

 Among the new selective P-gp modulators reported, compound 12a almost completely restores doxorubicin cytotoxicity in resistant cancer cells.
Among the new selective P-gp modulators reported, compound 12a almost completely restores doxorubicin cytotoxicity in resistant cancer cells.
Abstract
P-glycoprotein (P-gp, MDR1) is a membrane transporter expressed in several regions of our body. It plays a crucial defense role as it mediates the efflux of hundreds of potentially toxic substances. However, P-gp is one of the main causes of failure in cancer chemotherapy, as a number of chemotherapeutic agents are P-gp substrates. Another interesting implication concerns the correlation between P-gp expression impairment and the onset of several central nervous system pathologies such as Alzheimer's and Parkinson's diseases. In view of these considerations, in the present study, a new series of P-gp modulators have been designed, synthesized and evaluated for their activity towards P-gp and two other sister proteins (BCRP and MRP1). The compounds, structurally correlated to the potent but non-selective P-gp inhibitor MC70 [4′-(6,7-dimethoxy-3,4-dihydro-1H-isoquinolin-2-ylmethyl)biphenyl-4-ol], proved fairly selective towards P-gp, with a potency in the micromolar range. Compounds 5a, 5d and 12d proved capable of restoring doxorubicin toxicity in resistant cancer cells.
Introduction
P-glycoprotein (P-gp, also known as MDR1 or ABCB1) is a membrane protein belonging to the ATP-binding cassette (ABC) superfamily. It is a complex molecular machinery able to recognize and mediate the efflux of hundreds of structurally uncorrelated substances exploiting energy derived from ATP hydrolysis. It exhibits strategic localization in a number of organs and tissues, thus playing a crucial defence role against toxic substances, both of endogenous and exogenous origin, and constituting an essential component of several biological barriers.1 P-gp has attracted great attention since several years ago, owing to its involvement in multidrug resistance (MDR), a phenomenon that represents one of the main causes of cancer chemotherapy failure. In some tumors, MDR occurs without previous exposure to anti-cancer drugs, whilst in other cases, its onset occurs during treatment, quite often as a consequence of selection of resistant over sensitive cells in heterogeneous tumor tissue.2 P-gp is overexpressed in cancer cells and is responsible for the efflux of several chemotherapeutic agents, such as taxanes, vinca alkaloids, doxorubicin, etoposide, topotecan, methotrexate, imatinib, dasatinib, and gefitinib among others.3 The wide expression of P-gp on the surface of endothelial cells at the blood–brain barrier makes it a gatekeeper for the central nervous system (CNS), as it prevents potentially harmful substances from entering but also hampers many structurally and functionally uncorrelated drugs, compromising the success of the pharmacological treatment of different CNS disorders and tumours. More recently, an interesting connection has emerged between P-gp and the onset of Alzheimer's disease (AD), Parkinson's disease (PD), epilepsy and other CNS diseases: an impairment in the expression level of the protein has been observed in the early stages of these neurological disorders.4,5 Starting from this background, medicinal chemistry efforts to target P-gp have been undertaken, mainly with the aim of obtaining inhibitors which should be co-administered with chemotherapeutic agents subjected to P-gp mediated efflux, thus restoring therapy efficacy. According to this approach, a number of MDR reversal agents have been reported, which are usually classified into three generations; despite promising in vitro results, most of them failed in the clinical trial phase for several reasons: in the case of first generation agents which are drugs in clinical use for different indications, the main drawback was poor selectivity of action.6 Modulators of the second generation, which are supposed to be more selective, proved toxic.7 As for third generation agents, despite their high affinity for P-gp, they did not return satisfying results due to pharmacokinetic and safety concerns.8 Thus, the development of new agents able to impede P-gp mediated drug efflux is still an unmet need, and as MDR remains a prominent issue in cancer chemotherapy, it is still a field of interest in medicinal chemistry. In view of this, an alternative approach is the search of broad spectrum inhibitors targeting different transporters, which found a very recent exemplification in the study of Stefan et al. which reports on the design of 9-deazapurine-based inhibitors able to counteract drug efflux mediated by P-gp, multidrug resistance-associated protein 1 (MRP1, ABCC1) and breast cancer resistance protein (BCRP, ABCG2).9
Besides the design of P-gp modulators to be flanked by chemotherapy agents, recently, different approaches to counteract MDR have been undertaken by several groups. An important strategy is obviously the development of anti-cancer drugs that are not substrates of P-gp, both through the design of new molecules or modification of already known drugs.10 Some successful examples have been obtained, among others, with taxanes, with epothilones (a class of natural compounds structurally correlated to taxanes) and with vinca alkaloids.11 In the setting of this strategy, given the lack of structural insights into the mechanism of interaction of anti-cancer drugs with P-gp, it is difficult to envisage the nature and the position of the structural modifications required to evade P-gp efflux and retain desirable cytotoxicity.
