

 A novel category of BRD4 bromodomain inhibitors was discovered through AlphaScreen assay, and the binding mode was revealed by a crystallographic method.
A novel category of BRD4 bromodomain inhibitors was discovered through AlphaScreen assay, and the binding mode was revealed by a crystallographic method.
Abstract
As a member of the bromodomain and extra terminal domain (BET) protein family, BRD4 is closely related to cancers and other diseases. Small-molecule BRD4 inhibitors have already demonstrated promising potential for the therapy of BRD4-related cancers. In this study, we report the discovery and evaluation of a novel category of BRD4 inhibitors, which share a trimethoxy ring and target the first bromodomain of the human BRD4 protein. The IC50 value of the most potent compound, DC-BD-03, is 2.01 μM. In addition, a high-resolution crystal structure of the compound DC-BD-29 with the first bromodomain of BRD4 was determined, which revealed the binding mode and facilitated further structure-based optimization. These compounds exhibited anti-proliferation activity, caused cell cycle arrest, and induced apoptosis in human leukemia MV4-11 cells. Thus, the results presented in this study indicated the potential of this series of compounds as drug candidates for the therapy of BRD4-related cancers.
1. Introduction
Acetylation is an important form of post-translation modification for proteins, while the acetylation of histones is a mark of active gene transcription.1 The disorder of histone acetylation patterns often leads to aberrant transcription and promotes the occurrence and progression of cancers and other diseases.2 The human genome encodes 61 bromodomains (BRDs), which are present in 46 different proteins.3 As a branch of epigenetic readers, these BRD proteins play important roles in the regulation of gene expression, through recognition of the acetylated lysine residues on histone tails.1,4 Among these BRD proteins, the bromodomain and extra terminal domain (BET) family, including BRD2, BRD3, BRD4 and BRDT, is characterized by two N-terminal bromodomains.5,6
Under normal conditions, the BET family proteins function in the regulation of transcription. BRD4 interacts with the positive transcription elongation factor b (P-TEFb), which is recruited to the promoter and activates RNA polymerase II.5,7 Through retaining the P-TEFb at the promoter region, BRD4 is important for gene expression.8 While in tumor cells, BET family proteins regulate the expression of specific cancer-related genes, such as c-Myc and Bcl-2, which are key regulators for the proliferation and survival of tumor cells.6,9 Accumulative evidence indicates that BET family proteins are involved in various malignancies, and small-molecule inhibitors that target the BET family proteins show promising potential for the therapy of human cancers and other related diseases.9–13 Through interfering the epigenetic landscape of BRD4, small-molecule BET inhibitors showed wide activities against various malignancies.9,13,14 Pharmacological activity studies showed that the inhibition of BET family proteins decreased the expression of survival-related genes, which are essential for tumor cells.6 Recently, several categories of BET inhibitors have been reported,15–17 such as JQ-1 (ref. 4) and I-BET-762.12 These small-molecule inhibitors provided pharmacological tools to investigate the functions of BET family proteins, in different diseases or under different conditions.4,9
Despite these reported BET inhibitors, it is worthwhile to discover and develop novel BET inhibitors to diversify the categories of BET inhibitors. Small-molecule inhibitors that target the same protein often demonstrate different pharmacological profiles in cells, thus leading to different clinical applications. In this study, we report the discovery and evaluation of a novel category of BRD4 bromodomain inhibitors, which share a trimethoxy ring, through the application of high-throughput screening, protein thermal shift assay, protein crystallography, and flow cytometry. These novel BRD4 inhibitors effectively inhibited the proliferation of human leukemia MV4-11 cells, and the crystal structure determined by X-ray diffraction provided solid evidence for further structure-based optimization.
2. Results and discussion
2.1. Discovery and validation of novel BRD4 inhibitors
In this study, our in-house small-molecule library that contains more than 2000 compounds was screened, using high-throughput screening platforms. Several compounds that share a trimethoxy ring exhibited effective inhibition against the first bromodomain of BRD4 from substrate recognition, in a concentration-dependent manner. Different from known BRD4 bromodomain inhibitors, these hit compounds have novel scaffolds, from the perspective of chemical structure. The top 50 hit compounds from the first round of screening are listed in Table S1, ESI.‡ The first compound of this category discovered in this study was named DC-BD-03, with an IC50 value of 2.01 μM, and the curve is plotted in Fig. 1A. The IC50 value was calculated using the AlphaScreen assay. To confirm the binding of these compounds, a protein thermal shift assay was subsequently conducted to determine the impact of these compounds on the thermal stability of the first bromodomain of BRD4. As shown in Fig. 1B, the melting temperature Tm of the first bromodomain of BRD4 was increased by compound DC-BD-03, in a dose-dependent manner.
Fig. 1. (A) The chemical structure of compound DC-BD-03 and its inhibitory activities against the first bromodomain of BRD4 at different concentrations. The signals were normalized and plotted as a function of compound concentrations. The IC50 value was calculated through fitting a four-parameter log-logistic model using the software R. (B) The melting curves of the first bromodomain of BRD4, with compound DC-BD-03 at different concentrations. Fluorescence signals from the protein thermal shift assay were normalized and plotted as a function of temperature, the ΔTm values were calculated using the software that comes with the instrument, and the curves were drawn using the software R.

