

 Synthesis of a series of Schiff base-bridged tetrahydroprotoberberine triazoles as a new type of potential antimicrobial agents, and preliminary interactions with DNA indicated the possible interaction mechanism.
Synthesis of a series of Schiff base-bridged tetrahydroprotoberberine triazoles as a new type of potential antimicrobial agents, and preliminary interactions with DNA indicated the possible interaction mechanism.
Abstract
A series of novel Schiff base-bridged tetrahydroprotoberberine (THPB) triazoles were designed, synthesized and characterized for the first time. Antimicrobial assay showed that some of the prepared compounds exerted stronger antibacterial and antifungal activities than the reference drugs. Especially, THPB triazole 7a gave low MIC values of 0.5, 1 and 2 μg mL–1 against B. yeast, M. luteus and MRSA, respectively. Further experiments indicated that the highly active molecule 7a was able to rapidly kill the MRSA strain and did not trigger the development of bacterial resistance even after 14 passages. The preliminary exploration for the antimicrobial mechanism revealed that compound 7a could effectively intercalate into calf thymus DNA to form a 7a–DNA supramolecular complex, and its Zn2+ complex had the ability to directly cleave pUC19 DNA, which suggested that compound 7a might be a potentially dual-targeting antibacterial molecule. It was also found that compound 7a could be efficiently stored and carried by human serum albumin (HSA), and the hydrophobic interactions and hydrogen bonds played important roles in the transportation of HSA to the active molecule 7a.
1. Introduction
The increasing resistance to clinical drugs especially current antimicrobial agents is triggering high morbidity and mortality, which has become one of the most critical health concerns worldwide.1 Currently, an urgent global task is to combat drug resistance, and it has been universally recognized that no action today means no cure tomorrow. The development of structurally novel molecules with distinct action mechanisms from the well-known class of antimicrobial agents is therefore of great importance to the medical community.2
Berberine has been commonly used in clinics with a long history of more than 2000 years in traditional Chinese herbs to treat intestinal infections such as acute gastroenteritis, bacillary dysentery and cholera.3 The unique structure of berberine with a quaternary nitrogen and a large desirable π-conjugated backbone enables berberine-based derivatives to readily bind with biomolecules like deoxyribonucleic acid (DNA) or an enzyme via noncovalent forces such as π–π stacking and electronic interactions, thus exerting potent biological activities. Notably, despite its long term clinical use, the incidence of resistance to berberine is reported rarely.4 This attracts special interest in investigating berberines for possible further antimicrobial use and other medicinal potentialities like anticancer, antiviral, anti-inflammatory, antiparasitic activities, etc.5 However, berberine exhibits poor solubility, low bioavailability in vivo and some severe side effects such as anaphylactic shock and drug eruption, and these have seriously limited its useful profile. Therefore much effort has been devoted towards the structural modification of berberine. A literature search revealed that structural changes such as disruption of the symmetry or molecular planarity were proved to be an effective way to improve its intrinsic water solubility.6 Tetrahydroprotoberberine (THPB) was found to be a good structural alternative with large medicinal potentiality.7 An increased exploitation is directed towards THPBs for their medicinal value, especially in anti-infective aspects.8
Azole compounds are well-known types of heterocycles with various bioactivities and have been extensively designed and synthesized as antimicrobial drugs.9 Particularly, the triazole nucleus is a poly-nitrogen electron-rich heterocycle present in many biologically active compounds. It is generally considered that the triazole fragment is stable to metabolic degradation and capable of forming hydrogen bonds, thus the introduction of the triazole nucleus is beneficial to improve binding with biomolecular targets and increase the water solubility of target compounds.10 Therefore the triazole moiety is prevalently employed in drug design. So far, a large number of predominantly triazole-based medicinal drugs have been successfully developed and prevalently used in clinics, especially in the antimicrobial aspect, such as fluconazole, itraconazole, voriconazole, posaconazole, efinaconazole, terconazole, etc.11 Their important clinical uses have been motivating extensive efforts to construct new bioactive molecules based on the triazole fragment.
Our previous work showed that the introduction of azoles such as imidazole, triazole and benzimidazole moieties into the C-9 or C-12 position of the berberine backbone was not only beneficial to enhance the antimicrobial activities but also widen the antimicrobial spectrum, including against methicillin-resistant Staphylococcus aureus (MRSA).12 However, to our best knowledge, the combination of azoles and the THPB backbone has been rarely reported. In view of the above consideration and as an extension of our ongoing research on bioactive heterocycles, it is of great interest for us to combine THPB with the triazole nucleus to generate a new structural type of potential antimicrobial agents via a Schiff base bridge which is a validated moiety to beneficially improve the water solubility and the antimicrobial efficacy (Fig. 1).13 It is expected that these hybrids of THPB with a triazole moiety could not only beneficially enhance the antimicrobial activities and broaden the antimicrobial spectrum, but also helpfully improve water solubility. A literature search revealed that lipophilic substituents on berberine especially at the C-9 position could significantly influence the bioactivities by modifying molecular flexibility and regulating the lipid–water partition coefficient.14 Rationally, various aliphatic chains with different lengths and substituted benzyl moieties including chloro, fluoro and nitro groups were incorporated into the C-9 position of THPB to investigate their effects on antimicrobial activities. The synthetic route of the target THPB triazoles is shown in Scheme 1. All the newly synthesized compounds were confirmed by spectral analysis and screened against Gram-positive bacteria including MRSA, Gram-negative bacteria and fungi. Aqueous solubility, a bactericidal kinetic assay and antibacterial resistance to the highly active molecule were also investigated and conducted. In order to explore the preliminary antimicrobial action mechanism, the interaction of the most active triazole derivative with DNA was further carried out by fluorescence and UV-vis absorption spectroscopy as well as agarose gel electrophoresis. Additionally, the binding behavior of the highly active molecule to human serum albumin (HSA) was investigated to preliminarily study its absorption, distribution, and metabolism.
Fig. 1. Design of novel Schiff base-bridged tetrahydroprotoberberine triazoles.

Scheme 1. Reagents and conditions: (i) berberine, 20 mm Hg, 190 °C, 15 min, CH3CH2OH–HCl; (ii) CH3OH, NaBH4, rt, 4 h; (iii) (a) HMTA/TFA, 120 °C, 3 h; (b) 10% H2SO4, 90–100 °C, 2 h; (iv) CH3CH2OH, CH3COOH, reflux, 5 h; (v) alkyl bromides and halobenzyl halides, K2CO3, DMF, 80 °C.