Another strategy that has been undertaken to overcome P-gp mediated efflux consists in enhancing drug uptake or modifying the mechanism of cellular uptake through non-covalent or covalent conjugation of chemotherapy agents with targeting systems such as nanoparticles, liposomes, micelles, polymeric conjugates, and antibodies among others. Particular attention has been paid to liposome formulation and a number of such systems are currently under clinical evaluation.12 A word of caution must be given regarding this approach, as the intracellular release of drugs obtained from these formulations may still result in P-gp efflux. Regarding covalent derivatives, strategies that gave interesting results consist in the modification of the drug through the introduction of guanidinium-rich tags, with consequently higher water solubility and modified recognition by P-gp,13,14 and of more complex cationic oligopeptide-based moieties, as in the case of Taxol-octaarginine conjugates.15
Besides the above reported approaches, based on mainly medicinal chemistry tools, in the recent past, some less explored strategies have been attempted to counteract MDR, aiming at the downregulation of ABCB1 gene expression or translation, such as antisense oligonucleotides and small interfering RNA.16
In addition to the MDR issue, the potential application in the early diagnosis through imaging techniques such as positron emission tomography (PET) of several neurological disorders has been proposed for P-gp ligands, in view of the involvement of the transporter in the onset of these CNS pathologies: in particular, radiolabeled substrates are useful to measure the in vivo function of the transporter at the blood brain barrier (BBB).17
Following the heavy effects of P-gp mediated MDR on cancer chemotherapy, despite the obstacles encountered so far, the development of new P-gp modulators still appears as a valuable and possibly straightforward route to counteract MDR. In this ongoing effort and also in view of the application of P-gp ligands in the early diagnosis of some CNS degenerative diseases, in the present study, the authors have carried out several modifications on MC70 (Fig. 1), a previously studied P-gp inhibitor.18,19 MC70 shows a good P-gp inhibiting potency (EC50 = 0.69 μM); on the other hand, it displays a non-selective profile towards P-gp.
Fig. 1. Structure and biological activity profile of MC70.
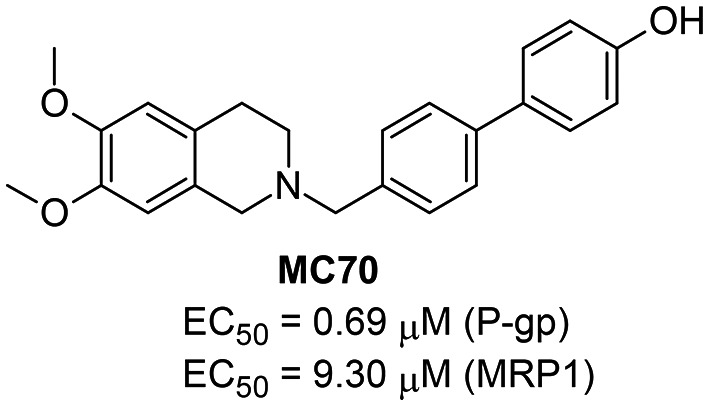
The authors have previously explored the structure activity relationships of MC70 derivatives; in particular, they focused on functionalization of the phenolic group with alkyl and oxyalkyl chains20 and with more complex moieties containing variously substituted furazan (1,2,5-oxadiazole) rings.21 In the present investigation, the structural modifications involve the biphenyl core. The aim of the study was to evaluate, in terms of inhibition potency and selectivity to P-gp, the effect of substituents able to modify the electronic properties and endowed with hydrogen bond donor or acceptor properties. Thus, at the four positions of the biphenyl moiety, different substituents were inserted: fluorine, a small strongly electronegative atom able to accept a hydrogen bond, a nitro group, a prototypical electron-withdrawing group with a strong resonance effect and hydrogen bond acceptor properties; a methoxy group, an electron-donating substituent with a resonance effect and hydrogen bond acceptor properties; an amino group, which is electron donating with hydrogen bond donor and acceptor properties.
