

 Synthesis and SAR of selective ligands of the pseudokinase domain of the Janus kinase (JAK) TYK2 are first reported herein.
Synthesis and SAR of selective ligands of the pseudokinase domain of the Janus kinase (JAK) TYK2 are first reported herein.
Abstract
As a member of the Janus (JAK) family of non-receptor tyrosine kinases, TYK2 mediates the signaling of pro-inflammatory cytokines including IL-12, IL-23 and type 1 interferon (IFN), and therefore represents an attractive potential target for treating the various immuno-inflammatory diseases in which these cytokines have been shown to play a role. Following up on our previous report that ligands to the pseudokinase domain (JH2) of TYK2 suppress cytokine-mediated receptor activation of the catalytic (JH1) domain, the imidazo[1,2-b]pyridazine (IZP) 7 was identified as a promising hit compound. Through iterative modification of each of the substituents of the IZP scaffold, the cellular potency was improved while maintaining selectivity over the JH1 domain. These studies led to the discovery of the JH2-selective TYK2 inhibitor 29, which provided encouraging systemic exposures after oral dosing in mice. Phosphodiesterase 4 (PDE4) was identified as an off-target and potential liability of the IZP ligands, and selectivity for TYK2 JH2 over this enzyme was obtained by elaborating along selectivity vectors determined from analyses of X-ray co-crystal structures of representative ligands of the IZP class bound to both proteins.
Background and introduction
The Janus family of kinases (JAK1, JAK2, JAK3 and TYK2) are non-receptor tyrosine kinases that bind to the intracellular portion of cell surface cytokine receptors. Upon stimulation of these receptors, the Janus members recruit, via phosphorylation of the receptor, and subsequently phosphorylate signal transducer and activator of transcription (STAT) proteins, which then dimerize, translocate to the nucleus and activate gene transcription. The Janus family of kinases have generated significant recent interest as targets for immunological disorders due to the involvement of the JAK/STAT pathway in inflammation.1 A pan-JAK inhibitor (tofacitinib) was approved for the treatment of rheumatoid arthritis (RA) in 2012, while ruxolitinib, a JAK1/JAK2 selective inhibitor, was approved for myelofibrosis. Currently, the development of more selective inhibitors is being broadly pursued due to concerns of dose-limiting side effects such as anemia, which have been attributed to JAK2 inhibition.2,3 The signaling of different cytokines and their receptors rely on pairs of Janus kinase family members. TYK2 in particular partners with JAK2 to mediate signaling by IL-12 and IL-23 (p40 subunit containing cytokines) and with JAK1 for the IFNα/β pathway. The TYK2-dependant pathways have been validated in treating human disease with antibody therapeutics. The IL-12/IL-23 antibody ustekinumab (Stelara®) is currently marketed for the treatment of psoriasis, with clinical development underway for treatment of Crohn's Disease.4,5 The anti-type 1 interferon receptor antibody anifrolumab has been reported to provide benefit for the treatment of systemic lupus erythematosus (SLE).6
Genome wide association studies (GWAS) have identified TYK2 single nucleotide polymorphisms (SNPs) that are tied to autoimmune disease.7 Unlike JAK1 deficient mice,8 TYK2 deficient mice are viable, and the TYK2 deficiency has been shown to be protective in various models of experimental autoimmunity.9–11 Given this, there has been some effort to identify selective inhibitors of TYK2 (Fig. 1).12,13 However, owing to the high sequence homology within the JAK family kinase Homology 1 (JH1) domains, achieving selectivity for TYK2 over other JAK family members has proved challenging. This is evidenced by the nanomolar potencies of TYK2 inhibitors 1 and 2 against the other JAK family members.
Fig. 1. Known TYK2 JH1 ligands.

The hallmark structural feature of the JAK family, and reason for its namesake being the two-headed Roman god Janus, is the pseudokinase (JH2) domain immediately N-terminal to the catalytic domain (JH1). Although the JH2 domain shares the overall fold of a typical catalytic domain, a series of individual residue and conformational differences between the TYK2 JH1 and JH2 domains likely explains the lack of catalytic activity of the JH2 domain (Fig. 2).14
Fig. 2. Janus family kinase architecture and structure of TYK2 kinase and pseudokinase domains. (a) Schematic illustrating the entire structure of the Janus kinase family (JAKs). (b) Superposition of TYK2 JH2 domain structure (green) PDB code 4WOV with the TYK2 JH1 domain structure complexed with ADP (magenta ribbons and ADP carbons in cyan), PDB code ; 4GVJ. (c) TYK2 pseudokinase domain residues corresponding to those of protein kinases normally involved in catalytic machinery are shown in stick. Key residues of the ATP-pocket are differentiated from the JH2 to the JH1 domains, see ref. 14 for additional details.

The JH2 domains of the JAK family have been shown to regulate the function of the JH1 domains, though their precise regulatory roles and mechanisms may differ between the family members.15 The precise molecular mechanism of regulation of TYK2 kinase signaling in particular has not been fully elucidated, although the biological literature and recently obtained crystal structures suggest a possible interplay between ATP and the JH1 and JH2 domains, and between full length kinase and the intracellular portion of cytokine receptors.16–18 The overall body of evidence is consistent with the TYK2 pseudokinase domain being auto-inhibitory, stabilizing the inactivated state of the kinase domain, and that small molecule ligands can stabilize this auto-inhibitory conformation, thereby preventing protein function in an allosteric manner.14
An advantage of targeting the JH2 domain is an increased likelihood of identifying inhibitors that are highly selective relative to those targeting the JH1 domain. Pseudokinases represent an attractive class of relatively untapped targets. Since they possess many, but not all, of the structural features of kinases, screens against them are likely to yield selective hits from collections of traditional kinase inhibitors.19 Given the challenges associated with identifying a selective orthosteric TYK2 inhibitor, coupled with knowledge that the TYK2 JH2 domain represents a fairly unique structural feature possessing regulatory function, we decided to focus our efforts on an allosteric approach to the inhibition of TYK2 by pursuing ligands of the JH2 domain.
As reported previously, an allosteric inhibitor of TYK2 (3) with submicromolar potency for inhibiting IL-23/IFNα stimulated reporter assays was identified from a phenotypic screen of a diverse, annotated collection of kinase inhibitors using an IL-23 stimulated cellular assay (Fig. 3).14 The activity of 3 was determined to be a consequence of its affinity for the TYK2 JH2 domain, which results in inhibition of the activation of the TYK2 kinase. Other ligands (4–6) to the JH2 domain have been reported in the context of co-crystal structures;16,17 however, functional potency and selectivity have not been disclosed, with only an equilibrium binding constant for indole 6 having been reported as a measure of binding affinity (Fig. 4). Optimization of the scaffold of 3 to further improve cell potency proved difficult, as did obtaining selectivity over IKKβ, an off-target against which the series of ligands represented by 3 showed consistently high affinity. The IL-23 stimulated assay that allowed identification of 3 also provided the imidazo[1,2-b]pyridazine (IZP) 7 as a unique hit compound. While less potent than 3, 7 was highly selective (with only 0.5% of kinases in the 386 kinase panel being inhibited by greater than 67% at 1 μM compound concentration). Importantly, 7 lacked potency against IKKβ (IC50 > 2 μM). Thus, optimization of the IZP scaffold required the identification of SAR that would provide improved affinity for the TYK2 JH2 domain while maintaining kinase selectivity.