2. Chemistry
All the target compounds were conveniently prepared from commercial berberine chloride, 4H-1,2,4-triazol-4-amine, alkyl bromides and halobenzyl halides. As depicted in Scheme 1, berberrubine 2 was easily obtained in 84.6% yield by the 9-demethylation of berberine chloride 1 at 190 °C under vacuum,15 and was further reduced with NaBH4 to give THPB 3 at 0 °C in 63.1% yield. The formylation of compound 3 by hexamethylenetetramine (HMTA) in trifluoroacetic acid under reflux produced the important intermediate 4 in a high yield of 76.5%,16 which proceeded more facilely and efficiently than a literature method.17 The latter was condensed with 4H-1,2,4-triazol-4-amine in ethanol under reflux using a catalytic amount of glacial acetic acid to afford the target Schiff base 5 in 88.3% yield. Further structural modification of compound 5 by alkyl or substituted aromatic halides generated the alkyl derivatives 6a–f and aralkyl 7a–h with yields from 15.2 to 35.4% in DMF at 80 °C using potassium carbonate as the base. All the new compounds were confirmed by IR, 1H NMR, 13C NMR and HRMS spectroscopy (ESI‡).
3. Results and discussion
3.1. Antimicrobial activities
The obtained results in Table 1 showed that almost all the synthesized THPBs displayed better in vitro biological activity than the precursor berberine against most of the tested strains. Towards M. luteus and B. typhi, the intermediate 4 with a formyl group at the 12-position of THPB gave comparable or superior antibacterial efficacy to chloromycin and norfloxacin. Generally, the target THPB triazoles gave better inhibitory potency than the precursor berberine and the corresponding intermediate 4, and some of them were much more active than the reference drugs. It is revealed that the triazole fragment at the 12-position of THPB should play an important role in exerting antibacterial potency.
Table 1. Antibacterial data as MIC (μg mL–1) for compounds 4–7 a b .
| Compounds | Gram-positive bacteria |
Gram-negative bacteria |
||||||||
| S. aureus | MRSA | B. subtilis | M. luteus | E. coli JM109 | E. coli DH52 | S. dysenteriae | P. aeruginosa | B. proteus | B. typhi | |
| 4 | 128 | 256 | 32 | 2 | 256 | 256 | 512 | 16 | 64 | 2 |
| 5 | 64 | 16 | 512 | 1 | 128 | 128 | 256 | 128 | 64 | 128 |
| 6a | 256 | 8 | 128 | 16 | 256 | 512 | 512 | 256 | 32 | 256 |
| 6b | 16 | 8 | 16 | 1 | 16 | 32 | 8 | 16 | 16 | 16 |
| 6c | 8 | 32 | 8 | 16 | 4 | 32 | 8 | 32 | 16 | 2 |
| 6d | 8 | 16 | 16 | 16 | 32 | 16 | 64 | 512 | 16 | 16 |
| 6e | 32 | 32 | 32 | 64 | 128 | 64 | 128 | 128 | 64 | 16 |
| 6f | 256 | 512 | 256 | 512 | 512 | 256 | 512 | 512 | 256 | 256 |
| 7a | 8 | 2 | 32 | 1 | 32 | 16 | 32 | 8 | 16 | 4 |
| 7b | 256 | 512 | 512 | 32 | >512 | 256 | 256 | 16 | 64 | 512 |
| 7c | 32 | 8 | 32 | 2 | 32 | 16 | 16 | 8 | 128 | 4 |
| 7d | 16 | 16 | 32 | 256 | 4 | 64 | 16 | 2 | 512 | 1 |
| 7e | 256 | 256 | 512 | 512 | 0.5 | 512 | 64 | 512 | 512 | 256 |
| 7f | 8 | 32 | 16 | 16 | 8 | 32 | 64 | 4 | 8 | 8 |
| 7g | 16 | 8 | 32 | 128 | 16 | 256 | 512 | 32 | 32 | 32 |
| 7h | 512 | 16 | 256 | 2 | 128 | 512 | 64 | 64 | 512 | 4 |
| Berberine | 512 | 128 | >512 | >512 | — | >512 | 256 | 256 | — | — |
| Chloromycin | 16 | 16 | 32 | 8 | 32 | 32 | 32 | 32 | 32 | 32 |
| Norfloxacin | 0.5 | 8 | 4 | 2 | 1 | 1 | 4 | 16 | 8 | 4 |
aMinimal inhibitory concentrations were determined by the micro broth dilution method for microdilution plates.
b S. aureus, Staphylococcus aureus ATCC25923; MRSA, methicillin-resistant Staphylococcus aureus N315; B. subtilis, Bacillus subtilis ATCC6633; M. luteus, Micrococcus luteus ATCC4698; E. coli JM109, Escherichia coli JM109; E. coli DH52, Escherichia coli DH52; S. dysenteriae, Shigella dysenteriae; P. aeruginosa, Pseudomonas aeruginosa ATCC27853; B. proteus, Bacillus proteus ATCC13315; B. typhi, Bacillus typhi.
The preliminary structure–activity relationships (SARs) study demonstrated the significant effect of the substituents at the 9-position of THPB on biological activities. In contrast with the clinical drugs chloromycin and norfloxacin, the unsubstituted compound 5 at the 9-position of the THPB nucleus exhibited weaker activity against all the tested strains except for M. luteus which was the most sensitive to compound 5 with a low MIC value of 1 μg mL–1.
In general, most of the alkyl-substituted compounds could effectively inhibit the growth of all the tested bacterial strains in vitro. Among the alkyl derivatives 6a–f, the butyl compound 6b and the hexyl analogue 6c gave broader antibacterial spectrum and better activities with MIC values of 1–32 μg mL–1. Noticeably, compound 6b exhibited the best anti-M. luteus activity with a MIC value of 1 μg mL–1, which was superior to the standard drugs chloromycin (MIC = 8 μg mL–1) and norfloxacin (MIC = 2 μg mL–1). Towards E. coli JM109 and B. typhi strains, compound 6c displayed 8- and 16-fold more potent inhibitory activity (MIC = 4 and 2 μg mL–1, respectively) than chloromycin. It also demonstrated effective anti-S. aureus, anti-B. subtilis and anti-S. dysenteriae activities with MIC values of 8 μg mL–1, being more potent than chloromycin. Moreover, when the alkyl substituents were extended to decyl and dodecyl groups, compounds 6e and 6f showed poor or even no activity against most of the tested bacterial strains. However, the ethyl derivative 6a was also less beneficial for the antibacterial efficiency even at high concentration. These results revealed that either decrease or increase of the alkyl chain length was unfavorable for the bioactivity, and only an appropriate length chain in THPBs exerted an important influence in enhancing bioactivity. The short alkyl chains with poor lipophilicity and long alkyl chains with good lipophilicity in these THPBs might make them unfavorable for being delivered to the binding sites.