2.2. Structure and activity relationship
The analogues of compound DC-BD-03 were searched in the database of Specs (http://www.specs.net), based on the shared substructure, and a series of derivative compounds were purchased and evaluated. The structures of these compounds and their activities against the first bromodomain of BDR4 from binding with acetylated lysines are listed in Tables 1 and S2, ESI.‡ As the trimethoxy group was shared by this series of compounds, the difference in activities lied in the tails of these compounds. Compared to the leading compound DC-BD-03, the tails of compounds DC-BD-10, DC-BD-20, DC-BD-33 and DC-BD-35 were bulky, which may prevent these compounds from proper binding with the residues in the pocket of BRD4. On the other hand, smaller tails may reduce the binding energy, such as compounds DC-BD-01, DC-BD-04, DC-BD-13, DC-BD-18 and DC-BD-27. The activity of compound DC-BD-14 was poor, indicating that rings were favored as tails of this series of compounds, in comparison with flexible chains. Compared to compound DC-BD-18, a decrease in activity was observed for compound DC-BD-25, which might be due to the clash with residue atoms in the binding pocket. Compared to compound DC-BD-28, an increase in activity was observed for compound DC-BD-02, indicating that a hydrogen bond donor was better than an acceptor at this position. It was worth noting that although the tail of compound DC-BD-12 was not ring structures, its activity was better than those of other analogue compounds, apart from the leading compound DC-BD-03. Then, ring structures might be introduced to this compound during further optimization.
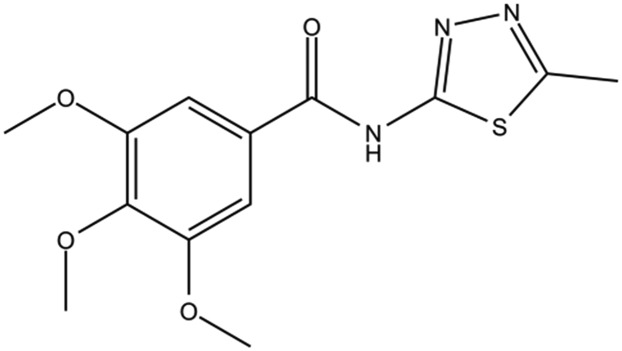
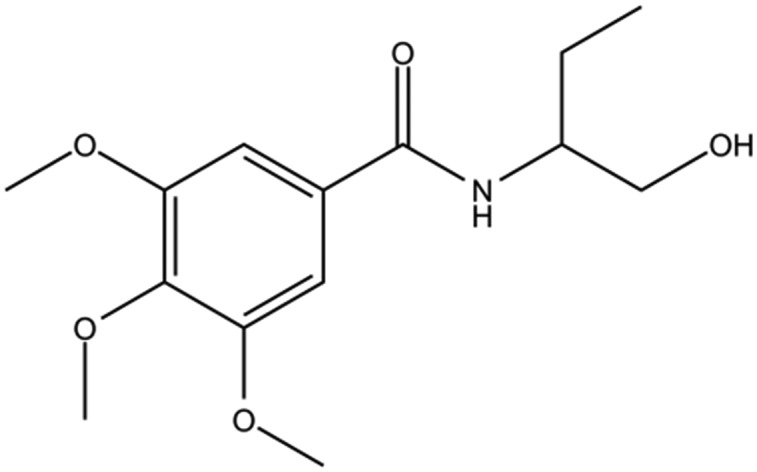
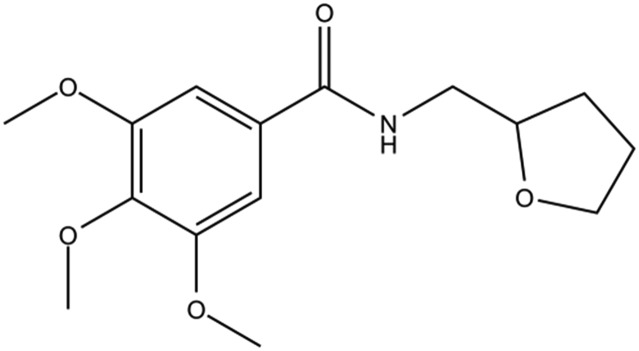
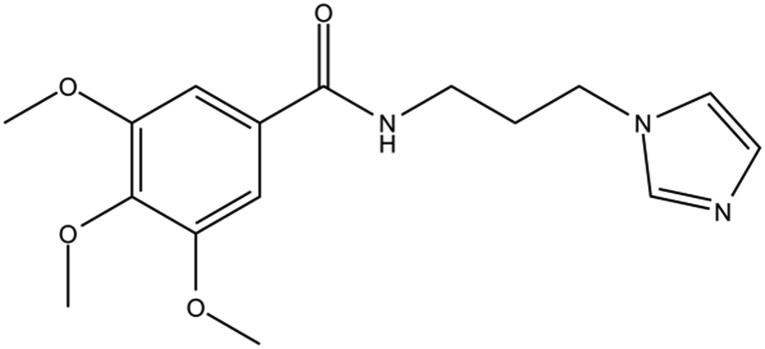
Table 1. The structures of DC-BD-03 series of compounds and their inhibitory activities against the first bromodomain of BRD4, from binding with acetylated lysines.