The compounds have been synthesized through an inexpensive and straightforward route; compared to MC70, the potency was slightly decreased, the EC50 values being in the micromolar range for most compounds, but all the ligands display high selectivity towards P-gp, being essentially inactive against MRP1 and BCRP. Three of them have been further evaluated by co-administration with doxorubicin and proved able to restore the intracellular concentration of the anthracycline to a different extent, without any effects on P-gp expression. A sensible insight into the binding pose of a selected compound has been gained through a molecular docking study.
Results and discussion
Synthesis of target compounds
The target compounds were synthesized according to the methods reported in Schemes 1 and 2. In brief, the key step is a Suzuki coupling reaction,22 carried out with palladium on activated charcoal in a ligand free fashion in an aqueous environment, starting from the 4-bromobenzylalcohols 1a–f (Scheme 1) or the 4-bromophenols 8a–e (Scheme 2). The substrates were coupled with 4-hydroxyphenylboronic acid or 4-hydroxymethylphenylboronic acid, respectively. The coupling products were then reacted with an excess of p-toluenesulfonyl chloride in the presence of triethylamine, to give the di-tosylate derivatives 3a–f (Scheme 1) or 10a–e (Scheme 2). The latter was used without purification to achieve benzylation of 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline. Finally, the removal of the phenyl tosylate group was achieved through hydrolysis with NaOH in a THF/CH3OH mixture.
Scheme 1. Synthesis of target compounds 5a–f. Reagents and conditions: a) 4-hydroxybenzeneboronic acid, KOH, Pd/C cat., H2O, 130 °C, 3 hours; b) p-toluenesulfonyl chloride, Et3N, DMAP cat., CH2Cl2, room temperature, 6 hours; c) 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride, DBU, CH3CN, 70 °C, 18 hours; d) NaOH, THF/CH3OH 2/1, 65 °C, 1 hour; e) BH3–THF complex, THF, N2, 18 hours, room temperature.

Scheme 2. Synthesis of target compounds 12a–e, 16, and 17a and b. Reagents and conditions: a) 4-hydroxymethylbenzeneboronic acid, KOH, Pd/C cat., H2O, 130 °C, 3 hours; b) p-toluenesulfonyl chloride, Et3N, DMAP cat., CH2Cl2, room temperature, 6 h; c) 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride, DBU, CH3CN, 70 °C, 18 hours; d) NaOH, THF/CH3OH 2/1, 65 °C, 1 hour; e) 4-hydroxymethylbenzeneboronic acid, K2CO3, Pd[(C6H5)3P]4, 1,4-dioxane/water 2/1, N2, 90 °C, 18 hours; f) BBr3, CH2Cl2, 18 hours, room temperature; g) H2, Pd/C cat., CH3OH, room temperature, 3 hours.

The two benzylic alcohols 1c and 1f were synthesized starting from the corresponding benzoic acids through reduction with the BH3–THF complex (Scheme 1).
As for compound 16, the procedure reported above for the coupling reaction starting from 1-bromo-4-methoxy-2-nitrobenzene 13 did not afford the desired intermediate, probably due to the poor solubility of 13. For this reason, the Suzuki coupling reaction was carried out in a more “traditional” manner (Scheme 2) using palladium tetrakis triphenylphosphine as the catalyst in a 1,4-dioxane/water mixture; intermediate 14 was subjected to hydrolysis of the methoxy group with BBr3, resulting in the concomitant bromination of benzylic alcohol. The bromo derivative 15 was then reacted with 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline, yielding compound 16.
Finally, the two amino substituted compounds 17a and 17b were obtained through catalytic hydrogenation of the nitro derivatives 12c and 16 (Scheme 2).