In comparison with the alkyl derivatives, most of the aralkyl-substituted ones (7a–h) exerted relatively better activities in inhibiting the growth of the tested strains. Notably, compound 7a without substitution on the phenyl ring gave the best antibacterial efficiencies with MIC values of 1–32 μg mL–1 towards the corresponding bacteria. It was found that M. luteus, MRSA and P. aeruginosa were sensitive to THPB triazole 7a with MIC values of 1, 2 and 8 μg mL–1, respectively, which were superior to those of chloromycin and norfloxacin. Excitedly, the target compounds 6a, 6b, 7a, 7c and 7g exerted good biological activities against MRSA with MIC values of 2–8 μg mL–1, which were more active than chloromycin (MIC = 16 μg mL–1). Especially compound 7a exhibited 8- and 4-fold more activity towards MRSA with lower concentration (MIC = 2 μg mL–1) than chloromycin and norfloxacin, respectively. These indicated that compound 7a had the potency to be a lead molecule in the development of more effective antimicrobial agents with a broad spectrum. A continual study revealed that the ortho- and para-substituted benzyl derivatives 7c, 7d and 7f were more effective against most of the tested bacteria than the meta-substituted 7b and 7e. Particularly, 2-chlorobenzyl derivative 7d gave potent inhibition against P. aeruginosa (MIC = 2 μg mL–1) and B. typhi (MIC = 1 μg mL–1), which was more active than chloromycin and norfloxacin. Unfortunately, the 4-nitrobenzyl compound 7h displayed relatively poor activity against most of the tested bacteria, which suggested that the introduction of an electron withdrawing group like NO2 might not be helpful for antibacterial activity. Among all the prepared compounds, the 3-chlorobenzyl derivative 7e gave the strongest activity against E. coli JM109 with a MIC value of 0.5 μg mL–1. This showed that this compound has potential to be developed as a special anti-E. coli JM109 agent.
The antifungal evaluation in vitro showed no remarkable activities for 12-formyl THPB 4 and 9-unsubstituted THPB triazole 5 against the tested fungi except for B. yeast which was highly sensitive to compound 4 (MIC = 1 μg mL–1). However, the structural modification at the 9-position of the THPB nucleus generated THPB triazole derivatives which displayed potent antifungal activities towards most of the tested fungal strains. It was noticed that the antifungal abilities of the alkyl compounds displayed similarity to their antibacterial efficiencies. The suitable length of an alkyl chain seemed to be the hexyl chain: the hexyl derivative 6c exerted the best antifungal efficacy with MIC values ranging from 2 to 32 μg mL–1 against all the tested fungi, better than other alkyl derivatives with a shorter or longer chain length. Among the aralkyl THPB triazoles 7a–h, the unsubstituted benzyl derivative 7a gave more effective antifungal potency with MIC values of 0.5–32 μg mL–1 than the other aralkyl-substituted compounds. Particularly, the same low MIC values of 0.5 μg mL–1 were observed for THPB triazole 7a towards B. yeast, 7b towards C. albicans and 7f towards C. utilis, and this suggested that these compounds should be much more active than the reference drug fluconazole, and there is a large possibility for them to be new antifungal agents and worthy to be the subject of deeper investigation (ESI‡ Table S1).
3.2. Effect of clog P values and aqueous solubility of selected compounds
THPB triazoles 5, 6b, 6c, 6f, 7a, 7c and 7f were further selected for solubility profiling. The water solubility (mg mL–1) was evaluated at physiological pH and the obtained results are depicted in Table 2. In general, the THPB triazoles were more soluble in water than berberine. Particularly, the THPB triazole 7f bearing a 4-chlorobenzyl substituent showed the largest water solubility in this series, 7 times higher than berberine. The highly bioactive derivative 7a possessed equivalent solubility to its fluorinated counterpart 7c with 2-fold enhanced soluble potency compared with berberine. Remarkably, the alkyl derivatives were less soluble in water than the benzyl ones. Among the alkylated THPBs 6b, 6c and 6f, the hexyl derivative 6c was endowed with more potent aqueous solubility, more than five times as soluble as berberine. Unfortunately, the replacement of the hexyl group by a dodecyl moiety dramatically led to a decrease in solubility. These results indicated that the insertion of a triazole moiety in a THPB backbone should be beneficial for the improvement of aqueous solubility.
Table 2. clog P values and evaluation of solubility of selected compounds (mean ± SD, n = 3) a b .
| Compounds | clog P | Aqueous solubility |
| 4 | 2.99 | 0.079 ± 0.002 |
| 5 | 0.58 | 0.392 ± 0.024 |
| 6b | 2.65 | 0.121 ± 0.016 |
| 6c | 3.70 | 0.442 ± 0.028 |
| 6f | 6.88 | 0.097 ± 0.006 |
| 7a | 2.83 | 0.178 ± 0.027 |
| 7c | 2.97 | 0.185 ± 0.013 |
| 7f | 3.54 | 0.613 ± 0.034 |
| Berberine | — | 0.085 ± 0.005 |
aclog P values were calculated by ChemDraw Ultra 14.0.
bIn mg mL–1, determined in pH 7.4 phosphate buffer, at 30 °C.
Hydrophobic/lipophilic properties play an important role in predicting the pharmacokinetic properties of a drug and its interaction with macromolecular targets. The calculated lipid/water partition coefficients (clog P) of some target compounds and berberine are shown in Table 2. The synthesized compounds 6b–c, 7a, 7c and 7f with moderate clog P values exhibited better antimicrobial activities, indicating that the compounds with suitable lipophilicity were favorable to permeate through biological membranes and to be delivered to the binding sites.
3.3. Bactericidal kinetic assay
The ability of an antibacterial agent to rapidly eradicate MRSA would decrease when this bacterium shows resistance towards that agent.18 To examine the antibacterial potency of the promising compound, a time–kill assay was performed by checking the viability of exponentially growing MRSA against the highly active compound 7a. As shown in Fig. 2, compound 7a showed more than 3 log (CFU mL–1) reduction in the number of viable bacteria within an hour at a concentration of 4 × MIC. Fig. 2 indicates the rapid killing effect of compound 7a against MRSA.
Fig. 2. Time–kill kinetics of compound 7a (4 × MIC) against MRSA.

