): δ ppm 167.38, 164.82, 162.61, 153.34, 147.27, 146.37, 134.54, 133.43, 129.88, 129.2, 125.04, 118.17, 113.78, 112.49, 111.19, 95.24, 66.2, 63.17 60.88, 60.03, 55.95, 55.38, 50.55. MS (ESI): m/z 531 (M + Na).
10c. MS (ESI): m/z 665 (M + Na).
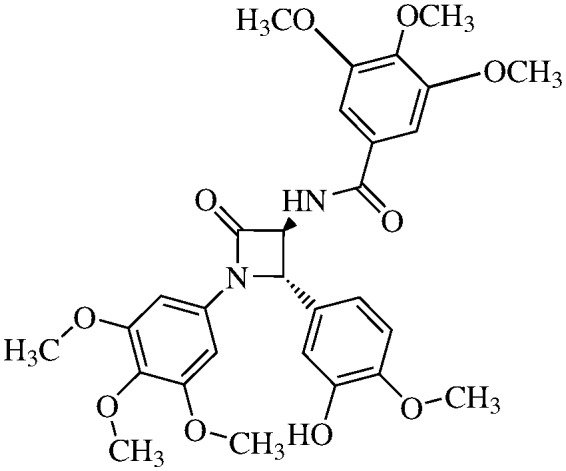
N-(3,4,5-Trimethoxybenzoyl)-3,4-trans-3-amino-1-(3,4,5-trimethoxyphenyl)-4-(3-hydroxy-4-methoxyphenyl)-azetidin-2-one
11d (60% yield). 1H NMR (400 MHz, CDCl3): δ ppm 8.34 (d, 1H, J = 6.5 Hz), 7.11 (s, 2H), 6.87 (d, 1H, J 0 1.0 Hz), 6.82 (dd, 1H, J = 8.8, 1.0 Hz), 6.78 (d, 1H, J = 8.8 Hz), 6.50 (s, 2H), 5.02 (d, 1H, J = 2.2 Hz), 4.71 (dd, 1H, J = 6.5), 3. 82 (s, 3H), 3. 81 (s, 3H), 3.80 (s, 6H), 3.73 (s, 3H), 3.69 (s, 3H), 3.62 (s, 3H). 13C (300 MHz, CDCl3): δ ppm 167.87, 165.82, 154.07, 153.99, 152.34, 148.09, 147.21, 135.08, 134.22, 129.99, 129.74, 128.58, 124.45, 118.75, 113.14, 111.94, 108.12, 105.47, 95.74, 93.33, 66.88, 63.34, 61.55, 56.77, 56.67, 56.70, 51.00. MS (ESI): m/z 591 (M + Na).
10d. MS (ESI): m/z 785 (M + Na).
N-(2-Furoyl)-3,4-trans-3-amino-1-(3,4,5-trimethoxyphenyl)-4-(3-hydroxy-4-methoxiphenyl)-azetidin-2-one
11e (76% yield). 1H NMR (400 MHz, CDCl3): δ ppm 8.07 (sdd, 1 h, J = 1.8, 0.9 Hz), 7.53 (dd, 1H, J = 3.5, 0.9 Hz), 7.92 and 7.91 (AA′ system, 2H, J = 7.3 Hz), 6.90 (s, 1H), 6.79 (dd, 1H, J = 3.5, 1.7 Hz), 6.61 (s, 2H), 5.10 (s, 1H), 4.72 (s, 1H), 3.87 (s, CH3), 3.72 (s, CH3), 3.70 (s, 2× CH3). 13C (300 MHz, CDCl3): δ ppm 165.52, 163.7, 153.91, 153.89, 147.87, 147.02, 146.41, 144.36, 135.02, 133.98, 129.76, 122.15, 118.89, 112.92, 111.83, 109.12, 95.76, 66.11, 63.92, 61.53, 56.59, 56.51. MS (ESI): m/z 491 (M + Na).
10e. MS (ESI): m/z 585 (M + Na).