Fig. 3. High throughput screen hits for IL-23 inhibition.14.

Fig. 4. Previously reported TYK2 JH2 ligands.

Results and discussion
The syntheses of the various IZP TYK2 JH2 ligands were accomplished in a number of ways; however, the majority of the compounds presented herein followed one of the two synthetic paths described in Scheme 1. To facilitate examination of diversity at the C3 amide, 9, available from a nucleophilic displacement of the C8 halide group of 8 with a PMB-protected amine, was first coupled to the C6 aniline using palladium-catalyzed N-arylation conditions (Path A). The PMB protecting group was then removed under acidic conditions and the ester saponified to provide the penultimate acid, which could then be converted to the desired amide following standard amidation conditions. Alternatively, to examine the C6 aniline, the ester of 9 could first be converted to the corresponding amide and then coupled to the desired aniline (Path B).
Scheme 1.

An initial exploration of the C6 position provided compounds of increased binding affinity, as measured by scintillation proximity assay (SPA),14 compared to the screening hit 7 (Table 1). Without the trifluoromethyl and cyano groups at the meta positions (10), binding potency improved by an order of magnitude, with an equivalent improvement in cellular potency. Selectivity against the JAK catalytic domains was maintained. A number of similar anilines, including bis-meta substituted anilines 11 and 12 were equipotent to 10, while aliphatic amines had significantly lower affinities (13).
Table 1. Initial examination of C-6 amine SAR.
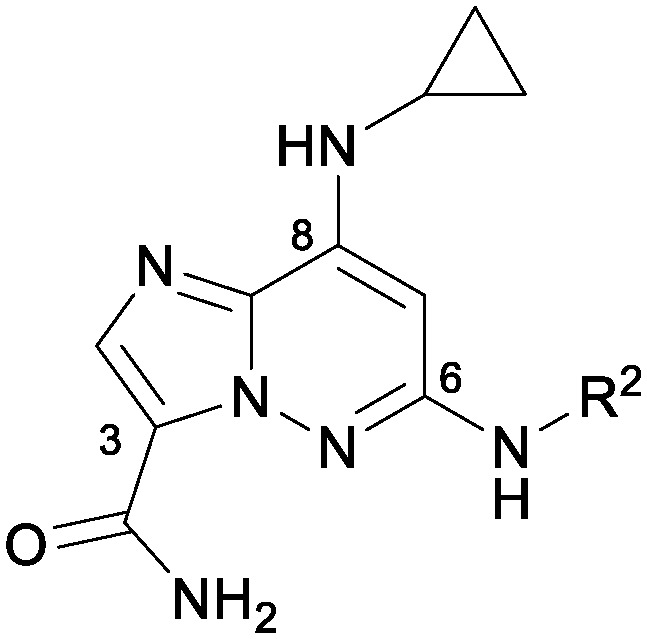
| ||||
| R2 | TYK2 JH2 (IC50, μM) | TYK2/JAK1/JAK2 JH1 (IC50, μM) | IL23/IFNα (IC50, μM) | |
| 7 |
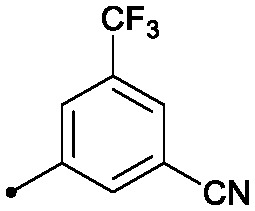
|
0.48 | >50 | 8.6/12.6 |
| 10 |
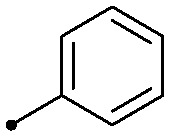
|
0.032 | >2 | 1.1/1.5 |
| 11 |
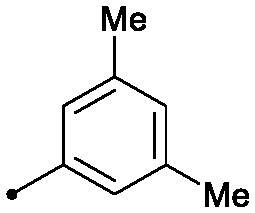
|
0.022 | >50/>2/>50 | 0.7/1.4 |
| 12 |
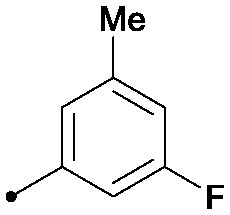
|
0.024 | >2 | 1.4/1.7 |
| 13 |
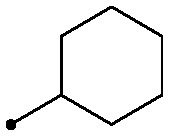
|
0.22 | >2 | 3.4/3.2 |
Next, a synthesis was designed to allow late stage examination of the C8 group (Scheme 2). Ethanethiol was used to mask the C8 position to enable selective coupling of 3,5-dimethylaniline with the C6 chloride. Subsequent oxidation of the thioether to the sulfoxide enabled nucleophilic displacement at the C8 position to introduce the desired amines. Hydrolysis of the cyano group then provided the primary amide.
Scheme 2.

As shown in Table 2, there was a clear relationship between the size of the R1 group and the binding affinity of the ligand. Replacing the cyclopropylamine of 7 with methylamine (18) provided a three-fold improvement in binding affinity and a 10–20 fold increase in cellular potency, with sub-100 nM potency achieved for the first time with a TYK2 JH2 ligand in the IFNα-stimulated reporter assay. Ethylamine (19) was only modestly more potent than the cyclopropylamine, and cyclobutyl and cyclopentyl groups led to further reductions in potency. Small aryl amines, such as 22, had modest affinity, but again were more than an order of magnitude less potent than 18.
Table 2. Optimization of C-8 amine.