3.4. Resistance study
The emergence of bacterial resistance towards most of the clinical drugs currently is a serious problem, especially MRSA strain towards norfloxacin.19 Hence, it would be important to evaluate the propensity of THPB triazole 7a to induce bacterial resistance. This work investigated the ability of the susceptible pathogen MRSA to develop resistance against compound 7a, and norfloxacin was used as a positive control. The standard strain of MRSA was exposed to increasing concentrations of compound 7a from MIC for the sustained passages, and the new MIC values were determined against each passage of MRSA. Fig. 3 exhibits no obvious change in the MIC for compound 7a even after 14 passages, whereas norfloxacin gave a significant increase in the MIC after 5 passages against MRSA. The obtained results suggested that MRSA was more difficult to develop resistance against compound 7a than the clinical drug norfloxacin.
Fig. 3. Evaluation of resistance development against compound 7a of the bacterial strain MRSA.

3.5. Molecular modeling
To rationalize the observed antibacterial activity and investigate the anti-MRSA action mechanism of THPB triazole 7a, a molecular docking study was performed between compound 7a and MRSA DNA (PDB ID: 2XCS). As shown in Fig. 4, the OCH2O group of the THPB 7a was adjacent to the ARG-1122 and MET-1121 residues, forming three hydrogen bonds with a distance of 2.1, 2.4 and 2.9 Å, respectively. Besides, the ARG-1122 and GLY-1072 residues could also form hydrogen bonds with the methoxy group of the THPB fragment and an N atom of the triazole ring. The hydrogen bonds might be favorable to stabilize the 7a–DNA complex, which might be responsible for the good inhibitory efficacy of compound 7a against MRSA.
Fig. 4. Molecular modeling of compound 7a and MRSA DNA.

3.6. Interactions with calf thymus DNA
DNA is the main cellular target for studies with small molecules of biological importance, and as a therapeutic target it has been extensively employed in rational design and construction of new and effective drugs.20 Calf thymus DNA is commonly selected as a DNA model due to its medical importance, low-cost and readily available properties. To explore the possible mechanism of the antimicrobial action, the interaction of the highly active compound 7a with DNA at the molecular level was investigated by UV-vis spectroscopy.
3.6.1. Absorption spectra of DNA in the presence of compound 7a
In absorption spectroscopy, hyperchromism and hypochromism are perceived as important spectral features to distinguish the change of the DNA double-helix structure. As reported in the literature, hypochromism indicates a close proximity of the aromatic chromophore to the DNA bases due to the interaction between the electronic states of the intercalating chromophore and that of the DNA base.21
With a fixed concentration of DNA, UV-vis absorption spectra were recorded with sequentially increasing amounts of compound 7a. As shown in Fig. 5, the UV-vis spectra gave a proportional increase at 260 nm for the maximum absorption peak of DNA and a slight red shift with the increasing concentration of compound 7a. Meanwhile, the absorption value of simply the sum of free DNA and free compound 7a was a little higher than the measured value of the 7a–DNA complex (inset of Fig. 5). This suggested a hypochromic effect between DNA and compound 7a. Furthermore, the intercalation of the chromophore fragment of compound 7a into the DNA helix and the strong overlap of π–π* states of the large π-conjugated system with the electronic states of DNA bases were in accordance with the observed spectral changes.
Fig. 5. UV absorption spectra of DNA with different concentrations of compound 7a (pH = 7.4, T = 303 K). Inset: comparison of absorption at 260 nm between the 7a–DNA complex and the sum values of free DNA and free compound 7a. c(DNA) = 5.20 × 10–5 mol L–1, and c(compound 7a) = 0–2.0 × 10–5 mol L–1 for curves a–i, respectively, at 0.25 × 10–5 mol L–1 increments.

On the basis of variations in the absorption spectra of DNA upon binding to compound 7a, the binding constant K can be calculated by the following equation:
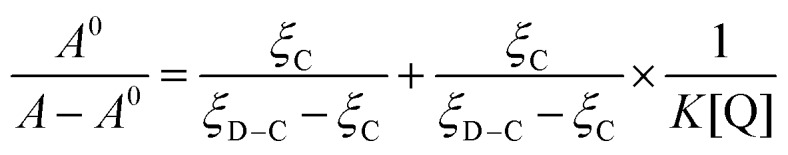 |
1 |
A 0 and A represent the absorbance of DNA in the absence and presence of compound 7a at 260 nm, ξC and ξD–C are the absorption coefficients of compound 7a and the compound 7a–DNA complex, respectively. The plot of A0/(A – A0) versus 1/[compound 7a] is constructed by using the absorption titration data and linear fitting (ESI,‡ Fig. S1), yielding the binding constant, K = 2.27 × 104 L mol–1, (R = 0.999, SD = 0.110). The experimental results revealed that THPB triazole 7a possessed a good DNA-binding ability with calf thymus DNA, which was also consistent with the above mentioned hypochromic effect.
3.6.2. Absorption spectra of the NR interaction with DNA
To further understand the interaction between compound 7a and DNA, the absorption spectra of the competitive interaction of compound 7a were also recorded. Neutral red (NR) is a planar phenazine dye, which possesses lower toxicity, higher stability and convenient application in comparison with other common probes. It has been sufficiently demonstrated that the binding of NR with DNA is an intercalation binding.22 Therefore, NR was employed as a spectral probe to investigate the binding mode of compound 7a with DNA in the present work. The absorption spectra of the NR dye upon the addition of DNA are shown in Fig. S2 (ESI‡). The absorption peak of NR at around 460 nm gradually decreased with the increasing concentration of DNA, and a new band at around 530 nm was generated, which could be ascribed to the formation of the new DNA–NR complex. The isosbestic point at 500 nm also provided evidence for the DNA–NR complex formation.
3.6.3. Absorption spectra of the competitive interaction of compound 7a and NR with DNA
Fig. 6 displays the absorption spectra of the competitive binding between compound 7a and NR with DNA. As shown, an apparent intensity increase was observed around 460 nm with the increasing concentration of compound 7a. In comparison with the absorption around 460 nm of free NR in the presence of the increasing concentrations of DNA (ESI‡ Fig. S2), the absorbance at the same wavelength exhibited the reverse process (inset of Fig. 6). The results suggested that compound 7a could intercalate into the double helix of DNA by substituting with NR in the DNA–NR complex.
Fig. 6. UV absorption spectra of the competitive reaction between compound 7a and NR with DNA. c(DNA) = 4.20 × 10–5 mol L–1, c(NR) = 2 × 10–5 mol L–1, and c(compound 7a) = 0–2.0 × 10–5 mol L–1 for curves a–i, respectively, at 0.25 × 10–5 mol L–1 increments. (Inset) Absorption spectra with the increasing concentration of 7a in the wavelength range of 400–550 nm of the competitive reaction between compound 7a and NR with DNA.