N-(2-Naphthoyl)-3,4-trans-3-amino-1-(3,4,5-trimethoxyphenyl)-4-(3-hydroxy-4-methoxyphenyl)-azetidin-2-one
11f (60% yield). 1H NMR (400 MHz, CDCl3): δ ppm 8.39 (s, 1H), 8.08 (d, 1H, J = 5.9 Hz, NH), 7.90–7.82 (4H), 7.55 (t, 1H, J = 7.4 Hz), 7.50 (t, 1H, J = 7.4 Hz), 6.97 (s, 1H), 6.89 (d, 1H, J = 7.5 Hz), 6.81 (d, 1H, J = 7.5 Hz), 6.52 (s, 2H), 5.04 (s, 1H), 4.93 (d, 1H, J = 5.9 Hz), 3.85 (s, 3H), 3.73 (s, 3H), 3.65 (s, 6H). 13C (400 MHz, CDCl3): 168.28, 165.35, 153.98, 147.81, 147.02, 135.58, 135.08, 134.17, 133.15, 130.69, 130.00, 129.79, 129.07, 128.94, 128.58, 128.34, 127.40, 124.25, 118.96, 113.02, 111.79, 95.81, 95.69, 66.87, 63.82, 61.59, 56.63, 56.59, 56.56. MS (ESI): m/z 551 (M + Na).
10f. MS (ESI): m/z 705 (M + Na).
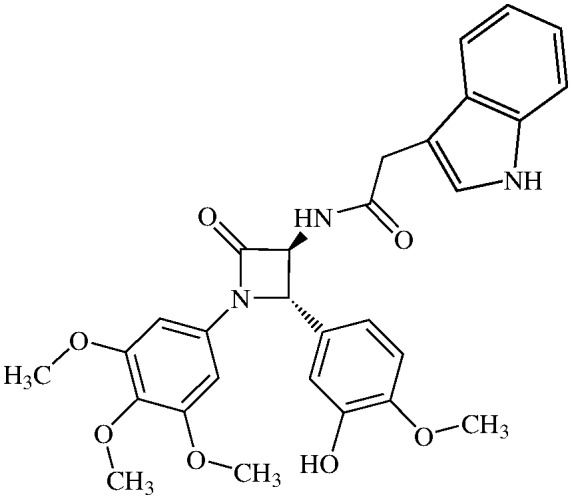
N-(Indolo-3-acetyl)-3,4-trans-3-amino-1-(3,4,5-trimethoxyphenyl)-4-(3-hydroxy-4-methoxyphenyl)-azetidin-2-one
11g (75% yield). 1H NMR (400 MHz, CDCl3): δ ppm 7.60 (d, 1H, J = 7.34 Hz), 7.41 (d, 1H, J = 7.34 Hz), 7.28 (S, 1H), 7.26 (t, 1H, J = 7.34 Hz), 7.21 (t, 1H, J = 7.34 Hz), 7.16 (s, 1H, NH), 6.89 (s, 1H), 6.83 (s, 2H), 6.51 (s, 2H), 6.37 (d, 1H, J = 6.13 Hz, NH), 4.86 (s, 1H), 4.51 (d, 1H, J = 6.13 Hz), 3.89 (s, 3H), 3.80 (s, 2H), 3.76 (s, 3H), 3.69 (s, 6H). 13C (400 MHz, CDCl3): 172.69, 164.67, 154.07, 147.61, 146.69, 137.08, 135.3, 134.10, 130.12, 127.57, 123.44, 120.96, 119.24, 118.80, 112.87, 112.72, 112.27, 111.72, 108.87, 95.93, 66.60, 63.50, 61.58, 56.74, 56.70, 33.90. MS (ESI): m/z 554 (M + Na).
10g. MS (ESI): m/z 711 (M + Na).
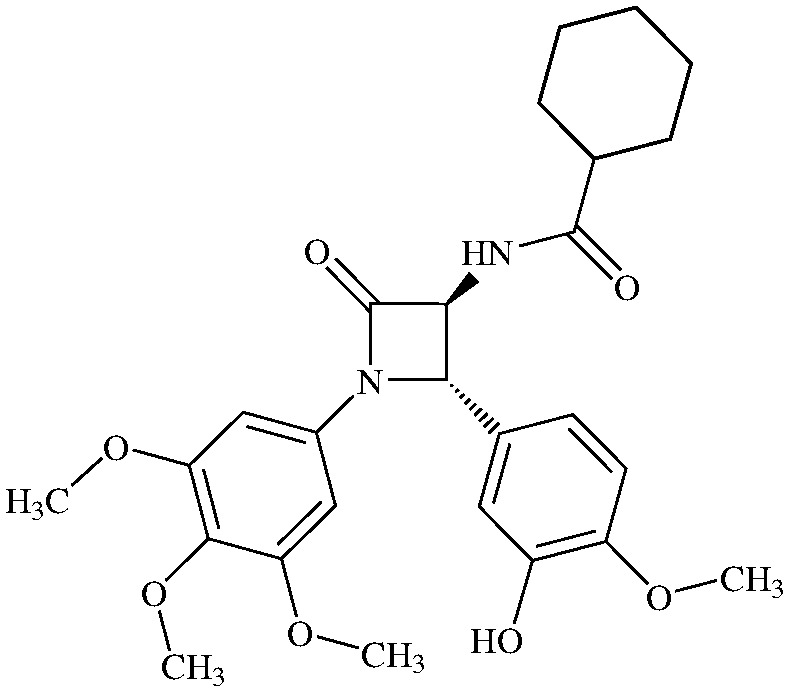
N-(Cyclohexanoyl)-3,4-trans-3-amino-1-(3,4,5-trimethoxyphenyl)-4-(3-hydroxy-4-methoxyphenyl)-azetidin-2-one