| Compound ID | Compound structure | IC50 (μM) | Inhibition (50 μM) |
| DC-BD-01 |
|
90% | |
| DC-BD-02 |
|
94% | |
| DC-BD-04 |
|
94% | |
| DC-BD-05 |
|
90% | |
| DC-BD-06 |
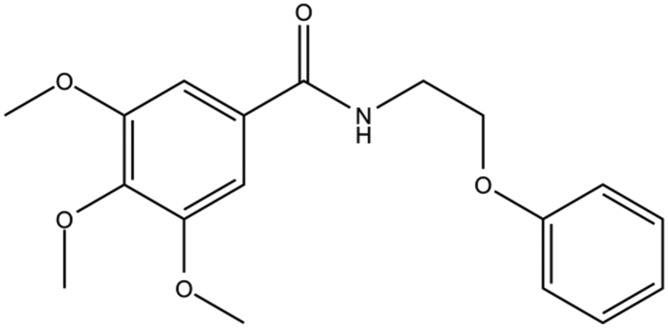
|
6.6 | |
| DC-BD-11 |
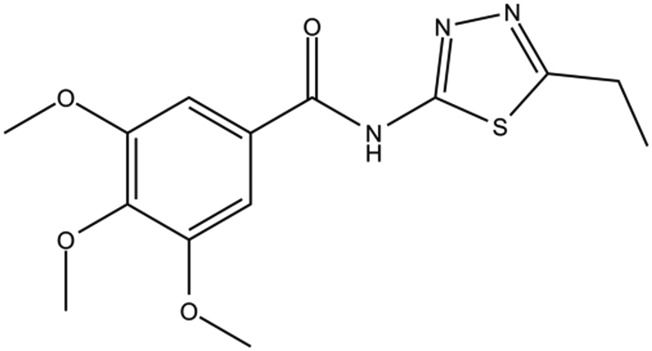
|
90% | |
| DC-BD-12 |
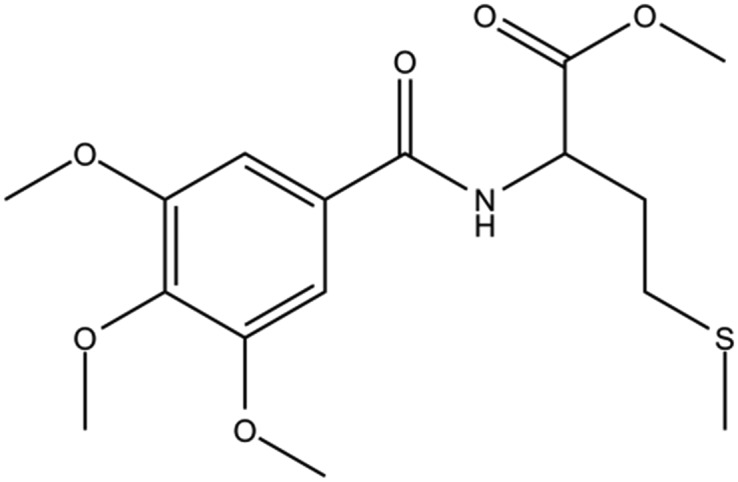
|
2.1 | |
| DC-BD-15 |
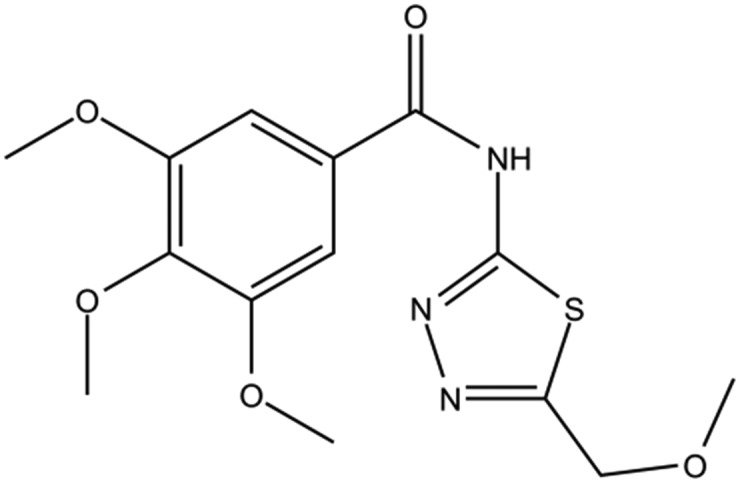
|
11 | |
| DC-BD-18 |
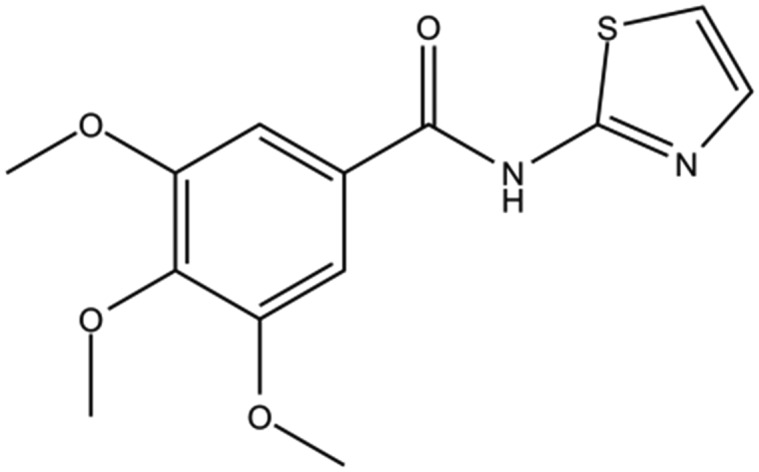
|
4.3 | |
| DC-BD-25 |
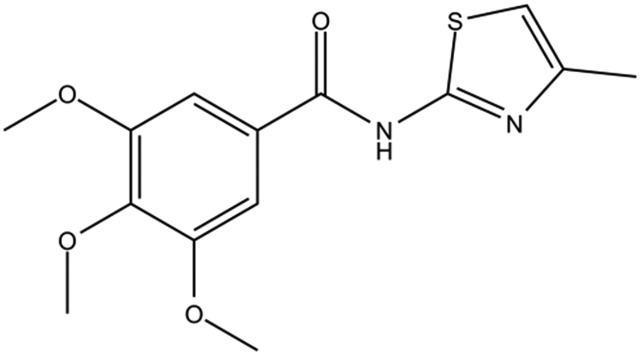
|
96% | |
| DC-BD-27 |
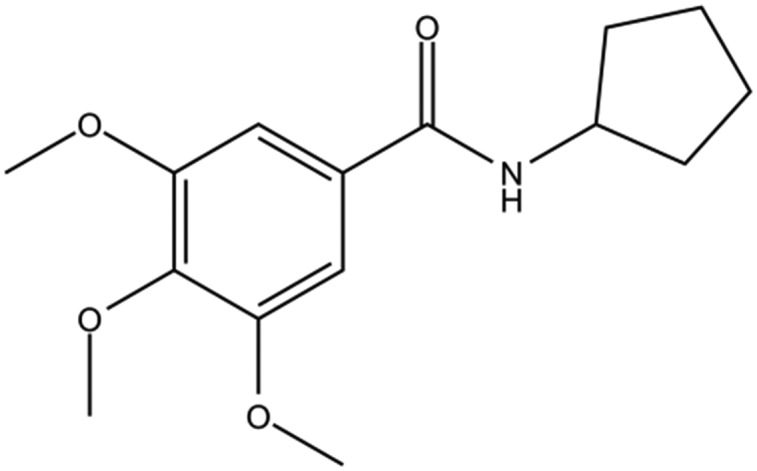
|
90% | |
| DC-BD-29 |
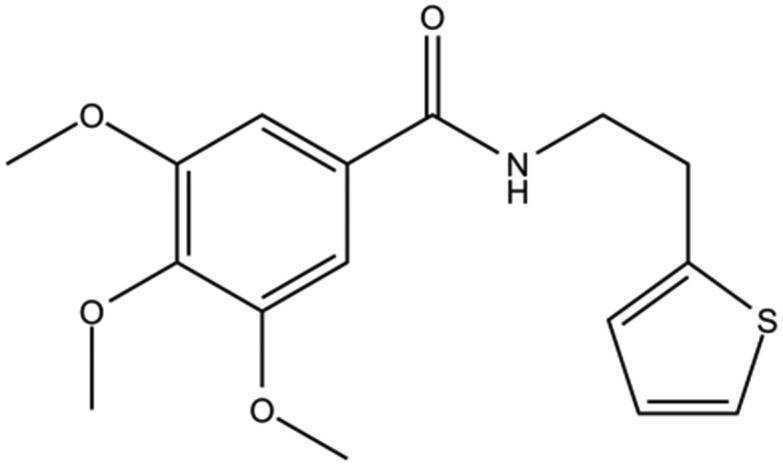
|
6.4 | |
| DC-BD-32 |
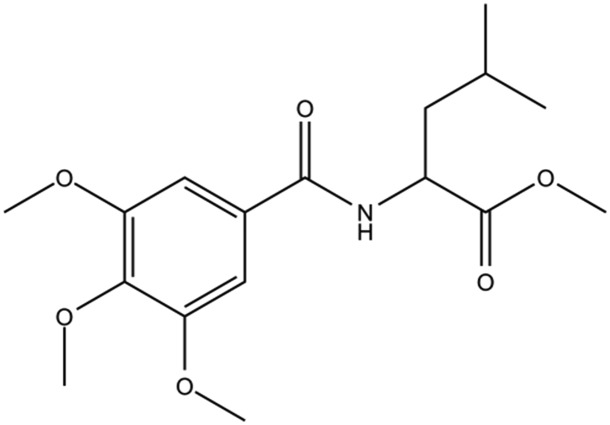
|
8.5 |
In addition, the selectivity data of compound DC-BD-03 against other BET bromodomains and BRD proteins are provided in Tables S3 and S4, ESI.‡ The inhibitory activity data of other compounds with determined IC50 values are provided in Fig. S1, ESI.‡
2.3. Crystal structure determination
To reveal the binding mechanism, as well as to facilitate further structure-based optimization, attempts were made to determine the complex structures of the first bromodomain of BRD4 with these compounds. However, the solubility of some compounds in this category was not good enough for diffraction-quality crystals. Thus, only the crystal structure of compound DC-BD-29 with the first bromodomain of BRD4 was solved, with enough electron density to determine the binding mode. The electron density map of the tail of the ligand was not intact, when compared with the deeply buried trimethoxy ring, indicating that the tail of the compound in crystals was relatively flexible.
As shown in Fig. 2 and S2,‡ compound DC-BD-29 formed direct and water-bridged hydrogen bonds, as well as hydrophobic contacts, with surrounding residues within the binding pocket of the first bromodomain of BRD4. The trimethoxy ring of compound DC-BD-29 formed a pair of effective hydrogen bonds with the conserved asparagine N140, which was found critical previously in the recognition of acetyl-lysine, as well as in the binding of other BRD4 bromodomain inhibitors of different chemotypes.11,18,19 In addition, this compound formed a pair of water-bridged hydrogen bonds with residue Y97, indicating the important role of water within the binding pocket, which was also observed in the binding of other BRD4 bromodomain inhibitors.15,18–20 On the other hand, compound DC-BD-29 formed hydrophobic contacts with residues W81, P82, F83, V87, L92, and I146, which contributed to the affinity of this category of BRD4 bromodomain inhibitors.