3.7. Cleavage ability towards pUC19 DNA
DNA cleavage is controlled by relaxation of the supercoiled form of DNA (form I). Plasmid pUC19 DNA, a widely used DNA cleavage substrate, is a circular double stranded DNA (form I). The cleavage agent can convert form I to the nicked form (form II) or to the linear form of DNA (form III). In gel electrophoresis, the migration rate of the three DNA forms is usually in the order form I > form III > form II. In order to assess the DNA cleavage activities, the cleavage of plasmid pUC19 DNA assay was conducted via agarose gel electrophoresis. The degradation of plasmid pUC19 DNA from form I to form II and form III in Fig. S3 (ESI‡) suggested that the compound 7a–Zn2+ complex might cleave DNA effectively at a low concentration of about 1.25 × 10–5 mol L–1, and it must be pointed out that in Fig. S3 (ESI‡), berberine, compound 7a and Zn2+ ion could not cleave DNA individually. It could be deduced that compound 7a was able to form a complex with the Zn2+ ion, which might further destroy DNA directly and thus exert its antibacterial activity.23
3.8. Interactions of compound 7a with HSA
HSA is an important extracellular protein with extensive distribution in the circulatory system, transports various exogenous and endogenous molecules, and thus significantly affects absorption, distribution, and metabolism of numerous therapeutic drugs.24 Therefore, the molecular characterization of drug–HSA interactions is not only beneficial to understand pharmacokinetic properties, but also instructive to design new drug molecules.
3.8.1. UV-vis absorption spectral study
The UV-vis absorption spectroscopic method is a convenient technique applicable to measure the structural change of a protein and to identify complex formation. The UV-vis absorption measurement to study the interaction of compound 7a with HSA was carried out and the results are shown in Fig. 7. The absorption peak at 278 nm was attributed to the aromatic rings of tryptophan (Trp-214), tyrosine (Tyr-411) and phenylalanine (Phe) residues in HSA. With the addition of compound 7a, the peak intensity increased, indicating that compound 7a could interact with HSA. However, the maximum absorption wavelength remained unchanged, suggesting the interactions of compound 7a and HSA were noncovalent interactions. These occurred via π–π stacking between the aromatic rings of compound 7a and the Trp, Tyr and Phe residues, which possess conjugated π-electrons and are located in the binding cavity of HSA.25
Fig. 7. Effect of compound 7a on HSA UV-vis absorption, c(HSA) = 1.0 × 10–5 mol L–1; c(compound 7a)/(10–5 mol L–1): 0, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75 (T = 298 K, pH = 7.4). The inset corresponds to the absorbance at 278 nm with different concentrations of compound 7a.

3.8.2. Fluorescence quenching mechanism
Fluorescence quenching is considered as an effective approach to investigate the transportation ability of HSA to small molecules. The fluorescence intensity of Trp-214 may change when HSA interacts with other small molecules, which could be reflected in the fluorescence spectra of HSA in the UV region. With a fixed amount of HSA, the fluorescence changes of HSA (T = 298 K, λex = 295 nm) in the presence of different amounts of compound 7a were determined. The blue line in Fig. 8 is the only emission spectrum of the active molecule 7a, which implied that its fluorescence intensity was very weak and could be negligible in comparison with the fluorescence of HSA at the excitation wavelength. The maximum emission peak of HSA at 352 nm in the fluorescence spectra exhibited a progressive decrease in the fluorescence intensity. However, the maximum emission wavelength of HSA remained unchanged, which revealed that Trp-214 did not undergo any change in polarity, and hence compound 7a was likely to interact with HSA via the hydrophobic region located in HSA.
Fig. 8. Emission spectra of HSA with various concentrations of compound 7a. c(HSA) = 1.0 × 10–5 mol L–1; c(compound 7a)/(10–5 mol L–1), a–h: from 0.0 to 1.17 at increments of 0.167; blue and red lines are the emission spectrum of compound 7a only and HSA only, respectively; T = 298 K, λex = 295 nm.

The fluorescence quenching data can be elucidated by the well-known Stern–Volmer equation:
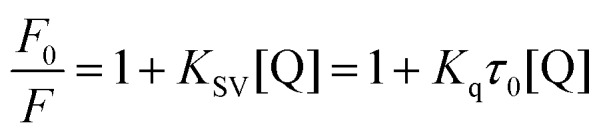 |
2 |
where F0 and F are the fluorescence intensities in the absence and presence of compound 7a, respectively. KSV (L mol–1) is the Stern–Volmer quenching constant, [Q] is the concentration of compound 7a, Kq is the bimolecular quenching rate constant (L mol–1 s–1). τ0 is the fluorescence lifetime of the fluorophore in the absence of the quencher, assumed to be 6.4 × 10–9 s for HSA. Hence, the Stern–Volmer plots of HSA in the presence of compound 7a at different concentrations and temperatures could be calculated and are shown in Fig. S4 (ESI‡).
The fluorescence quenching mechanisms are usually classified as either dynamic quenching or static quenching, which could be distinguished by their different dependence on temperature and viscosity. For dynamic quenching, higher temperatures result in faster diffusion and larger amounts of collisional quenching. As a result, the quenching constants are expected to increase with a gradually increasing temperature in dynamic quenching, but the reverse effect would be observed for static quenching. The calculated values of KSV and Kq for the interaction of compound 7a with HSA at different temperatures are listed in Table S3 (ESI‡). The KSV values are inversely correlated with the temperature, which indicates that the probable fluorescence quenching of HSA is initiated by the formation of the 7a–HSA complex rather than by dynamic collisions. Furthermore, the obtained larger Kq values (2.55 × 1013, 2.05 × 1013 and 1.70 × 1013 L mol–1 s–1 at 288 K, 298 K, 310 K, respectively) far exceeded the diffusion controlled rate constants of various quenchers with a biopolymer (2.0 × 1010 L mol–1 s–1), which suggested that the quenching was not initiated by a dynamic diffusion process but occurred in the static formation of the 7a–HSA complex.26
The difference in absorption spectroscopy was used to obtain spectra to reconfirm that the probable fluorescence quenching mechanism of HSA by compound 7a was mainly initiated by ground-state complex formation. The UV-vis absorption spectrum of HSA (ESI‡ Fig. S5, curve C) and the difference spectrum between the HSA–compound 7a 1 : 1 complex and compound 7a (ESI‡ Fig. S5, curve D) could not be superposed. This result reconfirmed that the probable fluorescence quenching mechanism of HSA by compound 7a might be a static quenching process.27
3.8.3. Binding constant and sites
For a static quenching procedure, quenching data were analyzed according to the modified Stern–Volmer equation:
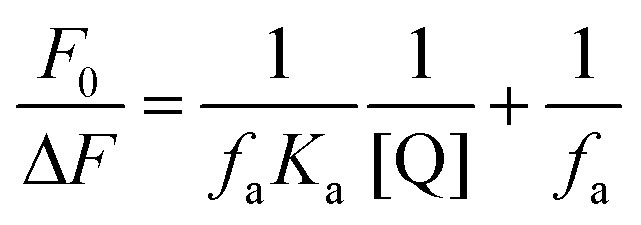 |
3 |
where ΔF denotes the difference in fluorescence in the absence and presence of compound 7a at concentration [Q], fa is the fraction of accessible fluorescence, and Ka is the effective quenching constant for the accessible fluorophores, which are analogous to the associative binding constants for quencher–acceptor systems. The dependence of F0/ΔF on the reciprocal value of the quencher concentration [Q]–1 is linear with the slope equal to the value of (faKa)–1. The value fa–1 is fixed on the ordinate. The constant Ka is a quotient of the ordinate fa–1 and the slope (faKa)–1. The modified Stern–Volmer plots are shown in Fig. S6 (ESI‡), and the calculated results are listed in Table S4 (ESI‡).