11h (69% yield). 1H NMR (400 MHz, CDCl3): δ ppm 6.92 (d, 1H, J = 6.7 Hz, NH), 6.88 (s, 1 H), 6.83 (d, 1H, J = 8.4 Hz), 6.80 (d, 1H, J = 8.4 Hz), 6.44 (s, 2H), 4.87 (d, 1H, J = 1.5 Hz) 4.54 (dd, 1H, J = 6.7, 1.5 Hz), 3.84 (s, 3H), 3.76 (s, 3H), 3.66 (s, 6H), 2.15 (m, 1H), 1.86 (m, 2H), 1.75 (m, 2H), 1.64 (m, 2H), 1.43 (m, 2H), 1.25 (m, 2H). 13C (400 MHz, CDCl3): 177.63, 165.29, 153.98, 147,85, 147.02, 135.06, 134.20, 130.06, 118.87, 113.04, 111.84, 95.83, 66.41, 63.79, 61.53, 56.74, 56.68, 56.61, 45.44, 41.17, 30.06, 26.29, 26.24. MS (ESI): m/z 507 (M + Na).
11h. MS (ESI): m/z 617 (M + Na).
N-(Acetyl)-3,4-trans-3-amino-1-(3,4,5-trimethoxyphenyl)-4-(3-hydroxy-4-methoxyphenyl)-azetidin-2-one
11i (80% yield). 1H NMR (400 MHz, CDCl3): δ ppm 6.95 (s, 1H), 6.89 and 6.87 (AB system, 2H, J = 7.4 Hz), 6.55 (s, 2H), 6.33 (d, 1H, J = 7.5 Hz, NH), 4.91 (d, 1H, J = 1.5 Hz), 4.62 (dd, 1H, J = 7.4, 1.5 Hz), 3.91 (s, 3H), 3.86 (s, 3H), 3.72 (s, 6H), 2.10 (s, 3H). 13C (400 MHz, CDCl3): 171.52, 165.24, 153.95, 153.93, 147.83, 147.02, 134.89, 134.18, 129.88, 118.79, 112.98, 111.80, 95.74, 66.18, 63.80, 61.57, 56.61, 23.36. MS (ESI): m/z: 439 (M + Na).
N-(S-2-amino-propanoyl)-3,4-trans-3-amino-1-(3,4,5-trimethoxyphenyl)-4-(3-hydroxy-4-methoxyphenyl)-azetidin-2-one
11j (80% yield). 1H NMR (300 MHz, CDCl3/CD3OD): δ ppm 8.26 (bs, 2H), 6.91 (s, 1H), 6.84 and 6.79 (AB system, 2H, J = 8.2 Hz), 6.52 (s, 2H), 4.92 (s, 1H), 4.59 (s, 1H), 3.84 (s, 3H), 3.75 (s, 3H), 3.66 (s, 6H), 3.65 (m, 1H), 1.35 (d, 3H, J = 7.5 Hz). 13C (300 MHz, CDCl3/CD3OD): 170.01, 168.29, 154.07, 147.79, 147.10, 135.31, 134.18, 130.32, 118.43, 112.94, 111.80, 96.06, 95.86, 69.87, 66.89, 61.54, 56.67, 56.64, 51.17, 49.99, 20.60. MS (ESI): m/z 446 (M + 1).
N-Boc-11j. 1H NMR (300 MHz, CDCl3/CD3OD): 7.64 (s, 1H), 6.88 (s, 1H9), 6.88 and 6.86 (AB system, 2H, J = 7.4 Hz), 6.57 (s, 2H), 4.95 (s, 1H), 4.59 (s, 1H), 3.86 (s, 3H), 3.72 (s, 3H), 3.70 (s, 6H), 3.6 (q, 1H, J = 7.4 Hz), 1.45 (s, 9H), 1.34 (d, 3H, J = 7.4 Hz). MS (ESI): m/z 568 (M + Na).