| |||
| R1 | TYK2 JH2 (IC50, μM) | IL23/IFNα (IC50, μM) | |
| 11 |
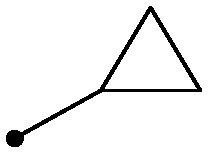
|
0.022 | 0.7/1.4 |
| 18 |
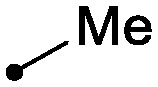
|
0.007 | 0.1/0.05 |
| 19 |
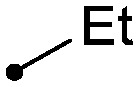
|
0.017 | 0.6/0.6 |
| 20 |
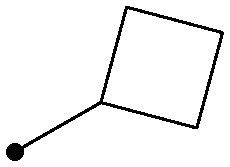
|
0.11 | 6.2/4.1 |
| 21 |
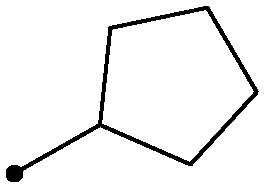
|
0.50 | 7.3/4.8 |
| 22 |
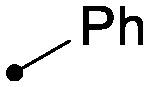
|
0.05 | 4.9/3.7 |
A co-crystal structure of 18 (PDBID 5TKD) bound to the TYK2 JH2 was solved and helps rationalize the SAR findings of Tables 1 and 2 (Fig. 5). First, limited room between C8 and the hinge is seen, consistent with the loss in affinity seen with groups larger than methylamino at this position. There were also hydrogen bonds revealed between the NH of the C8 methylamine and from N1 of the IZP core to the ‘hinge’ (Val690). Additional hydrogen bonds are observed from the oxygen of the C3 amide to Lys642 and to the hinge carbonyl of Glu688 through a bridging water molecule. The pocket proximal to the C3 amide of the TYK2 JH2 domain contains a combination of residues which are largely unique relative to the kinome14 such as a small residue (Ala671) under the “gatekeeper” (Thr687) and the replacement of the highly kinase-conserved DFG motif by DPG which alters the positioning of the conserved catalytic Lys642 and Asp759. The ability of the C3 amide to fit and bind to this pocket is believed to be a key source of kinome selectivity for compound 18. In contrast to the steric crowding proximal to C8, the crystal structure revealed a fair degree of space off of the C3 amide as well as the C6 aniline, the latter of which is solvent exposed on one half of its ring. Therefore, the C3 and C6 positions were examined with a variety of combinations of R2 and R3 groups accessible following the chemistry described in Scheme 1 (Table 3).
Fig. 5. X-ray crystal structure representation of 18 co-complexed with TYK2 JH2 (PDBID 5TKD). (a) Close-up of the binding site. Hydrogen bond interactions are shown with dotted lines with key residues and observed water labelled. TYK2 JH2 ribbon and carbons in green. Carbons of 18 in magenta. (b) Surface representation of the protein binding site highlighting space off of the C3 amide and part of the C6 aniline (yellow arrows). Hydrogens added for clarity.

Table 3. Combining R2 and R3 SAR.
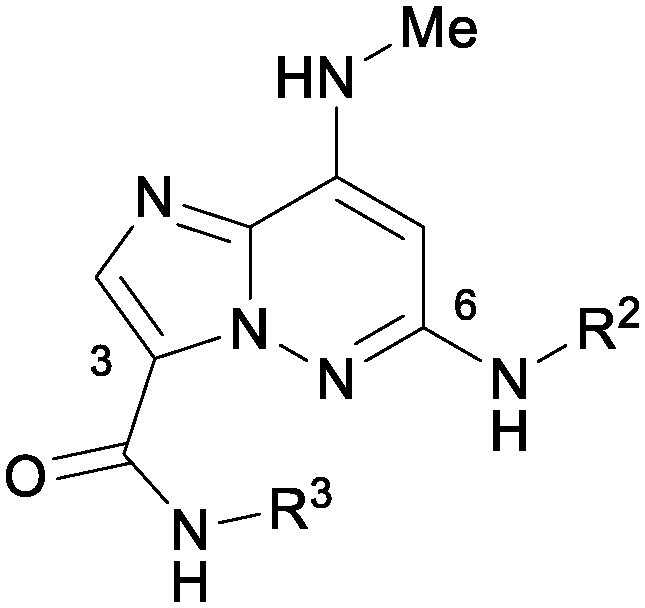
| ||||||
| R2 | R3 | TYK2 JH2 (IC50, μM) | IL23/IFNα (IC50, μM) | PDE4 (IC50, μM) | LM a % rem (h/r/m) | |
| 18 |
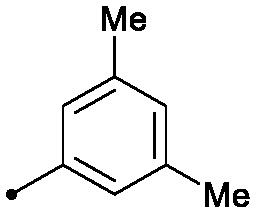
|
|
0.007 | 0.10/0.05 | 0.062 | 15/21/6 |
| 23 |
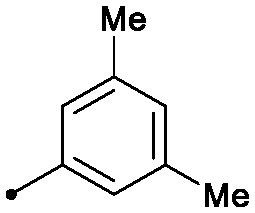
|

|
0.008 | 0.11/0.10 | 0.006 | 11/14/1 |
| 24 |
|
|
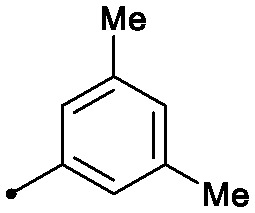
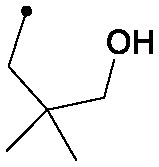
0.002 | 0.30/0.11 | 0.015 | 20/4/0 |
| 25 |
|
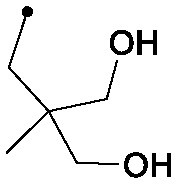
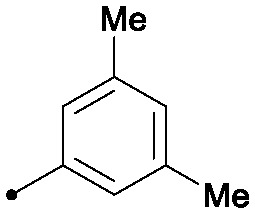
|
0.015 | 0.48/0.33 | 0.018 | 70/16/0 |
| 26 |
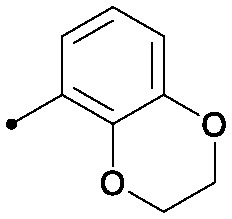
|

|
0.006 | 0.02/0.01 | 0.024 | 52/9/0 |
| 27 |
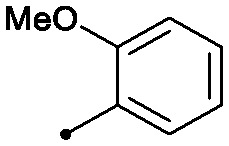
|
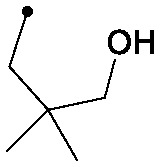
|
0.005 | 0.15/0.12 | 0.088 | 85/40/2 |
| 28 |
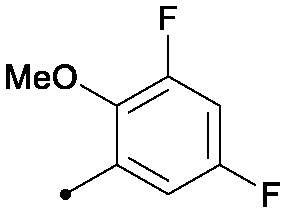
|
|
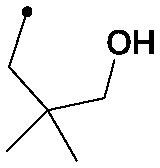
0.006 | 0.07/0.03 | 0.032 | 100/34/24 |
| 29 |
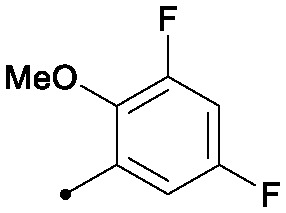
|
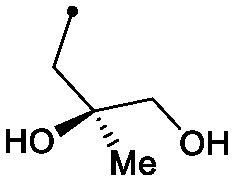
|
0.004 | 0.24/0.16 | 0.043 | 99/76/44 |
| 30 |
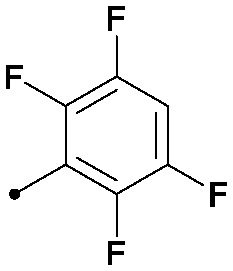
|
|
0.008 | 0.30/0.45 | 0.013 | 100/40/26 |
aLM % rem (h/r/m) refers to liver microsomal stability in human/rat/mouse as measured by % remaining.20
A variety of secondary C3 amides were nearly equipotent to the primary amide 18; thus, the R3 group was used as a handle to improve metabolic stability. The introduction of more polar substituents, especially those with multiple hydroxyl groups, provided better liver microsomal stability, as seen by comparing 23 to 24 and 25. A methoxy substituent at the ortho-position provided improved cellular potency (e.g., 27). Other ortho-alkoxy groups were also effective in this regard (26). Introduction of appropriately placed fluorines to the C6 aniline provided additional improvement to metabolic stability, as can be seen by comparing 28 to 27. Combining these observations resulted in the identification of compound 29, a potent TYK2 JH2 ligand with improved metabolic stability that possessed, despite its high polarity, reasonable cell permeability (Caco-2 AB = 33 nm s–1, efflux ratio = 15). As expected 29 showed complete selectivity over the Janus JH1 domains (>500× fold) and had an IC50 ≥ 1 μM for 229 out of 231 tested kinases in an fluorescent probe displacement panel, including an IC50 = 27 μM against the JAK1 JH2 domain (Fig. 6).