The Scatchard equation could be used to estimate the equilibrium binding constant (Kb) and the number of binding sites (n); it is described as:
| log[(F0/F) – 1] = log Kb + n log[Q] | 4 |
Fig. 9 displays the plots of log(F0 – F)/F versus log[Q] for the interaction between HSA and compound 7a at various temperatures. Kb and n obtained from the Scatchard plots are listed in Table S4 (ESI‡). The decreasing trend of Ka and Kb with the increase in temperature was in accordance with the temperature dependence of the KSV values. The value of the binding site approximates to 1, indicating that only one high affinity binding site was present in the interaction of compound 7a with HSA. The results also showed that the binding constants were moderate and the effects of temperatures were not obvious, thus compound 7a might be stored and carried by this protein.
Fig. 9. Scatchard plots of the 7a–HSA system at different temperatures.

3.8.4. Binding mode and thermodynamic parameters
Generally, the major forces involved in small molecule and biomolecule interactions include hydrogen bonds, electrostatic interactions, van der Waals forces, and hydrophobic interactions.28 Thermodynamic parameters like the enthalpy change (ΔH) and entropy change (ΔS) of the binding reaction are the main evidence for confirming the interactions between small molecules and a protein. If ΔH does not vary significantly in the temperature range studied, both its value and ΔS can be estimated from the van't Hoff equation as follows:
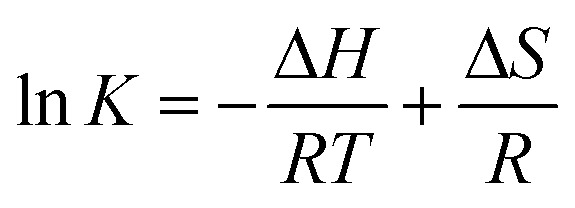 |
5 |
where K is analogous to the associative binding constants at the corresponding temperature and R is the gas constant. To elucidate the binding model between compound 7a and HSA, the thermodynamic parameters were calculated from the van't Hoff plots. The enthalpy change (ΔH) was estimated from the slope of the van't Hoff relationship (ESI‡ Fig. S7). The free energy change (ΔG) was then calculated from the following equation:
| ΔG = ΔH – TΔS | 6 |
Table S5 (ESI‡) summarizes the values of ΔH, ΔG and ΔS. The negative values of the free energy ΔG of the interactions between compound 7a and HSA indicated that the binding process was spontaneous. The negative values of ΔH suggested that the binding was predominantly enthalpy driven and involved an exothermic reaction. The positive ΔS value is frequently taken as a typical evidence for hydrophobic interactions, which is consistent with the above discussion. The negative ΔH value (–9.498 kJ mol–1) observed cannot be mainly attributed to electrostatic interactions since the ΔH values of electrostatic interactions are very small, almost zero. Therefore, ΔH < 0 and ΔS > 0 obtained in this case indicated that both hydrophobic interactions and hydrogen bonds played a major role in the binding of compound 7a to HSA, and electrostatic interactions might also be involved in the binding process.
4. Conclusion
In conclusion, a series of novel Schiff-base bridged THPB triazoles as antimicrobial agents have been successfully developed for the first time by an easy, convenient and economical procedure, and their structures were characterized by HRMS, NMR and IR spectroscopy. The in vitro biological evaluation revealed that some of the prepared compounds exerted good to better antibacterial and antifungal activities in comparison with the reference drugs. Noticeably, THPB triazole 7a could effectively inhibit the growth of B. yeast, M. luteus and MRSA with MIC values of 0.5, 1 and 2 μg mL–1, respectively. Further research suggested that compound 7a could rapidly kill MRSA and induce bacterial resistance more slowly than norfloxacin. Molecular docking indicated that target compound 7a could bind with MRSA DNA through hydrogen bonds. The SAR study revealed that the combination of the triazole fragment and THPB was beneficial to exert biological activity, and the substituents on the benzyl moiety as well as the length of the alkyl chain at the 9-position of THPB influenced the antibacterial potency. The binding investigation of compound 7a with HSA revealed that this molecule could be effectively transported by HSA. The preliminary exploration for the antimicrobial mechanism disclosed that compound 7a could not only form a stable 7a–DNA complex with calf thymus DNA by an intercalating mode, but also directly cleave pUC19 DNA by formation of a complex with a Zn2+ ion, thus exerting antibacterial activity. Therefore, this work revealed that compound 7a might be a potentially dual DNA-targeting antibacterial molecule with better water solubility than berberine, which should be a promising start as a novel antibacterial agent to overcome drug resistance.
Supplementary Material
Acknowledgments
This work was partially supported by the National Natural Science Foundation of China (No. 21672173, 21372186), the Research Fellowship for International Young Scientists from the International (Regional) Cooperation and Exchange Program of the Chinese National Natural Science Foundation (81650110529), the Chongqing Research Program of Basic Research and Frontier Technology (No. cstc2013jcyjA50012, cstc2016jcyjA0508), and a China Postdoctoral Science Foundation funded project (2014M562326, 2016T90851).
Footnotes
†The authors declare no competing interests.