Biology
Reagents
RPMI-1640 medium, fetal bovine serum (FBS), l-glutamine, penicillin, and streptomycin were obtained from Lonza (Basel, Switzerland). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Cell cultures
Colorectal adenocarcinoma SW48 cells were purchased from ATCC, were cultured using RPMI-1640 medium supplemented with 10% (v/v) FBS, 2 mM l-glutamine, 100 U mL–1 penicillin, and 100 μg mL–1 streptomycin and were maintained at 37 °C in a humidified 5% CO2 incubator.
Growth inhibition assay
The antiproliferative activity of the azetidinone derivatives was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. 8000 cells were seeded in 96-well plates and treated with different concentrations of each compound (dissolved in DMSO). After 72 h, the MTT solution was added and then plates were incubated for 2 h at 37 °C. The purple formazan crystals were solubilized and the plates were read on a Model 550 microplate reader (BioRad Laboratories, Hercules, CA) at 570 nm. Assays were performed in triplicate in three independent experiments and data were analyzed using Sigma Plot software (using the four parameter logistic equation) to estimate IC50 values, defined as the concentration of drug causing 50% inhibition in absorbance compared to control cells (in which 0.1% DMSO was used).
Tubulin polymerization assay
The effect of compounds on tubulin polymerization was determined spectrophotometrically, essentially as previously described.24 Lyophilized purified porcine brain tubulin (Cytoskeleton, Denver, CO) was resuspended in assembly buffer (80 mM PIPES, pH 6.9, 1 mM MgCl2, 2 mM EGTA, 10% glycerol) at 2.5 mg mL–1 and mixed with 1 mM GTP and varying concentrations of the indicated compounds. DMSO (0.2% v/v) was used as a vehicle control. Tubulin assembly was monitored at 340 nm at 37 °C for 15 minutes on a Jasco V-530 spectrophotometer (Jasco Europe, Italy). The IC50 values are the compound concentrations required to inhibit tubulin polymerization by 50% and were estimated using the Sigma Plot software (using the four parameter logistic equation).
Conflicts of interest
The authors declare no competing interest.
Supplementary Material
Acknowledgments
This work was supported by grants to P. C. and F. O. from MIUR and “Piano Sostegno Ricerca 2015-17-Linea 2-Azione B” from the University of Milano.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00147b
References
- Yue Q.-X., Liu X., Guo D.-A. Planta Med. 2010;76:1037–1043. doi: 10.1055/s-0030-1250073. [DOI] [PubMed] [Google Scholar]
- Lin C. M., Ho H. H., Pettit G. R., Hamel E. Biochemistry. 1989;28:6984–6991. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- McGown A. T., Fox B. W. Cancer Chemother. Pharmacol. 1990;26:79–81. doi: 10.1007/BF02940301. [DOI] [PubMed] [Google Scholar]
- Tron G. C., Pirali T., Sorba G., Pagliai F., Busacca S., Genazzani A. A. J. Med. Chem. 2006;49:3033–3044. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- Pettit G. R., Singh S. B., Hamel E., Lin C. M., Alberts D. S., Garcia-Kendall D. Experientia. 1989;45:209–211. doi: 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- Nam N.-H. Curr. Med. Chem. 2003;10:1697–1722. doi: 10.2174/0929867033457151. [DOI] [PubMed] [Google Scholar]
- Quan H., Xu Y., Lou L. Int. J. Cancer. 2008;122:1730–1737. doi: 10.1002/ijc.23262. [DOI] [PubMed] [Google Scholar]
- Gaspari R., Prota A. E., Bargsten K., Cavalli A., Steinmetz M. O. Chem. 2017;2:102–113. doi: 10.1016/j.chempr.2016.12.005. [DOI] [Google Scholar]
- West C. M. L., Price P. Anti-Cancer Drugs. 2004;15:179–187. doi: 10.1097/00001813-200403000-00001. [DOI] [PubMed] [Google Scholar]
- www.clinicaltrials.gov .