Fig. 6. Selectivity profile of 29. Selectivity was determined from binding data generated using a fluorescent probe displacement assay. Illustration reproduced courtesy of Cell Signaling Technology, Inc.

To assess the pharmacokinetic properties of 29, the compound was dosed in Balb/C mice as an oral solution (25 mg kg–1) and as an intravenous solution (3 mg kg–1) (Table 4). Bioavailability was reasonable (27%), but overall exposures were modest, consistent with clearance (45 ml min–1 kg–1) and modest microsomal stability.
Table 4. In vivo pharmacokinetic characterization of 29 in mice.
|
29
|
||
| 3 mg kg–1 IV a | 25 mg kg–1 p.o. b | |
| C max (μM) | 2.8 | |
| AUClast (μM h) | 2.3 | 5.1 |
| t 1/2 (h) | 5.0 | 1.0 |
| Cl (mL min–1 kg–1) | 45 | |
| V ss (L kg–1) | 6.3 | |
| F | 27% | |
a5% DMAC/76% PEG400/19% water; Balb/C; male.
b5% DMAC/9.5% EtOH/76% PEG300/9.5% TPGS; Balb/C; male.21
However, as shown in Table 3, compound 29 and the rest of the compounds described were determined to be potent inhibitors of phosphodiesterase 4 (PDE4). A co-crystal structure of tetrafluoroaniline 30 with PDE4 (PDBID 5TKB) provided insight as to how selectivity may be obtained over PDE4 (Fig. 7). As shown in Fig. 7, the meta positions of the C6 aniline represented likely selectivity vectors. Concordant with this, bulky substituents at the meta position of the C6 aniline were not tolerated by PDE4, and provided the desired selectivity (Table 5). Large aliphatic groups provided selectivity (31), but were also metabolically labile, and thus substituted meta-biphenyl rings were evaluated (vide infra). A simple meta-biphenyl (32) provided reasonable PDE4 selectivity with modest metabolic stability. The phenyl substituent was optimized for stability by adding electron withdrawing groups, providing 33. A screen of C3 amide substituents to improve potency then provided 34, which was highly selective over PDE4 and reasonably potent, showing good metabolic stability in human and rat liver microsomes.
Fig. 7. X-ray crystal structure representation of 30 co-complexed with PDE4 (PDBID 5TKB). Surface representation of the protein binding site is shown highlighting that space off of both meta positions of the C6 aniline are hindered (yellow arrows). Carbons of PDE4 are coloured green and carbons of 30 in magenta. Hydrogens added for clarity.

Table 5. Improving PDE4 selectivity.
| ||||||
| R2 | R3 | TYK2 JH2 (IC50, μM) | IL23/IFNα (IC50, μM) | PDE4 (IC50, μM) | LM % rem (h/r/m) | |
| 31 |
|
|
0.013 | 0.51/0.41 | 2.4 | 41/4/3 |
| 32 |
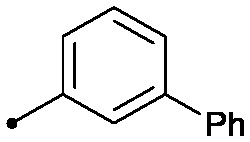
|
|
0.004 | 0.33/0.20 | 0.38 | 51/16/0 |
| 33 |
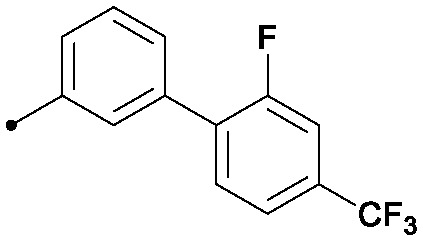
|

|
0.066 | 1.2/1.1 | >100 | 89/86/65 |
| 34 |
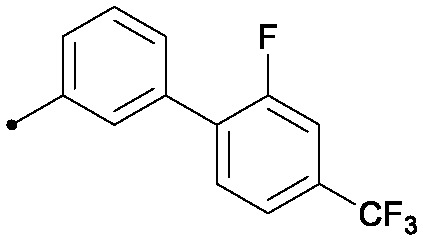
|
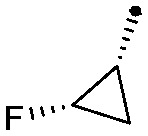
|
0.025 | 0.41/0.32 | >33 | 96/80/50 |
| 35 |
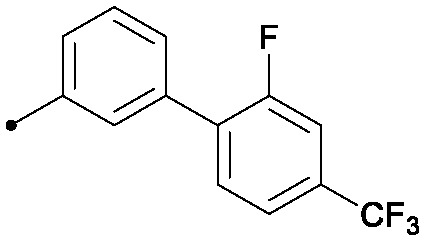
|
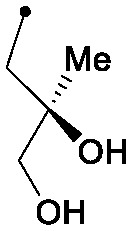
|
0.008 | 0.78/0.75 | 5.6 | 75/75/34 |
As shown in Table 6, 34 lacked potency in a human whole blood IFNα stimulated assay (IC50 > 10 μM), despite reasonable potency in the cellular IFNα reporter assay (0.32 μM). The disconnect between cellular and whole blood potency may be attributed to the high protein binding of the lipophilic biphenyl analogue. This could be partially overcome by increasing the hydrophilic character of the amide substituent. For example, 35, which possesses a pendant diol off of the C3 amide, shows a noticeable improvement, in terms of both whole blood potency and protein binding, from 34, while maintaining high selectivity over PDE4.