‡Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00688d
References
- (a) Brown E. D., Wright G. D. Nature. 2016;529:336–343. doi: 10.1038/nature17042. [DOI] [PubMed] [Google Scholar]; (b) Baym M., Stone L. K., Kishony R. Science. 2016;351:aad3292. doi: 10.1126/science.aad3292. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang L., Peng X. M., Damu G. L. V., Geng R. X., Zhou C. H. Med. Res. Rev. 2014;34:340–437. doi: 10.1002/med.21290. [DOI] [PubMed] [Google Scholar]; (d) Peng X. M., Damu G. L. V., Zhou C. H. Curr. Pharm. Des. 2013;19:3884–3930. doi: 10.2174/1381612811319210013. [DOI] [PubMed] [Google Scholar]
- (a) Moloney M. G. Trends Pharmacol. Sci. 2016;37:689–701. doi: 10.1016/j.tips.2016.05.001. [DOI] [PubMed] [Google Scholar]; (b) Gong H. H., Addla D., Lv J. S., Zhou C. H. Curr. Top. Med. Chem. 2016;16:3303–3364. doi: 10.2174/1568026616666160506145943. [DOI] [PubMed] [Google Scholar]; (c) Cheng Y., Wang H., Addla D., Zhou C. H., Chin. J. Org. Chem., 2015, 36 , 1 –42 , (in Chinese) . [Google Scholar]; (d) He S. C., Jeyakkumar P., Avula S. R., Wang X. L., Zhang H. Z., Zhou C. H., Zhongguo Kexue: Huaxue, 2016, 46 , 823 –847 , (in Chinese) . [Google Scholar]
- Silva L. N., Zimmer K. R., Macedo A. J., Trentin D. S. Chem. Rev. 2016;116:9162–9236. doi: 10.1021/acs.chemrev.6b00184. [DOI] [PubMed] [Google Scholar]
- (a) Ball A. R., Casadei G., Samosorn S., Bremner J. B., Ausubel F. M., Moy T. I., Lewis K. ACS Chem. Biol. 2006;1:594–600. doi: 10.1021/cb600238x. [DOI] [PubMed] [Google Scholar]; (b) Tillhon M., Ortiz L. M. G., Lombardi P., Scovassi A. I. Biochem. Pharmacol. 2012;84:1260–1267. doi: 10.1016/j.bcp.2012.07.018. [DOI] [PubMed] [Google Scholar]
- (a) Kumar A., Ekavali, Chopra K., Mukherjee M., Pottabathini R., Dhull D. K. Eur. J. Pharmacol. 2015;761:288–297. doi: 10.1016/j.ejphar.2015.05.068. [DOI] [PubMed] [Google Scholar]; (b) Preeti S., Prabhat U., Shardendu M., Ananya S., Suresh P. World J. Pharm. Pharm. Sci. 2015;4:547–573. [Google Scholar]
- (a) Ishikawa M., Hashimoto Y. J. Med. Chem. 2011;54:1539–1554. doi: 10.1021/jm101356p. [DOI] [PubMed] [Google Scholar]; (b) Takeuchi T., Oishi S., Kaneda M., Ohno H., Nakamura S., Nakanishi I., Yamane M., Sawada J., Asai A., Fujii N. ACS Med. Chem. Lett. 2014;5:566–571. doi: 10.1021/ml500016j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Mo J., Guo Y., Yang Y. S., Shen J. S., Jin G. Z., Zhen X. Curr. Med. Chem. 2007;14:2996–3002. doi: 10.2174/092986707782794050. [DOI] [PubMed] [Google Scholar]; (b) Chu H. Y., Jin G. Z., Friedman E., Zhen X. C. Cell. Mol. Neurobiol. 2008;28:491–499. doi: 10.1007/s10571-007-9179-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ge H. X., Zhang J., Chen L., Kou J. P., Yu B. Y. Bioorg. Med. Chem. 2013;21:62–69. doi: 10.1016/j.bmc.2012.11.002. [DOI] [PubMed] [Google Scholar]; (d) Guo D. L., Li J., Lin H., Zhou Y., Chen Y., Zhao F., Sun H. F., Zhang D., Li H. L., Shoichet B. K., Shan L., Zhang W. D., Xie X., Jiang H. L., Liu H. J. Med. Chem. 2016;59:9489–9502. doi: 10.1021/acs.jmedchem.6b01217. [DOI] [PubMed] [Google Scholar]
- Cheng P., Wang B., Liu X. B., Liu W., Kang W. S., Zhou J., Zeng J. G. Nat. Prod. Res. 2014;28:413–419. doi: 10.1080/14786419.2013.867344. [DOI] [PubMed] [Google Scholar]
- (a) Zhang H. Z., Gan L. L., Wang H., Zhou C. H. Mini-Rev. Med. Chem. 2017;17:122–166. doi: 10.2174/1389557516666160630120725. [DOI] [PubMed] [Google Scholar]; (b) Peng X. M., Cai G. X., Zhou C. H. Curr. Top. Med. Chem. 2013;13:1963–2010. doi: 10.2174/15680266113139990125. [DOI] [PubMed] [Google Scholar]; (c) Zhang H. Z., Damu G. L. V., Cai G. X., Zhou C. H. Curr. Org. Chem. 2014;18:359–406. [Google Scholar]; (d) Fang X. J., Jeyakkumar P., Avula S. R., Zhou Q., Zhou C. H. Bioorg. Med. Chem. Lett. 2016;26:2584–2588. doi: 10.1016/j.bmcl.2016.04.036. [DOI] [PubMed] [Google Scholar]
- (a) Kaur R., Dwivedi A. R., Kumar B., Kumar V. Anti-Cancer Agents Med. Chem. 2016;16:465–489. doi: 10.2174/1871520615666150819121106. [DOI] [PubMed] [Google Scholar]; (b) Zhou C. H., Wang Y. Curr. Med. Chem. 2012;19:239–280. doi: 10.2174/092986712803414213. [DOI] [PubMed] [Google Scholar]; (c) Wang Y., Zhou C. H., Zhongguo Kexue: Huaxue, 2011, 41 , 1429 –1456 , (in Chinese) . [Google Scholar]
- (a) Allen D., Wilson D., Drew R., Perfect J. Expert Rev. Anti-Infect. Ther. 2015;13:787–798. doi: 10.1586/14787210.2015.1032939. [DOI] [PubMed] [Google Scholar]; (b) Cao X. F., Sun Z. S., Cao Y. B., Wang R. L., Cai T. K., Chu W. J., Hu W. H., Yang Y. S. J. Med. Chem. 2014;57:3687–3706. doi: 10.1021/jm4016284. [DOI] [PubMed] [Google Scholar]