Fig. 2. (A) The crystal structure (PDB code: 5H21) of compound DC-BD-29 in complex with the first bromodomain of BRD4. Crystal water molecules are shown as red spheres. (B) The 2Fo–Fc electron density map for compound DC-BD-29, in its complex crystal structure with the first bromodomain of BRD4. The contour level was set to 1.0 sigma.

Taken together, these crystallographic results helped to explain the structure and activity relationship of these compounds. As the tails of these compounds were relatively shallow within the binding pocket, and no effective hydrogen bonds were formed as well, the contribution of the shared trimethoxy ring was predominant for the potency of this category of BRD4 bromodomain inhibitors. This mechanism was consistent with the fact that a wide variation was not observed in terms of the potency of these compounds. In addition, the IC50 values of several compounds such as DC-BD-10, DC-BD-14, DC-BD-33, DC-BD-34, and DC-BD-35 in this category (Table S2‡) were above 50 μM, which might be attributed to the solubility problem or severe clashes with the receptor atoms in the binding pocket.
2.4. DC-BD-03 inhibited the proliferation of human leukemia MV4-11 cells
As BRD4 plays important roles in the occurrence and progression of leukemia,6,9,10 the inhibitory activity of this novel category of BRD4 inhibitors against human leukemia cells was determined. After treatment with compound DC-BD-03, a reduction in viable cells was observed in the MLL-AF4-expressing acute leukemia cell line MV4-11, in a dose-dependent manner. As shown in Fig. 3A, the IC50 value was 41.92 μM for three days after treatment and 31.36 μM for seven days, demonstrating that compound DC-BD-03 inhibited the proliferation of MV4-11 cells.
Fig. 3. (A) Compound DC-BD-03 inhibited the proliferation of MV4-11 cells, with IC50 values of 41.92 μM for three days and 31.36 μM for seven days after treatment. (B) Compound DC-BD-03 inhibited the transcription of BRD4 downstream genes Bcl-2, c-Myc, and CDK6, at concentrations of 10 μM and 30 μM, after incubation for 6 hours. (C) Compound DC-BD-03 decreased the protein abundance of c-Myc, in a dose-dependent manner, after incubation for 6 hours. The compound I-BET151 was used as the positive control.

The proliferation data for positive control I-BET151 are provided in Fig. S3, ESI.‡
2.5. DC-BD-03 decreased the expression of downstream genes of BRD4
To determine the effect of these compounds on the expression of BRD4 downstream genes, MV4-11 cells were treated with compound DC-BD-03 and incubated for 24 h. Then, quantitative real-time fluorescence PCR was used to measure the expression of c-Myc, Bcl-2, and CDK6,10 which were previously reported to be driven by BRD4, after the cells were treated with compound DC-BD-03 of different concentrations and incubated for 6 hours. As shown in Fig. 3B, compound DC-BD-03 decreased the expression of these target genes, as compared to the untreated control, in a concentration-dependent manner. The PCR data for positive control I-BET151 are provided in Fig. S4, ESI.‡ In addition, western blot results showed that compound DC-BD-03 decreased the abundance of c-Myc protein, in a concentration-dependent manner, as shown in Fig. 3C. The western blot results for Bcl-2 and CDK6 are provided in Fig. S5, ESI.‡ These results indicated that compound DC-BD-03 decreased the expression as well as protein abundance of BRD4 downstream genes.
2.6. DC-BD-03 caused cell cycle arrest and apoptosis in MV4-11 cells
To evaluate the effect of this novel category of BRD4 inhibitors on cell cycle and apoptosis, MV4-11 cells treated with DMSO and 50 μM DC-BD-03 were subjected to flow cytometry. As shown in Fig. 4, compound DC-BD-03 induced effective cell cycle arrest and apoptosis in MV4-11 cells.
Fig. 4. (A) Compound DC-BD-03 (50 μM) caused cell cycle arrest in MV4-11 cells, after 24 hours of incubation. (B) Compound DC-BD-03 (50 μM) caused apoptosis in MV4-11 cells, after 72 hours of incubation.

3. Conclusion and perspective
In this study, a novel category of BRD4 inhibitors was discovered and evaluated. These inhibitors target the first bromodomain of the human BRD4 protein and share a trimethoxy ring in terms of chemical structure, and the first compound of this series that was discovered was named DC-BD-03. Among these compounds, the crystal structure of compound DC-BD-29 with the first bromodomain of BRD4 was determined through X-ray diffraction. These crystallographic results revealed the binding mechanism of this category of BRD4 inhibitors and provided solid basis for structure-based optimization in the future. At the cellular level, these compounds effectively inhibited the proliferation of human leukemia MV4-11 cells and caused cell cycle arrest and apoptosis, which validated the biological activities of this novel category of BRD4 inhibitors as anti-tumor agents. However, structural modifications could be made for further development, when aligned and compared with I-BET151, as shown in Fig. 5. The tails of these compounds could be modified into less flexible groups and hydrophobic contacts could be increased by introducing a bulky group near one of the trimethoxy rings or the amide groups. Taken together, this novel category of BRD4 inhibitors was promising to be further developed into drug candidates for the therapy of BRD4-driven cancers and other related diseases.