Table 6. Relationship of protein binding to whole blood potency.
| TYK2 JH2 (IC50, μM) | IL23/IFNα (IC50, μM) | % unbound (h/r/m) | hWB IFNa IP-10 (IC50, μM) | |
| 29 | 0.004 | 0.24/0.16 | 27/31/30 | 0.37 |
| 34 | 0.025 | 0.41/0.32 | 0.4/0.5/0.4 | >10 |
| 35 | 0.008 | 0.78/0.75 | 1.0/1.2/1.2 | 4.2 |
Conclusions
Having previously determined that ligands of the pseudokinase domain of TYK2 stabilize an un-phosphorylated, un-activated state of the protein and inhibit receptor-mediated kinase activation, we sought to improve upon a single imidazopyridazine screening hit. The initial screening hit 7 was highly selective, with weak affinity for the TYK2 pseudokinase. Evaluation of SAR at the C8 position led to identification of methylamino as an optimal substituent. Tuning the electronics at C6 provided 18, that was fully selective over the Janus catalytic domains with excellent ligand and lipophilic ligand efficiency (LE = 0.35, LLE = 6.2). From this starting point we identified 2-alkoxy substituted anilines at C6, as a means to improve cellular potency, while the C3 amide was recognized as occupying an accommodating pocket for the optimization of physical properties. An off-target liability, PDE4, was later identified, but SBDD allowed identification of a vector that enabled the identification of highly selective compounds. The most significant remaining concern was the poor metabolic stability of the scaffold; however, this was partially addressable using electron-withdrawing groups on the C6 aniline and pendant hydroxyl groups on the C3 amide. Pharmacokinetic testing of 29 in mice showed that reasonable systemic exposures can be achieved with oral dosing of this chemotype. Future efforts will be directed towards further improving potency and metabolic stability, while also maintaining the selectivity over PDE4 demonstrated by 34, in order to identify a compound well suited for evaluation in clinical studies for the treatment of autoimmune diseases.
Experimentals
Compounds 8 and 14 were prepared following previously described procedures.22 All reagents and starting materials were obtained from commercial suppliers and used without further purification unless otherwise stated. Reaction progress was monitored using a variety of LC instruments equipped with electrospray positive ionization detectors. The purity of all final compounds was established to be ≥95% using two separate LC/MS injections with the following conditions: injection 1: column: Waters Acquity UPLC BEH C18, 2.1 × 50 mm, 1.7 μm particles; mobile phase A: 5 : 95 acetonitrile : water with 10 mM ammonium acetate; mobile phase B: 95 : 5 acetonitrile : water with 10 mM ammonium acetate; temperature: 50 °C; gradient: 0–100% B over 3 minutes, then a 0.75 minute hold at 100% B; flow: 1.11 mL min–1. Injection 2: column: Waters Acquity UPLC BEH C18, 2.1 × 50 mm, 1.7 μm particles; mobile phase A: 5 : 95 acetonitrile : water with 0.05% TFA; mobile phase B: 95 : 5 acetonitrile : water with 0.05% TFA; temperature: 50 °C; gradient: 0–100% B over 3 minutes, then a 0.75 minute hold at 100% B; flow: 1.11 mL min–1. Data was collected as a single point (n = 1). Representative procedures below.
General procedures for generation of R2 and R3 libraries
Displacement of C8 halide with basic amine
A solution of 8 (7.35 g, 28.3 mmol), 1-(4-methoxyphenyl)-N-methylmethanamine (4.74 g, 31.4 mmol) and triethylamine (6.73 mL, 48.3 mmol) in dioxane (75 mL) was heated in an oil bath at 90 °C for 2.5 hours. The reaction was cooled to room temperature and concentrated to provide a sludge that was triturated with water to provide a solid which was filtered, rinsed with water and then collected with dichloromethane. The solution was dried over anhydrous sodium sulfate, filtered and concentrated to provide the product (8.95 g, 84% yield). 1H NMR (400 MHz, CHLOROFORM-d) δ 8.13 (s, 1H), 7.17 (d, J = 8.6 Hz, 2H), 6.87 (d, J = 8.6 Hz, 2H), 6.12 (s, 1H), 5.50 (s, 2H), 4.46 (q, J = 7.1 Hz, 2H), 3.81 (s, 3H), 3.18 (s, 3H), 1.74–1.58 (m, 1H), 1.44 (t, J = 7.2 Hz, 3H). MS (E+) m/z: 375.3 (MH+).
Path A
b) Nitrogen was bubbled through a solution of ethyl 6-chloro-8-((4-methoxybenzyl)(methyl)amino)imidazo[1,2-b]pyridazine-3-carboxylate (1.00 g, 2.67 mmol) in DMA (10 mL) for 15 minutes and then 3,5-dimethylaniline (0.647 g, 5.34 mmol) was added to the reaction followed by Pd2dba3 (489 mg, 0.534 mmol), Xantphos (617 mg, 1.07 mmol) and cesium carbonate (3.48 g, 10.67 mmol). The reaction was sealed and heated to 100 °C for 4 hours, after which the reaction was cooled to room temperature and ethyl acetate and water were added. The slurry was filtered and the filtrate layers were separated, the organic layer was rinsed three times with brine, dried over sodium sulfate, filtered and concentrated to provide the crude product, which was then purified using automated chromatography providing the product (710 mg, 58% yield). 1H NMR (400 MHz, CHLOROFORM-d) δ 8.04 (s, 1H), 7.15 (d, J = 8.8 Hz, 2H), 7.10 (s, 2H), 6.84 (d, J = 8.6 Hz, 2H), 6.69 (s, 1H), 6.35 (s, 1H), 5.78 (s, 1H), 5.35 (s, 2H), 4.45 (q, J = 7.0 Hz, 2H), 3.79 (s, 3H), 3.10 (s, 3H), 2.31 (d, J = 0.4 Hz, 6H), 1.42 (t, J = 7.0 Hz, 3H). MS (E+) m/z: 460.4 (MH+).
c) To a solution of ethyl 6-((3,5-dimethylphenyl)amino)-8-((4-methoxybenzyl)(methyl)amino)imidazo[1,2-b]pyridazine-3-carboxylate (710 mg, 1.545 mmol) in dioxane (2 mL) was added hydrochloric acid (4 M in dioxane, 3.86 mL, 15.45 mmol). The solution was stirred at room temperature for two hours and then concentrated in vacuo. Dichloromethane was added and the solution was re-concentrated, and then more dichloromethane was added and the solution was concentrated again (repeated 5 times). The intermediate was then dissolved in methanol (5 mL) and sodium hydroxide (1 M in water, 3.09 mL, 3.09 mmol) was added. The reaction was stirred overnight and then the reaction was diluted with water and the methanol was removed in vacuo. Hydrochloric acid (1 M in water) was added until the pH measured ∼3. The product was extracted with ethyl acetate (twice) and then once with dichloromethane. The combined organic layers were dried over sodium sulfate, filtered, and concentrated providing the crude acid (480 mg, 90% yield), which was used without further purification. MS (E+) m/z: 312.1 (MH+).