- (a) Zhang L., Chang J. J., Zhang S. L., Damu G. L. V., Geng R. X., Zhou C. H. Bioorg. Med. Chem. 2013;21:4158–4169. doi: 10.1016/j.bmc.2013.05.007. [DOI] [PubMed] [Google Scholar]; (b) Zhang S. L., Chang J. J., Damu G. L. V., Fang B., Zhou X. D., Geng R. X., Zhou C. H. Bioorg. Med. Chem. Lett. 2013;23:1008–1012. doi: 10.1016/j.bmcl.2012.12.036. [DOI] [PubMed] [Google Scholar]; (c) Wen S. Q., Jeyakkumar P., Avula S. R., Zhang L., Zhou C. H. Bioorg. Med. Chem. Lett. 2016;26:2768–2773. doi: 10.1016/j.bmcl.2016.04.070. [DOI] [PubMed] [Google Scholar]
- (a) Liao Z. Q., Dong C., Carlson K. E., Srinivasan S., Nwachukwu J. C., Chesnut R. W., Sharma A., Nettles K. W., Katzenellenbogen J. A., Zhou H. B. J. Med. Chem. 2014;57:3532–3545. doi: 10.1021/jm500268j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gong H. H., Baathulaa K., Lv J. S., Cai G. X., Zhou C. H. Med. Chem. Commun. 2016;7:924–931. [Google Scholar]
- (a) Lo C. Y., Hsu L. C., Chen M. S., Lin Y. J., Chen L. G., Kuo C. D., Wu J. Y. Bioorg. Med. Chem. Lett. 2013;23:305–309. doi: 10.1016/j.bmcl.2012.10.098. [DOI] [PubMed] [Google Scholar]; (b) Huang L., Luo Z. H., He F., Shi A. D., Qin F. F., Li X. S. Bioorg. Med. Chem. Lett. 2010;20:6649–6652. doi: 10.1016/j.bmcl.2010.09.013. [DOI] [PubMed] [Google Scholar]
- Jeyakkumar P., Zhang L., Avula S. R., Zhou C. H. Eur. J. Med. Chem. 2016;122:205–215. doi: 10.1016/j.ejmech.2016.06.031. [DOI] [PubMed] [Google Scholar]
- Zhou C. H., Jayakumar P. and Peng X. M., CN Pat., 2016/105218537, 2016.
- Kametani T., Fukumoto K., Terui T., Yamaki K., Taguchi E. J. Chem. Soc. C. 1971:2709–2711. [Google Scholar]
- (a) Hoque J., Konai M. M., Gonuguntla S., Manjunath G. B., Samaddar S., Yarlagadda V., Haldar J. J. Med. Chem. 2015;58:5486–5500. doi: 10.1021/acs.jmedchem.5b00443. [DOI] [PubMed] [Google Scholar]; (b) Cheng Y., Avula S. R., Gao W. W., Addla D., Tangadanchu V. K. R., Zhang L., Lin J. M., Zhou C. H. Eur. J. Med. Chem. 2016;124:935–945. doi: 10.1016/j.ejmech.2016.10.011. [DOI] [PubMed] [Google Scholar]
- Zhang L., Kumar K. V., Rasheed S., Geng R. X., Zhou C. H. Chem. Biol. Drug Des. 2015;86:648–655. doi: 10.1111/cbdd.12532. [DOI] [PubMed] [Google Scholar]
- (a) Denning E. J., MacKerellJr A. D. J. Am. Chem. Soc. 2011;133:5770–5772. doi: 10.1021/ja201213b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Addla D., Wen S. Q., Maddili S. K., Zhang L., Zhou C. H. Med. Chem. Commun. 2016;7:1988–1994. [Google Scholar]; (c) Zhang L., Kumar K. V., Rasheed S., Zhang S. L., Geng R. X., Zhou C. H. Med. Chem. Commun. 2015;6:1303–1310. [Google Scholar]
- (a) Zhang G., Fu P., Wang L., Hu M., Agric J. Food Chem. 2011;59:8944–8952. doi: 10.1021/jf2019006. [DOI] [PubMed] [Google Scholar]; (b) Peng X. M., Kumar K. V., Damu G. L., Zhou C. H. Sci. China: Chem. 2016;59:878–894. [Google Scholar]
- (a) Liu F., Wang X. L., Han X., Tan X. X., Kang W. J. Int. J. Biol. Macromol. 2015;77:92–98. doi: 10.1016/j.ijbiomac.2015.03.017. [DOI] [PubMed] [Google Scholar]; (b) Dai L. L., Zhang H. Z., Nagarajan S., Rasheed S., Zhou C. H. Med. Chem. Commun. 2015;6:147–154. [Google Scholar]
- Peng X. M., Peng L. P., Li S., Rao A. S., Kumar K. V., Zhang S. L., Tam K. Y., Zhou C. H. Future Med. Chem. 2016;8:1927–1940. doi: 10.4155/fmc-2016-0002. [DOI] [PubMed] [Google Scholar]
- (a) Guo Y., Yang F., Liang H. Curr. Top. Med. Chem. 2016;16:996–1008. doi: 10.2174/1568026615666150825142908. [DOI] [PubMed] [Google Scholar]; (b) Peng L. P., Nagarajan S., Rasheed S., Zhou C. H. Med. Chem. Commun. 2015;6:222–229. [Google Scholar]
- Suryawanshi V. D., Anbhule P. V., Gore A. H., Patil S. R., Kolekar G. B. Ind. Eng. Chem. Res. 2012;51:95–102. [Google Scholar]
- (a) Yeggoni D. P., Rachamallub A., Subramanyam R. RSC Adv. 2016;6:40225–40237. [Google Scholar]; (b) Zhang S. L., Chang J. J., Damu G. L. V., Geng R. X., Zhou C. H. Med. Chem. Commun. 2013;4:839–846. [Google Scholar]
- (a) Cui S. F., Addla D., Zhou C. H. J. Med. Chem. 2016;59:4488–4510. doi: 10.1021/acs.jmedchem.5b01678. [DOI] [PubMed] [Google Scholar]; (b) Hu Y. J., Liu Y., Xiao X. H. Biomacromolecules. 2009;10:517–521. doi: 10.1021/bm801120k. [DOI] [PubMed] [Google Scholar]
- (a) Rehman M. T., Shamsi H., Khan A. U. Mol. Pharmaceutics. 2014;11:1785–1797. doi: 10.1021/mp500116c. [DOI] [PubMed] [Google Scholar]; (b) Yin B. T., Yan C. Y., Peng X. M., Zhang S. L., Rasheed S., Geng R. X., Zhou C. H. Eur. J. Med. Chem. 2014;71:148–159. doi: 10.1016/j.ejmech.2013.11.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.