Biological evaluation
All the new synthesized compounds have been tested to establish their P-gp interacting mechanism as the substrate or inhibitor by three assays: 1) the inhibition of the transport of a fluorescent or a pro-fluorescent substrate of the transporter;23 2) the determination of the apparent permeability value (Papp);23 3) the determination of the ATP cell level depletion.23 The results are reported in Table 1.
Table 1. Biological characterization of target compounds.
| Compound | Structure | MDR1 EC50 a (μM) | MRP1 EC50 a (μM) | BCRP EC50 a (μM) | ATP consumption | P app b |
| 5a |

|
1.60 ± 0.27 | na c | na | NO d | 4.5 |
| 5b |
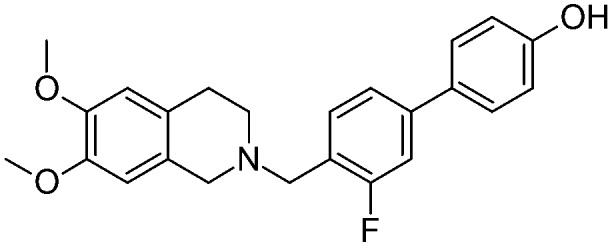
|
4.73 ± 0.90 | na | na | NO | 4.1 |
| 5c |
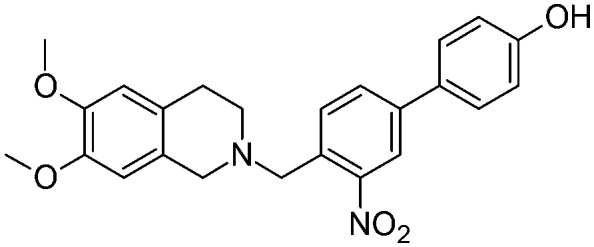
|
8.4 ± 1.58 | na | na | NO | 5.8 |
| 5d |

|
1.51 ± 0.30 | 38 ± 7.6 | na | NO | 4.4 |
| 5e |
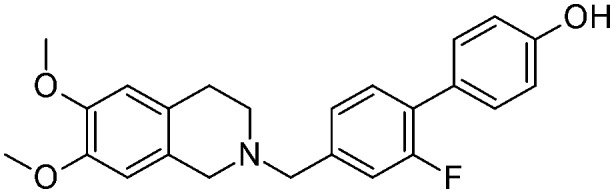
|
6.2 ± 1.24 | na | na | NO | 3.7 |
| 5f |

|
15.2 ± 3.00 | na | na | NO | 2.9 |
| 12a |
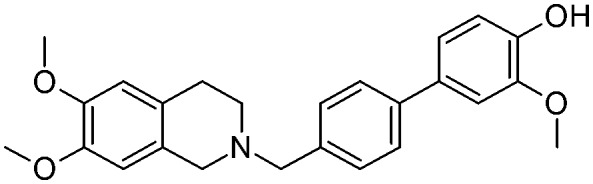
|
2.0 ± 0.40 | na | na | NO | 3.9 |
| 12b |

|
2.8 ± 0.56 | na | na | NO | 4.9 |
| 12c |

|
12.7 ± 2.50 | 28.9 | na | NO | 6.3 |
| 17a |
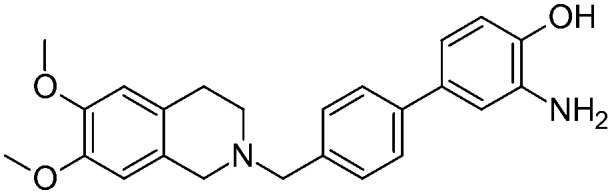
|
6.9 ± 1.32 | na | na | NO | 7.5 |
| 12d |

|
1.86 ± 0.37 | 62 | na | NO | 3.5 |
| 12e |
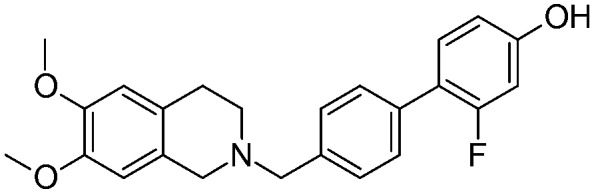
|
2.17 ± 0.43 | na | na | NO | 4.5 |
| 16 |
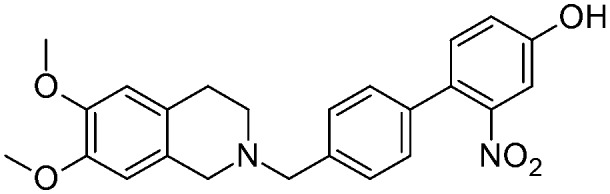
|
2.54 ± 0.48 | na | na | NO | 11.2 |
| 17b |
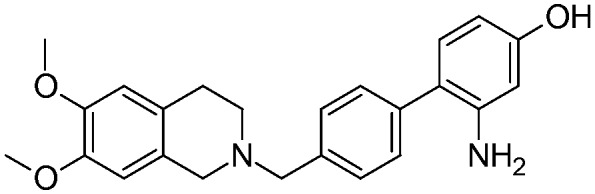
|
5.4 ± 1.00 | na | na | NO | 4.5 |
| MC70 |

|
0.69 | 9.30 | 73 | NO | 1.3 |
aThe values are the means of three independent experiments; samples analyzed in duplicate.