d) To a solution of 6-((3,5-dimethylphenyl)amino)-8-(methylamino)imidazo[1,2-b]pyridazine-3-carboxylic acid (30 mg, 0.096 mmol) and ammonium chloride (10.31 mg, 0.193 mmol) in DMF (1 mL) was added HATU (44.0 mg, 0.116 mmol) and 4-methylmorpholine (48.7 mg, 0.482 mmol) and the reaction was stirred for 1 hour. The reaction was then filtered through a micropore filter, diluted with DMF and purified by preparative LC to provide 18 (6.7 mg, 9% yield). 1H NMR (500 MHz, DMSO-d6) δ 8.97 (s, 1H), 8.34 (d, J = 2.5 Hz, 1H), 7.93 (d, J = 2.5 Hz, 1H), 7.81 (s, 1H), 7.44 (d, J = 5.0 Hz, 1H), 7.06 (s, 2H), 6.62 (s, 1H), 5.74 (s, 1H), 2.87 (d, J = 4.5 Hz, 3H), 2.25 (s, 6H). MS (E+) m/z: 311.2 (MH+).
Path B
e) To a solution of ethyl 6-chloro-8-((4-methoxybenzyl)(methyl)amino)imidazo[1,2-b]pyridazine-3-carboxylate (1.20 g, 3.20 mmol) in methanol (15 mL) and tetrahydrofuran (15 mL) was added 0.5 M (aqueous) lithium hydroxide (25.6 mL, 12.81 mmol) and the reaction was stirred overnight. The reaction was diluted with water and then the methanol was removed in vacuo, the resulting solution was adjusted to pH ∼4 using hydrochloric acid (1 M aqueous) the product was extracted with dichloromethane and the combined organic layers were dried over sodium sulfate, filtered, and concentrated to provide the acid (1.00 g, 81% yield), which was used without further purification. MS (E+) m/z: 347.0 (MH+).
f) A mixture of 6-chloro-8-((4-methoxybenzyl)(methyl)amino)imidazo[1,2-b]pyridazine-3-carboxylic acid (536 mg, 1.546 mmol), cyclopropanamine (0.321 mL, 4.64 mmol), and triethylamine (0.646 mL, 4.64 mmol) in dimethylformamide (3 mL) was treated with BOP (752 mg, 1.700 mmol), and the reaction was stirred at room temperature for 5 hours. The desired product was precipitated with water (5 mL), and collected by filtration. The solids were rinsed twice with water, once with a small amount of methanol, and dried under vacuum to yield 6-chloro-N-cyclopropyl-8-((4-methoxybenzyl)(methyl)amino)imidazo[1,2-b]pyridazine-3-carboxamide (480 mg, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.55 (d, J = 3.5 Hz, 1H), 8.06 (s, 1H), 7.19 (d, J = 8.6 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 6.39 (s, 1H), 5.52 (br. s., 2H), 3.73 (s, 3H), 3.31 (s, 3H), 2.88 (tq, J = 7.2, 3.8 Hz, 1H), 0.89–0.75 (m, 2H), 0.63–0.51 (m, 2H). MS (E+) m/z: 386.0 (MH+).
g) 26 in a sealable vial, a mixture of 6-chloro-N-cyclopropyl-8-((4-methoxybenzyl)(methyl)amino)imidazo[1,2-b]pyridazine-3-carboxamide (25 mg, 0.065 mmol), 2,3-dihydrobenzo[b][1,4]dioxin-5-amine (20 mg, 0.13 mmol), and cesium carbonate (42 mg, 0.13 mmol) in DMA (1 mL) was degassed with bubbling nitrogen. After 5 minutes, Xantphos (7.5 mg, 0.013 mmol) and Pd2(dba)3 (5.9 mg, 0.0065 mmol) were added, and degassing was continued for 5 minutes. The vial was capped, and the reaction was heated at 125 °C until completion (40 minutes), and then allowed to come to room temperature. The reaction was diluted with ethyl acetate (20 mL) and filtered, the filtrate was washed twice with water, and once with 10% lithium chloride solution, then dried over sodium sulfate and concentrated in vacuo. The residue was taken up in dichloromethane (1 mL) and then treated with 4 M HCl in dioxane (1 mL). The reaction was stirred at room temperature until complete deprotection was observed (1 h). The crude product was then filtered through a micropore frit and purified using preparative LC to provide 26 (isolated as TFA salt) (9.1 mg, 28% yield). 1H NMR (500 MHz, METHANOL-d4) δ 7.91 (s, 1H), 7.20 (dd, J = 8.2, 1.2 Hz, 1H), 6.85 (t, J = 8.2 Hz, 1H), 6.65 (dd, J = 8.2, 1.2 Hz, 1H), 5.86 (s, 1H), 4.34–4.30 (m, 2H), 4.30–4.26 (m, 2H), 2.99 (s, 3H), 2.82 (tt, J = 7.3, 3.8 Hz, 1H), 0.80–0.72 (m, 2H), 0.50–0.36 (m, 2H). MS (E+) m/z: 381.2 (MH+).
The following compounds were prepared using the above procedures:
7 (as HCl salt) 1H NMR (400 MHz, DMSO-d6) δ 10.12 (1 H, s), 8.06–8.41 (3 H, m), 7.76–8.01 (2 H, m), 6.31 (1 H, s), 2.62 (1 H, br. s.), 0.85 (2 H, d, J = 5.02 Hz), 0.68 (2 H, br. s.). MS (E+) m/z: 402.0 (MH+).
10 1H NMR (400 MHz, DMSO-d6) δ 9.16 (s, 1H), 8.32 (s, 1H), 8.28 (m, 1H), 7.82 (s, 1H), 7.84 (m, 1H), 7.46 (dd, J = 8.6, 1.0 Hz, 2H), 7.32 (m, 2H), 7.00 (t, J = 7.2 Hz, 1H), 6.17 (s, 1H), 2.56 (m, 1H), 0.82 (m, 2H), 0.68 (m, 2H). MS (E+) m/z: 309.2 (MH+).
11 1H NMR (400 MHz, DMSO-d6) δ 9.04 (s, 1H), 8.33 (m, 1H), 8.32 (s, 1H), 7.91 (m, 1H), 7.81 (s, 1H), 7.68 (s, 1H), 7.08 (s, 2H), 6.64 (s, 1H), 6.15 (s, 1H), 2.56 (m, 1H), 2.08 (s, 6H), 0.82 (m, 2H), 0.69 (m, 2H). MS (E+) m/z: 337.2 (MH+).
12 (as TFA salt) 1H NMR (400 MHz, METHANOL-d4) δ 7.98 (s, 1H), 7.12 (s, 1H), 7.02 (d, J = 11.2 Hz, 1H), 6.61 (d, J = 9.5 Hz, 1H), 6.29 (s, 1H), 5.49 (d, J = 1.3 Hz, 2H), 2.65 (td, J = 6.9, 3.6 Hz, 1H), 2.35 (s, 3H), 0.93 (d, J = 6.6 Hz, 2H), 0.78–0.63 (m, 2H). MS (E+) m/z: 341.1 (MH+).