Fig. 5. Compound DC-BD-29 aligned with I-BET151, according to their crystal structures (I-BET151 PDB code: 3ZYU).

4. Experiments
4.1. Protein expression and purification
Proteins were cloned, expressed and purified generally as previously described.4 The first bromodomain of the human BRD4 protein was cloned into a pET28a vector. The fusion protein was expressed in Escherichia coli BL21 (DE3) cells with an N-terminal 6× His-tag. The cultures were grown to an OD600 value of 0.6–0.8 at 37 °C, induced with 400 mM IPTG, and incubated for 14–16 h at 16 °C. The protein was first purified by nickel affinity chromatography (HisTrap FF, GE Healthcare) and then further purified by gel-filtration chromatography on a Superdex 75 10/300 column (GE Healthcare). Buffer A for the protein purification nickel column (GE 5 ml FF) consisted of 50 mM HEPES (pH 7.4), 100 mM NaCl, 0.1% β-mercaptoethanol, and 10 mM imidazole, and buffer B consisted of 50 mM HEPES (pH 7.4), 100 mM NaCl, 0.1% β-mercaptoethanol, and 500 mM imidazole. The purified protein was concentrated in a buffer containing 10 mM HEPES (pH 7.4), 100 mM NaCl, and 1 mM dithiothreitol (DTT) and was stored for subsequent assays and crystallization. The eluted protein was treated overnight at 4 °C with the tobacco etch virus (TEV) protease to remove the 6× His-tag if the protein was prepared for crystallization.
4.2. Compound screening
The amplified luminescence proximity homogeneous assay (ALPHA) peptide displacement assay for the first bromodomain of the human BRD4 protein was carried out as previously described.21 Purified proteins were incubated with different compounds of equal concentrations on 384-well plates (OptiPlate-384, PerkinElmer) for 10 min at room temperature with the assay buffer (0.1% Triton X-100, 1 mM DTT, 0.1% bovine serum albumin (w/v)), before 25 nM biotinylated peptide was added to every plate hole with compounds. The substrate peptide was synthesized by Shanghai China Peptide Corporation with the sequence SGRG-K(Ac)–GG-K(Ac)–GLG-K(Ac)–GGA-K(Ac)–RHRKVGG-K(Biotin) and was incubated at room temperature for 10 minutes. After the Ni2+ chelate acceptor beads (PerkinElmer) were added, the mixture was incubated for 30 minutes. The compound JQ-1 (Sigma) was used as the positive control. Finally, streptavidin coated donor beads (PerkinElmer) were added, and the mixtures were incubated in subdued light for 30 minutes at room temperature. The signals for all the 384-well plates were read in alpha mode using an EnVision reader (PerkinElmer).
4.3. Protein thermal shift assay
For the protein thermal shift assay used in this study, 5 μM purified proteins were incubated with 5× SYPRO® Orange (Molecular Probes) in 20 mM HEPES (pH 7.4) and 100 mM NaCl. Then, compounds of different concentrations or DMSO (0.2%) was mixed with the proteins on a fast 96-well optical plate (Applied Biosystems). The fluorescence signals were gathered with the slope of 2.5 °C min–1 from 25 °C to 95 °C using a QuantStudio™ 6 Flex real-time PCR machine (Applied Biosystems). The output data were analyzed and melting temperatures were calculated using the software Protein Thermal Shift™ v1.0 (Life Technologies), and curves were plotted using the software R.
4.4. Complex crystal structure of compound DC-BD-29 with BD1
Different temperature and pool conditions were tried to obtain crystals of the proteins, using the sitting drop vapor diffusion method. The first bromodomain of the BRD4 construct was concentrated to 10 mg mL–1 in the buffer consisting of 10 mM HEPES (pH 7.5) and 100 mM NaCl. Then, 1 μL proteins with an equal volume of reservoir solution were mixed and placed and protected from light. After two weeks, rice-shaped crystals were found in the 13th pool condition, while the size and shape were not good enough. Then, the concentrations of proteins as well as salts and glycerol were optimized. Finally, diffraction-quality crystals were obtained under the pool conditions of 10–15% glycerol and 4 M sodium formate. To obtain the crystals of compounds with the first bromodomain of BRD4, the grown protein crystals were transferred and soaked into the reservoir solution containing compounds, and the pool conditions were the same as described above. After incubation for one day, the crystals of proteins with compounds were harvested.
The X-ray diffraction data were collected at the Shanghai Synchrotron Radiation Facility on the beamline of BL18U at 100 K. Diffraction data were indexed and integrated using the program XDS22 and scaled using the Aimless23 module of the Collaborative Computational Project No. 4 (CCP4) program suite.24 The structure was solved by the method of molecular replacement using the Phaser25 module of Phenix,26 with the template of PDB code ; 2OSS. Then, the initial structure was rebuilt using the AutoBuild27 module of Phenix. Ligands were fit into the electron density map with the LigandFit28 module of Phenix and Coot.29 Then, complex structures were refined for several rounds using Phenix. Structure validation was performed using MolProbity.30 Data statistics are summarized in Table S5, ESI.‡
4.5. Cell viability assay
Cells used in this study were cultured in RPMI 1640 medium (Life Technologies) supplemented with 10% fetal bovine serum and 1% Pen/Strep (Life Technologies). Then, cells were cultured in a humidified incubator at 37 °C with 5% CO2. Alamar Blue assays were used to determine the effects of compounds on cell viability.31 For the cell viability assay, cells were grown on plates at the density of 1 × 105–3 × 105 and incubated for 30 minutes. Then, compounds of different concentrations or DMSO was added to the plates and incubated for three days or seven days. After 10 μL of Alamar Blue agent was added to every hole and the plates were incubated for 3 h at 37 °C, fluorescence signals were measured at an excitation wavelength of 544 nm and an emission wavelength of 590 nm using the reader (PE EnVison). The percentage of viability was calculated by normalizing the fluorescence signals to the control.