bThe apparent permeability was determined as the BA/AB ratio (the values are from two independent experiments).
cNot active at 100 μM.
dNo significant ATP consumption at the activity doses.
The first assay is performed in a cell line overexpressing P-gp (MDCK-MDR1 cells) and it evaluates the ability of the ligands to compete with the efflux of the pro-fluorescent P-gp substrate Calcein-AM towards P-gp; it is a measure of the potency of each compound towards the target. The second assay evaluates the basolateral-apical vs. apical-basolateral ratio (BA/AB), representative of two contributions, the passive diffusion (BA) and active transport (AB) in a system mimicking BBB, such as Caco-2 cells; if this ratio is < 2, the compound can be considered a P-gp inhibitor, otherwise (Papp > 2) it behaves as a substrate. The third assay measures the consumption of ATP in MDCK-MDR1 cells. Generally, a substrate, since transported by the pump, induces ATP consumption while an inhibitor, inhibiting the binding of ATP on its site on P-gp, is not transported and thus it does not induce a decrease in ATP cell level. The selectivity of all compounds towards the sister proteins BCRP and MRP1 has also been tested by measuring the ability of the compounds to interfere with the BCRP- or MRP1-mediated transport of the fluorescent BCRP substrate Hoechst 33342 or the pro-fluorescent MRP1 substrate Calcein-AM.
As depicted in Table 1, all of the compounds were found to be less active than the lead compound MC70 (EC50 = 0.69 μM), displaying EC50 values ranging from 1.51 to 15.2 μM, and among them 5a (EC50 = 1.60 μM), 5d (EC50 = 1.51 μM), and 12d (EC50 = 1.86 μM) displayed the best activity values. However, all the compounds were more selective towards P-gp, proving inactive towards BCRP and MRP1. Only compounds 5d and 12c showed moderate MRP1 activity (EC50 = 38 μM and 28.9 μM, respectively). All the ligands, having Papp values > 2 and not inducing ATP cell depletion, behaved as substrates of the category IIB3.18
As a whole, only slight differences can be observed among the series; nevertheless some general considerations are worth noting: the presence of a methoxy group, an electron-donor and a hydrogen bond acceptor, seems to give a slightly higher activity in the series, no matter what its position on the biphenyl core is (EC50 values ranging from 1.51 μM for 5d to 2 μM for 12a). In contrast, the presence of a strong electron-withdrawing substituent, such as a nitro group, determines a loss of activity which is more pronounced for compounds where the substituent influences to a greater extent the acidity of the phenol group (position 2′ and 3 of the biphenyl core) (EC50 = 15.2 μM for 5f and EC50 = 12.7 μM for 12c). The fluorine atom seems to not greatly influence the activity, with a slightly more detrimental effect when present on the benzylic ring (positions 2′ and 3′) (EC50 = 6.2 μM for 5e). Finally, the presence of an amino substituent, which is able to donate a hydrogen bond, bears a significant loss of activity (EC50 = 6.9 μM for 17a).