- Siebert A., Gensicka M., Cholewinski G., Dzierzbicka K. Anti-Cancer Agents Med. Chem. 2016;16(8):942–960. doi: 10.2174/1871520616666160204111832. [DOI] [PubMed] [Google Scholar]
- Madadi N. R., Penthala N. R., Howk K., Ketkar A., Eoff R. L., Borrelli M. J., Crooks P. A. Eur. J. Med. Chem. 2015;103:123–132. doi: 10.1016/j.ejmech.2015.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beale T. M., Allwood D. M., Bender A., Bond P. J., Brenton J. D., Charnock-Jones D. S., Ley S. V., Myers R. M., Shearman J. W., Temple J., Unger J., Watts C. A., Xian J. ACS Med. Chem. Lett. 2012;3:177–181. doi: 10.1021/ml200149g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Dong X., Xue N., Wu R., He Q., Yang B., Hu Y. Bioorg. Med. Chem. 2009;17:6279–6285. doi: 10.1016/j.bmc.2009.07.040. [DOI] [PubMed] [Google Scholar]
- Guan Q., Yang F., Guo D., Xu J., Jiang M., Liu C., Bao K., Wu Y., Zhang W. Eur. J. Med. Chem. 2014;87:1–9. doi: 10.1016/j.ejmech.2014.09.046. [DOI] [PubMed] [Google Scholar]
- Brown A. W., Fisher M., Tozer G. M., Kanthou C., Harrity J. P. A. J. Med. Chem. 2016;59:9473–9488. doi: 10.1021/acs.jmedchem.6b01128. [DOI] [PubMed] [Google Scholar]
- Simoni D., Grisolia G., Giannini G., Roberti M., Rondanin R., Piccagli L., Baruchello R., Rossi M., Romagnoli R., Invidiata F. P., Grimaudo S., Jung M. K., Hamel E., Gebbia N., Crosta L., Abbadessa V., Di Cristina A., Dusonchet L., Meli M., Tolomeo M. J. Med. Chem. 2005;48:723–736. doi: 10.1021/jm049622b. [DOI] [PubMed] [Google Scholar]
- Gurjar M. K., Wakharkar R. D., Singh A. T., Jaggi M., Borate H. B., Shinde P. D., Verma R., Rajendran P., Dutt S., Singh G., Sanna V. K., Singh M. K., Srivastava S. K., Mahajan V. A., Jadhav V. H., Dutta K., Krishnan K., Chaudhary A., Agarwal S. K., Mukherjee R., Burman A. C. J. Med. Chem. 2007;50:1744–1753. doi: 10.1021/jm060938o. [DOI] [PubMed] [Google Scholar]
- Ty N., Pontikis R., Chabot G. G., Devillers E., Quentin L., Bourg S., Florent J.-C. Bioorg. Med. Chem. 2013;21:1357–1366. doi: 10.1016/j.bmc.2012.11.056. [DOI] [PubMed] [Google Scholar]
- Smith D. M., Kazi A., Smith L., Long T. E., Heldreth B., Turos E., Dou Q. P. Mol. Pharmacol. 2002;61:1348–1358. doi: 10.1124/mol.61.6.1348. [DOI] [PubMed] [Google Scholar]
- Xing B., Rao J., Liu R. Mini-Rev. Med. Chem. 2008;8:455–471. doi: 10.2174/138955708784223558. [DOI] [PubMed] [Google Scholar]
- O'Boyle N. M., Carr M., Greene L. M., Bergin O., Nathwani S. M., McCabe T., Lloyd D. G., Zisterer D. M., Meegan M. J. J. Med. Chem. 2010;53:8569–8584. doi: 10.1021/jm101115u. [DOI] [PubMed] [Google Scholar]
- Malebari A. M., Greene L. M., Nathwani S. M., Fayne D., O'Boyle N. M., Wang S., Twamley B., Zisterer D. M., Meegan M. J. Eur. J. Med. Chem. 2017;130:261–285. doi: 10.1016/j.ejmech.2017.02.049. [DOI] [PubMed] [Google Scholar]
- Tripodi F., Pagliarin R., Fumagalli G., Bigi A., Fusi P., Orsini F., Frattini M., Coccetti P. J. Med. Chem. 2012;55:2112–2124. doi: 10.1021/jm201344a. [DOI] [PubMed] [Google Scholar]
- Valtorta S., Nicolini G., Tripodi F., Meregalli C., Cavaletti G., Avezza F., Crippa L., Bertoli G., Sanvito F., Fusi P., Pagliarin R., Orsini F., Moresco R. M., Coccetti P. Invest. New Drugs. 2014;32:1123–1133. doi: 10.1007/s10637-014-0148-8. [DOI] [PubMed] [Google Scholar]
- Suvi A. P., Rajamaki H. M., De Luca L., Capitta F. RSC Adv. 2016;6:38553–38557. [Google Scholar]
- Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M., Goodsell D. S., Halliday R. S., Huey R., Hart W. E., Belew R. K., Olson A. J. J. Comput. Chem. 1639;19:1639–1662. [Google Scholar]
- Humphrey W., Dalke A., Schulten K., J. Mol. Graphics, 1996, 14 , 33 –38 , , 27–28 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.