23 (as TFA salt) 1H NMR (500 MHz, DMSO-d6) δ 8.88 (s, 1H), 8.82 (d, J = 3.5 Hz, 1H), 8.30 (s, 1H), 7.80 (s, 1H), 7.44 (d, J = 5.0 Hz, 1H), 7.01 (s, 2H), 6.69 (s, 1H), 5.72 (s, 1H), 2.87–2.83 (m, 3H), 2.79 (td, J = 7.2, 3.5 Hz, 1H), 2.28 (s, 6H), 0.74–0.63 (m, 2H), 0.33–0.22 (m, 2H). MS (E+) m/z: 351.2 (MH+).
24 1H NMR (500 MHz, METHANOL-d4) δ 6.55 (s, 1H), 5.61 (s, 2H), 5.39 (s, 1H), 4.44 (s, 1H), 1.96 (d, J = 1.5 Hz, 6H), 1.87 (s, 2H), 1.72 (s, 2H), 1.63 (s, 3H), 0.94 (s, 6H). MS (E+) m/z: 397.0 (MH+).
25 1H NMR (500 MHz, CHLOROFORM-d/METHANOL-d4) δ 7.91 (s, 1H), 6.96 (s, 2H), 6.75 (s, 1H), 5.82 (s, 1H), 4.33 (br. s., 2H), 3.37 (s, 2H), 3.28 (d, J = 4.5 Hz, 3H), 2.98 (s, 3H), 2.31 (s, 6H), 0.58 (s, 3H). MS (E+) m/z: 413.2 (MH+).
27 1H NMR (500 MHz, METHANOL-d4) δ 7.94 (s, 1H), 7.70–7.65 (m, 1H), 7.15–7.07 (m, 1H), 7.03–6.95 (m, 2H), 5.90 (s, 1H), 4.31 (s, 1H), 3.90 (s, 3H), 3.41 (s, 2H), 3.00 (s, 3H), 1.14 (s, 6H). MS (E+) m/z: 385.2 (MH+).
28 1H NMR (500 MHz, DMSO-d6) δ 8.85–8.71 (m, 1H), 7.86 (s, 1H), 7.60 (q, J = 4.6 Hz, 1H), 7.57–7.49 (m, 1H), 6.97 (ddd, J = 11.4, 8.7, 3.2 Hz, 1H), 6.17 (s, 1H), 4.52 (t, J = 5.4 Hz, 1H), 3.80 (s, 3H), 3.18 (d, J = 6.4 Hz, 2H), 3.01 (d, J = 5.0 Hz, 2H), 2.89 (d, J = 4.5 Hz, 3H), 0.63 (s, 6H). MS (E+) m/z: 435.2 (MH+).
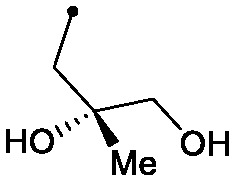
29 1H NMR (400 MHz, DMSO-d6) δ 8.73 (t, J = 6.4 Hz, 1H), 8.69 (s, 1H), 7.88 (s, 1H), 7.66 (dt, J = 11.2, 2.4 Hz, 1H), 7.55 (q, J = 4.8 Hz, 1H), 6.90 (ddd, J = 11.6, 8.7, 3.1 Hz, 1H), 6.23 (s, 1H), 3.83 (d, J = 0.7 Hz, 3H), 3.42–3.32 (m, 2H), 3.14–3.09 (m, 2H), 2.90 (d, J = 4.8 Hz, 3H), 0.91 (s, 3H). MS (E+) m/z: 437.2 (MH+). Enantiomeric excess determined to be 96.6% ee via chiral HPLC: [AD-H (.46 × 25cm), 25% MeOH w 0.1% DEA in CO2, 3 ml min–1, 35C, 100bars BPR, 220 nm] major @5.06 min minor @7.34 min. Assignment based on synthesis (aminolysis of commercial (2S)-(+)-2-methylglycidyl 4-nitrobenzoate).
30 1H NMR (500 MHz, DMSO-d6) δ 9.17 (br. s., 1H), 8.52 (t, J = 6.2 Hz, 1H), 7.84 (s, 1H), 7.72–7.56 (m, 2H), 5.86 (s, 1H), 4.59 (t, J = 6.2 Hz, 1H), 3.24–3.14 (m, 1H), 3.14–3.07 (m, 1H), 3.04 (dd, J = 5.7, 4.7 Hz, 2H), 2.93–2.86 (m, 3H), 0.82 (s, 3H). MS (E+) m/z: 443.1 (MH+). Enantiomeric excess not determined
31 1H NMR (500 MHz, DMSO-d6) δ 9.03 (s, 1H), 8.29 (br. s., 1H), 7.83 (s, 1H), 7.80 (br. s., 1H), 7.46 (d, J = 5.0 Hz, 1H), 7.39–7.34 (m, 1H), 7.30–7.16 (m, 2H), 7.13–6.94 (m, 1H), 5.77 (s, 1H), 2.87 (d, J = 4.5 Hz, 3H), 1.28 (s, 9H). MS (E+) m/z: 339.2 (MH+).
32 1H NMR (500 MHz, DMSO-d6) δ 9.20 (s, 1H), 8.30 (br. s., 1H), 7.84 (s, 1H), 7.77 (br. s., 1H), 7.67–7.60 (m, 3H), 7.54–7.45 (m, 4H), 7.43–7.34 (m, 2H), 7.27 (d, J = 7.9 Hz, 1H), 5.80 (s, 1H), 2.94–2.84 (m, 3H). MS (E+) m/z: 359.2 (MH+).
33 1H NMR (500 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.66 (d, J = 3.5 Hz, 1H), 7.88–7.78 (m, 3H), 7.70 (d, J = 7.9 Hz, 1H), 7.65–7.58 (m, 2H), 7.56–7.46 (m, 2H), 7.28 (d, J = 6.9 Hz, 1H), 5.78 (s, 1H), 2.88 (d, J = 5.0 Hz, 3H), 2.68 (tq, J = 7.4, 3.8 Hz, 1H), 0.62–0.53 (m, 2H), 0.31–0.20 (m, 2H). MS (E+) m/z: 485.2 (MH+).