Molecular docking study
A structure based study was then carried out to furnish fresh insights into the binding mode of the new compounds and to support the explication of the EC50 data. To fulfill this topic, our previously published P-gp receptor model21 served as a valuable tool to explain the ligand pose of the most active compound (5d). In our past study, we have postulated that the “inward-outward facing” of the P-gp scaffold should facilitate the “pulling out” of substrates as soon as they pass the cell bilayer, while active inhibitors would hamper the P-gp flipping depending on the molecular shape and pharmacophoric features of the molecules; in line with this view, molecular docking further supports this evidence.
As it might be perceived from Fig. 2, reporting the binding mode of 5d, the ligand scaffold properly fits the binding site space delimited by two six helical-transmembrane domains (TMDs) and deeply locks into the crevice comprising the intracellular moiety of the same domains, with a kind of “reversed wedge” pose that might be responsible for the hampering of P-gp flipping upon ligand binding.
Fig. 2. Binding mode of 5d into the MDR1 binding site. Water molecules are represented as red spheres, and the extracellular and intracellular sides are at the top and bottom of the image, respectively. The free energy of binding calculated with the hydration force field in AutoDock is –8.62 kcal mol–1, while the contact surface area measures 440 Å2.

Interestingly, the tetrahydroisoquinoline ring is placed close to one of the TMDs, and it occupies a mainly hydrophobic receptor slot, surrounded by Tyr310, Phe336, Phe728 and Phe983 generating extensive favourable contacts and π–π stacking, as well as stable binding through two water molecules enlacing the hydrogen bonding bridges with Tyr310 and Ser979. The rest of the aromatic moiety points to the opposite TMD with the phenolic group making polar contacts with the backbone of Met949 and the methoxy group interacting with Tyr953 throughout a water molecule coordinating with a hydrogen bond, and as far as this is concerned, the same methoxy substituent should accommodate, to some extent, the biphenyl motif in a non-planar conformation. Indeed, this steric hindrance, in combination with hydrogen bond formation, might clarify, at least within this series of derivatives, the better EC50 value of 5d.
Co-administration of doxorubicin with 5a, 5d and 12d
Compounds 5a, 5d, and 12d, displaying the best P-gp activity profile, have been evaluated in co-administration with doxorubicin in order to study their efficacy as doxorubicin-rehabilitating agents in the treatment of resistant tumours (Fig. 3).
Fig. 3. Co-administration of doxorubicin (10 μM) with compounds MC70, 5a, 5d, and 12d (10 μM). Antiproliferative effect of MC70, 5a, 5d, and 12d (10 μM) alone (gray bars) and co-administered with doxorubicin (10 μM) (black bars) at 48 h in the MDCK-MDR1 cell line. The ctr bar represents the administration of 10 μM doxorubicin alone.

Preliminary data demonstrated that the three compounds were not cytotoxic at 48 h and 72 h (data not shown), and doxorubicin at 10 μM was not able to induce cell death as the efflux is mediated by P-gp in the resistant tumour cell model overexpressing P-gp (MDCK-MDR1 cells). When MC70, 5a, 5d, and 12d were co-administered with doxorubicin, they restored its anti-neoplastic cytotoxicity. In fact, while the lead compound MC70 and 5a and 5d displayed a moderate ability to restore the doxorubicin effect (20% of cytotoxicity increase for MC70 and 15% for 5a and 5d), 12d was able to produce an increase of 60% in doxorubicin cytotoxicity, demonstrating its ability to almost completely rehabilitate the access of the anti-neoplastic drug in tumour cells. Considering the structural correlation and the similarity of the EC50 values of the three tested compounds, it is difficult to envisage the reason for the efficient restoration of doxorubicin toxicity elicited by 12d; the hypothesis that can be made is a possible reinforcement of activity at concentrations higher than EC50; this can be due to the peculiarity of the binding region of P-gp, which is large and comprises multiple and possibly overlapping binding sites. This feature has led several authors to postulate the possibility for some small molecules to bind simultaneously to different binding sites, and this has also been exploited through the design of dimeric modulators with enhanced potency.24
Immunoblotting experiments using MC70, 5a, 5d and 12d
The same compounds (MC70, 5a, 5d and 12d) which were evaluated in the co-administration with doxorubicin were further studied in immunoblotting experiments to verify a possible interference with P-gp expression under the same experimental conditions as in the cell viability tests. As can be seen in Fig. 4, no differences in P-gp expression were observed in treated cells compared to the control, thus supporting the hypothesis of a direct effect of the compounds on the transporter.