4.6. Flow cytometry
For the cell cycle assay, cells were grown on 6-well plates at the density of 3 × 105 and incubated for 30 minutes. Then, cells were treated with DC-BD-03 of different concentrations or DMSO and incubated for 24 h. Cells were centrifuged and collected at 25 °C and subsequently resuspended in 300 μL of PBS. Pre-cooled oscillating drops of 70% ethanol (700 μL) were added to the sample and the ethanol was removed by centrifugation after letting it stand for one night at 4 °C. After purging several times with PBS, the cells were resuspended at the density of 1 × 106 in 500 μL of PI/RNase solution (BD Pharmingen) for cell cycle analysis using BD Flow Cytometry. For the apoptosis assay, cells were grown on 6-well plates at the density of 3 × 105 and incubated for 30 minutes. Then, compounds of different concentrations or DMSO was added to the plates and incubated for 72 h. Cells were centrifuged and resuspended in 300 μL of 1× binding buffer. After mixing with 5 μL of Annexin V-FITC, the plates were protected from light and incubated for 15 minutes at room temperature. Then, cells were collected and stained using the Annexin V-FITC/PI Apoptosis Detection Kit (Vazyme). Finally, 5 μL of PI was added for dyeing 5 minutes before flow cytometry, and 200 μL of 1× binding buffer was added just before the apoptosis assay. Data analysis was performed using the software that was supplied with the instrument.
4.7. Western blot
For the western blot experiment, the MV4-11 cells were grown on 6-well plates at the density of 3 × 105 and incubated for 30 minutes. Then, compounds of different concentrations or DMSO was added and the plates were left to stand for 6 h. The cells were washed with PBS and centrifuged at 2000 rpm. After the supernatant was discarded, RIPA solution (50 mM Tris-HCl, pH 8.0; 150 mM NaCl; 1% NP-40; 0.5% sodium deoxycholate; 0.1% SDS) was added for cell lysis. Then, the cell lysis solution was heated for denaturation, and 12% SDS-PAGE was used for separation. Total proteins were transferred to the cellulose nitrate filter (GE Healthcare). The primary antibody was c-Myc (CST, D84C12) rabbit mAb (CST, 5605S), and the secondary antibody was anti-rabbit IgG (Jackson, 111-035-003). Rabbit IgG antibody (Beyotime A0208) and mouse IgG antibody (Beyotime, A0216) labeled with horseradish peroxidase (HRP) were used to recognize primary antibodies. Finally, HRP substrate (GE Healthcare) for chemical luminescence was used for detection, and FUJIFILM LAS-4000 was used for photograph and data processing.
Accession code
The X-ray coordinates of compound DC-BD-29 with the first bromodomain of BRD4 has been deposited to the Protein Data Bank (http://www.rcsb.org), with accession code ; 5H21.
Supplementary Material
Acknowledgments
The computation resources were supported by the Computer Network Information Center, Chinese Academy of Sciences and the Guangdong Supercomputing Center (nsfc2015-446 to C. L.). We thank the staff from the BL18U1 beamline at the Shanghai Synchrotron Radiation Facility (SSRF) and the staff from the BL19U1 beamline of the National Center for Protein Science Shanghai (NCPSS) at SSRF, for assistance during data collection. We are grateful to the National Center for Protein Science Shanghai (Shanghai Science Research Center, Protein Expression and Purification System) for their instrument support and technical assistance. We gratefully acknowledge financial support from the Ministry of Science and Technology of China (2015CB910304 to H. J.), the National Natural Science Foundation of China (21472208 to C. L. and 21210003 and 81230076 to H. J.), and the Fund of State Key Laboratory of Toxicology and Medical Countermeasures, Academy of Military Medical Science (TMC201505 to C. L.).
Footnotes
†The authors declare no competing interests.