Fig. 4. Immunoblotting experiment with compounds MC70, 5a, 5d, and 12d (10 μM). Proteins were extracted from MDCK-MDR1 cells after 48 hours of incubation. Actin levels were used as the protein loading control. Ctrl bands refer to untreated cells. The figure is representative of 1 out of 3 experiments with similar results.

Conclusion
A new series of P-gp modulators have been developed through the “decoration” of the biphenyl moiety of MC70 with substituents endowed with different electronic and hydrogen bond donor/acceptor properties. The compounds were synthesized exploiting a straightforward and inexpensive route. They displayed a slightly lower potency than the parent compound but proved highly selective, thus representing suitable candidates to be used in imaging techniques for the measurement of the P-gp function at the BBB.
A molecular docking simulation carried out for the most potent compound (5d) highlighted an additional anchorage for the biphenyl moiety through the methoxy substituent, which engages a hydrogen bond mediated by a water molecule. Besides the biochemical characterization aimed at clarifying the mechanism of interaction of the ligands with P-gp, the most potent compounds (5a, 5d, and 12d) and MC70 were evaluated in a co-administration assay with doxorubicin and proved efficient in restoring drug toxicity against doxorubicin resistant MDCK-MDR1 cells. A possible effect on P-gp expression was ruled out by performing immunoblotting experiments using the same selected compounds.
The present study thus represents an additional investigation of a previously started structure activity relationship study on tetrahydroisoquinoline derivatives and furnishes additional information about the mode of binding of this class of compounds which can be useful for future development of potent P-gp modulators.
Among the series, compound 12d deserves particular consideration; it represents a valuable candidate for the further development of P-gp mediated efflux reversal agents as it displayed an interesting efficacy (60%) in restoring doxorubicin cytotoxicity in resistant tumour cells. Moreover, this information probably reflects a peculiar behavior of the compound, which may interact with different binding sites simultaneously; this hypothesis deserves further in-depth analyses and, if confirmed, opens the way to the exploitation of a “dimeric analogue” approach to designing P-gp modulators based on the biphenyl-tetrahydroisoquinoline substructure.
Contributions
S. Guglielmo and N. Colabufo designed the study and wrote the manuscript.
S. Guglielmo planned the synthetic strategy.
M. Contino planned and performed the biological assays to define the P-gp interacting mechanism and participated in the editing of the manuscript.
M. G. Perrone performed the permeability assays.
R. Giampietro performed the co-administration assay.
A. Carrieri and D. Zaccaria performed the molecular docking study.
K. Chegaev and V. Borio carried out the synthesis of compounds.
B. Rolando carried out the structural characterization and purity assessment.
C. Riganti and K. Zabielska-Koczywąs carried out the immunoblotting experiments.
R. Fruttero carried out literature research and wrote the manuscript.
Conflicts of interest
The authors declare no conflict of interests.
Supplementary Material
Acknowledgments
This study was supported by the University of Turin - ”Ricerca Locale“ to SG and by MIUR (FIRB 2012 RBFR12SOQ1_002, FIRB 2012, grant RBFR12SOQ1). This work was also supported by My First AIRC Grant-MFAG2015 (Project Id.17566). We are thankful to Prof. A. Gasco for the fruitful discussion.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00075a
References
- Ambudkar S. V., Kimchi-Sarfaty C., Sauna Z. E., Gottesman M. M. Oncogene. 2003;22:7468–7485. doi: 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- Swanton C. Cancer Res. 2012;72:4875–4882. doi: 10.1158/0008-5472.CAN-12-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckford P. D. W., Sharom F. J. Chem. Rev. 2009;109:2989–3011. doi: 10.1021/cr9000226. [DOI] [PubMed] [Google Scholar]
- Löscher W., Potschka H. Prog. Neurobiol. 2005;76:22–76. doi: 10.1016/j.pneurobio.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Abuznait A. H., Kaddoumi A. ACS Chem. Neurosci. 2012;3:820–831. doi: 10.1021/cn300077c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas H., Coley H. M. Cancer Control. 2003;10:159–165. doi: 10.1177/107327480301000207. [DOI] [PubMed] [Google Scholar]
- Kuppens I. E. L. M., Witteveen E. O., Jewell R. C., Radema S. A., Paul E. M., Mangum S. G., Beijnen J. H., Voest E. E., Schellens J. H. M. Clin. Cancer Res. 2007;13:3276–3285. doi: 10.1158/1078-0432.CCR-06-2414. [DOI] [PubMed] [Google Scholar]
- Shukla S., Ohnuma S., Ambudkar S. V. Curr. Drug Targets. 2011;12:621–630. doi: 10.2174/138945011795378540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan K., Schmitt S. M., Wiese M. J. Med. Chem. 2017;60:8758–8780. doi: 10.1021/acs.jmedchem.7b00788. [DOI] [PubMed] [Google Scholar]
- Nobili S., Landini I., Mazzei T., Mini E. Med. Res. Rev. 2012;32:1220–1262. doi: 10.1002/med.20239. [DOI] [PubMed] [Google Scholar]
- Waghray D., Zhang Q., J. Med. Chem., 2017. 10.1021/acs.jmedchem.7b01457 , , Publication Date (Web): December 18, 2017 . [Google Scholar]
- Sercombe L., Veerati T., Moheimani F., Wu S. Y., Sood A. K., Hua S. Front. Pharmacol. 2015;6:286. doi: 10.3389/fphar.2015.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas J. R., Stanzl E. G., Teng N. N., Wender P. A. Mol. Pharmaceutics. 2014;11:2553–2565. doi: 10.1021/mp500161z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinlay C. J., Waymouth R. M., Wender P. A. J. Am. Chem. Soc. 2016;138:3510–3517. doi: 10.1021/jacs.5b13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A., Galliher W. C., Bhat N. M., Pillow T. H., Bieber M. M., Teng N. N. Gynecol. Oncol. 2012;126:118–123. doi: 10.1016/j.ygyno.2012.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbasi M., Lavasanifar A., Uludag H. Med. Res. Rev. 2013;33:33–53. doi: 10.1002/med.20244. [DOI] [PubMed] [Google Scholar]
- Colabufo N. A., Berardi F., Cantore M., Contino M., Inglese C., Niso M., Perrone R. J. Med. Chem. 2010;53:1883–1897. doi: 10.1021/jm900743c. [DOI] [PubMed] [Google Scholar]
- Colabufo N. A., Berardi F., Cantore M., Perrone M. G., Contino M., Inglese C., Niso M., Perrone R., Azzariti A., Simone G. M., Paradiso A. Bioorg. Med. Chem. 2008;16:3732–3743. doi: 10.1016/j.bmc.2008.01.055. [DOI] [PubMed] [Google Scholar]
- Colabufo N. A., Berardi F., Perrone M. G., Cantore M., Contino M., Inglese C., Niso M., Perrone R. ChemMedChem. 2009;4:188–195. doi: 10.1002/cmdc.200800329. [DOI] [PubMed] [Google Scholar]
- Guglielmo S., Contino M., Lazzarato L., Perrone M. G., Blangetti M., Fruttero R., Colabufo N. A. ChemMedChem. 2016;11:374–376. doi: 10.1002/cmdc.201500538. [DOI] [PubMed] [Google Scholar]
- Guglielmo S., Lazzarato L., Contino M., Perrone M. G., Chegaev K., Carrieri A., Fruttero R., Colabufo N. A., Gasco A. J. Med. Chem. 2016;59:6729–6738. doi: 10.1021/acs.jmedchem.6b00252. [DOI] [PubMed] [Google Scholar]
- Miyaura N., Suzuki A. J. Chem. Soc., Chem. Commun. 1979:866–867. [Google Scholar]
- Capparelli E., Zinzi L., Cantore M., Contino M., Perrone M. G., Luurtsema G., Berardi F., Perrone R., Colabufo N. A. J. Med. Chem. 2014;57:9983–9994. doi: 10.1021/jm501640e. [DOI] [PubMed] [Google Scholar]
- Emmert D., Campos C. R., Ward D., Lu P., Namanja H. A., Bohn K., Miller D. S., Sharom F. J., Chmielewski J., Hrycyna C. A. ACS Chem. Neurosci. 2014;5:305–317. doi: 10.1021/cn4002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.