‡Electronic supplementary information (ESI) available. See DOI: 10.1039/c7md00083a
References
- Kouzarides T. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Belkina A. C., Denis G. V. Nat. Rev. Cancer. 2012;12:465–477. doi: 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P., Picaud S., Mangos M., Keates T., Lambert J.-P., Barsyte-Lovejoy D., Felletar I., Volkmer R., Müller S., Pawson T., Gingras A.-C., Arrowsmith C. H., Knapp S. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W. B., Fedorov O., Morse E. M., Keates T., Hickman T. T., Felletar I., Philpott M., Munro S., McKeown M. R., Wang Y., Christie A. L., West N., Cameron M. J., Schwartz B., Heightman T. D., La Thangue N., French C. A., Wiest O., Kung A. L., Knapp S., Bradner J. E. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Vakoc C. R. Mol. Cell. 2014;54:728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J., Shi J., Wang E., Rappaport A. R., Herrmann H., Sison E. A., Magoon D., Qi J., Blatt K., Wunderlich M., Taylor M. J., Johns C., Chicas A., Mulloy J. C., Kogan S. C., Brown P., Valent P., Bradner J. E., Lowe S. W., Vakoc C. R. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang M. K., Mochizuki K., Zhou M., Jeong H.-S., Brady J. N., Ozato K. Mol. Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Yang Z., He N., Zhou Q. Mol. Cell. Biol. 2008;28:967–976. doi: 10.1128/MCB.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M. A., Prinjha R. K., Dittmann A., Giotopoulos G., Bantscheff M., Chan W.-I., Robson S. C., Chung C.-W., Hopf C., Savitski M. M., Huthmacher C., Gudgin E., Lugo D., Beinke S., Chapman T. D., Roberts E. J., Soden P. E., Auger K. R., Mirguet O., Doehner K., Delwel R., Burnett A. K., Jeffrey P., Drewes G., Lee K., Huntly B. J. P., Kouzarides T. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L.-L., Tian M., Li X., Li J.-J., Huang J., Ouyang L., Zhang Y., Liu B. Oncotarget. 2015;6:5501–5516. doi: 10.18632/oncotarget.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciceri P., Müller S., O'Mahony A., Fedorov O., Filippakopoulos P., Hunt J. P., Lasater E. A., Pallares G., Picaud S., Wells C., Martin S., Wodicka L. M., Shah N. P., Treiber D. K., Knapp S. Nat. Chem. Biol. 2014;10:305–312. doi: 10.1038/nchembio.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E., Jeffrey K. L., Schaefer U., Beinke S., Dewell S., Chung C.-W., Chandwani R., Marazzi I., Wilson P., Coste H., White J., Kirilovsky J., Rice C. M., Lora J. M., Prinjha R. K., Lee K., Tarakhovsky A. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asangani I. A., Dommeti V. L., Wang X., Malik R., Cieslik M., Yang R., Escara-Wilke J., Wilder-Romans K., Dhanireddy S., Engelke C., Iyer M. K., Jing X., Wu Y.-M., Cao X., Qin Z. S., Wang S., Feng F. Y., Chinnaiyan A. M. Nature. 2014;510:278–282. doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedeh G., Cerny-Reiterer S., Eisenwort G., Herrmann H., Blatt K., Hadzijusufovic E., Sadovnik I., Mullauer L., Schwaab J., Hoffmann T., Bradner J. E., Radia D., Sperr W. R., Hoermann G., Reiter A., Horny H. P., Zuber J., Arock M., Valent P. Leukemia. 2015;29:2230–2237. doi: 10.1038/leu.2015.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran X., Zhao Y., Liu L., Bai L., Yang C.-Y., Zhou B., Meagher J. L., Chinnaswamy K., Stuckey J. A., Wang S. J. Med. Chem. 2015;58:4927–4939. doi: 10.1021/acs.jmedchem.5b00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran X., Zhao Y. J., Liu L., Bai L. C., Yang C. Y., Zhou B., Meagher J. L., Chinnaswamy K., Stuckey J. A., Wang S. M. J. Med. Chem. 2015;58:4927–4939. doi: 10.1021/acs.jmedchem.5b00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L. L., Wang Y. Q., Cao D. Y., Chen T. T., Wang Q., Li Y. L., Xu Y. C., Zhang N. X., Wang X., Chen D. Q., Chen L., Chen Y. L., Xia G. X., Shi Z., Liu Y. C., Lin Y. J., Miao Z. H., Shen J. K., Xiong B. J. Med. Chem. 2015;58:1281–1297. doi: 10.1021/jm501504k. [DOI] [PubMed] [Google Scholar]
- Gosmini R., Nguyen V. L., Toum J., Simon C., Brusq J.-M. G., Krysa G., Mirguet O., Riou-Eymard A. M., Boursier E. V., Trottet L., Bamborough P., Clark H., Chung C.-W., Cutler L., Demont E. H., Kaur R., Lewis A. J., Schilling M. B., Soden P. E., Taylor S., Walker A. L., Walker M. D., Prinjha R. K., Nicodème E. J. Med. Chem. 2014;57:8111–8131. doi: 10.1021/jm5010539. [DOI] [PubMed] [Google Scholar]
- Hügle M., Lucas X., Weitzel G., Ostrovskyi D., Breit B., Gerhardt S., Einsle O., Günther S., Wohlwend D. J. Med. Chem. 2016;59:1518–1530. doi: 10.1021/acs.jmedchem.5b01267. [DOI] [PubMed] [Google Scholar]
- Hewings D. S., Fedorov O., Filippakopoulos P., Martin S., Picaud S., Tumber A., Wells C., Olcina M. M., Freeman K., Gill A., Ritchie A. J., Sheppard D. W., Russell A. J., Hammond E. M., Knapp S., Brennan P. E., Conway S. J. J. Med. Chem. 2013;56:3217–3227. doi: 10.1021/jm301588r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott M., Yang J., Tumber T., Fedorov O., Uttarkar S., Filippakopoulos P., Picaud S., Keates T., Felletar I., Ciulli A., Knapp S., Heightman T. D. Mol. BioSyst. 2011;7:2899–2908. doi: 10.1039/c1mb05099k. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011;67(4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L.-W., Read R. J., Adams P. D. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2008;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T. C., Klei H., Adams P. D., Moriarty N. W., Cohn J. D. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006;62:915–922. doi: 10.1107/S0907444906017161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W. G., Cowtan K. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B., Arendall W. B., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Nasiry S., Geusens N., Hanssens M., Luyten C., Pijnenborg R. Hum. Reprod. 2007;22:1304–1309. doi: 10.1093/humrep/dem011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.