34 1H NMR (500 MHz, DMSO-d6) δ 9.28 (s, 1H), 8.74 (d, J = 4.0 Hz, 1H), 7.87 (s, 1H), 7.84–7.76 (m, 2H), 7.70 (d, J = 7.9 Hz, 1H), 7.64–7.58 (m, 2H), 7.56 (d, J = 4.5 Hz, 1H), 7.48 (t, J = 7.9 Hz, 1H), 7.26 (d, J = 7.4 Hz, 1H), 5.81 (s, 1H), 4.74–4.57 (m, 1H), 2.89 (d, J = 4.5 Hz, 3H), 2.77 (dd, J = 9.2, 4.7 Hz, 1H), 1.08–0.95 (m, 1H), 0.75–0.57 (m, 1H). MS (E+) m/z: 503.2 (MH+). Enantiomeric excess not determined.
35 1H NMR (500 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.82 (t, J = 6.4 Hz, 1H), 7.86 (s, 1H), 7.84–7.76 (m, 2H), 7.74–7.66 (m, 2H), 7.62 (d, J = 1.0 Hz, 1H), 7.58–7.49 (m, 2H), 7.21 (d, J = 6.9 Hz, 1H), 5.83 (s, 1H), 3.19–3.02 (m, 4H), 2.93–2.87 (m, 3H), 0.90 (s, 3H). MS (E+) m/z: 533.2 (MH+). Enantiomeric excess not determined.
General procedures for generation of R1 libraries
15
To a suspension of 14 (6.1 g, 23.69 mmol) in tetrahydrofuran (39.5 ml) was added triethylamine (33.0 ml, 237 mmol) followed by ethanethiol (2.103 ml, 28.4 mmol). The mixture was heated to 50 °C for 10 minutes and then concentrated. The crude product was suspended in hexanes and then filtered, the powder was collected and then sonicated in ethyl acetate and filtered, rinsing with ethyl acetate. The filtrate was concentrated and purified by automated chromatography to provide 15 (5.56 g, 96% yield). 1H NMR (400 MHz, CHLOROFORM-d) δ 8.13 (s, 1H), 6.93 (s, 1H), 3.19 (q, J = 7.5 Hz, 2H), 1.53 (t, J = 7.5 Hz, 3H). MS (E+) m/z: 239.0 (MH+).
16
To a suspension of 15 (3.16 g, 13.24 mmol) in DMA (65 mL) was added 3,5-dimethylaniline (3.30 mL, 26.5 mmol), followed by Pd2dba3 (2.42 g, 2.65 mmol), cesium carbonate (17.2 g, 53.0 mmol), and Xantphos (3.1 g, 5.30 mmol) in a single portion. The reaction vessel was evacuated and backfilled with nitrogen (×3), sealed and then heated to 125 °C for 90 minutes. The reaction was cooled to room temperature and filtered through celite, eluting with ethyl acetate. The ethyl acetate layer was washed three times with water, dried over sodium sulfate, filtered, concentrated and then purified using flash chromatography to provide 16 (2.9 g, 61% yield). 1H NMR (400 MHz, CHLOROFORM-d) δ 8.13 (s, 1H), 6.93 (s, 1H), 3.19 (q, J = 7.5 Hz, 2H), 1.53 (t, J = 7.5 Hz, 3H). MS (E+) m/z: 324.2 (MH+).
17
To a solution of 16 (1.19 g, 3.67 mmol) in DCM (37 mL) was added mCPBA (75% by weight, 1.06 g, 4.59 mmol). The reaction was stirred for 20 minutes at room temperature and then concentrated. The crude product was suspended in ethyl acetate and filtered, rinsing with ethyl acetate to provide the crude racemic product (762 mg, 55% yield). 1H NMR (400 MHz, CHLOROFORM-d/METHANOL-d4) δ 7.99 (s, 1H), 7.37 (s, 1H), 3.50 (dq, J = 14.1, 7.3 Hz, 1H), 3.23 (dq, J = 14.1, 7.3 Hz, 1H), 1.28 (t, J = 7.3 Hz, 3H). MS (E+) m/z: 340.1 (MH+).
20
To a solution of 17 (7 mg, 0.021 mmol) in tetrahydrofuran (0.2 ml) was added cyclobutanamine (0.075 ml, 0.884 mmol). The reaction was sealed and heated to 85 °C overnight. The reaction was concentrated, the intermediate was dissolved in dimethylsulfoxide (DMSO, 0.20 mL), and then potassium hydroxide (5 M in water, 0.044 mL, 0.22 mmol) and hydrogen peroxide (33%, 0.041 mL, 0.44 mmol) were added. The reaction was run at room temperature for 14 minutes and then quenched via the addition of 1 M (aqueous) hydrochloric acid. The resulting solid was collected via filtration, dissolved in methanol and purified using preparative LC providing 20 as the trifluoroacetate salt (2.2 mg, 11% yield). 1H NMR (400 MHz, METHANOL-d4) δ 8.08 (br. s., 1H), 7.05 (s, 1H), 6.73 (s, 1H), 5.89 (s, 1H), 4.12 (quin, J = 7.7 Hz, 1H), 2.54 (dd, J = 11.4, 4.0 Hz, 2H), 2.30 (s, 6H), 2.21–2.03 (m, 2H), 2.02–1.82 (m, 2H). MS (E+) m/z: 351.2 (MH+).
The following two compounds were prepared in a manner analogous to 20.
19 1H NMR (500 MHz, CHLOROFORM-d/METHANOL-d4) δ 7.93 (s, 1H), 7.03 (s, 2H), 6.68 (s, 1H), 5.81 (s, 1H), 3.34–3.32 (m, 2H), 2.28 (s, 6H), 1.36 (t, J = 7.2 Hz, 3H). MS (E+) m/z: 325.2 (MH+).
21 1H NMR (500 MHz, CHLOROFORM-d/METHANOL-d4) δ 7.92 (s, 1H), 7.03 (s, 2H), 6.68 (s, 1H), 5.85 (s, 1H), 3.98–3.84 (m, 1H), 2.29 (s, 6H), 2.14–2.01 (m, 2H), 1.87–1.76 (m, 2H), 1.72–1.61 (m, 4H). MS (E+) m/z: 365 (MH+).
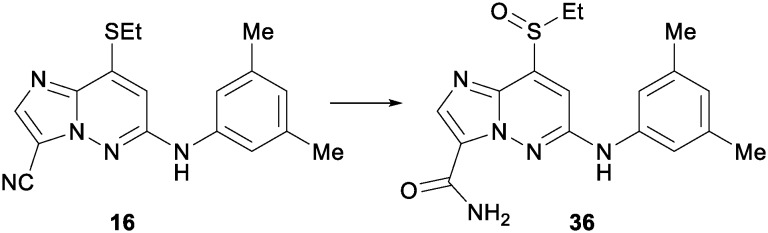
22 to a solution of 16 (0.9 g, 2.78 mmol) in dimethylsulfoxide (DMSO, 16 mL) was added potassium hydroxide (5 M in water, 2.78 mL, 13.91 mmol) followed by the careful addition of hydrogen peroxide (33%, 2.84 mL, 27.8 mmol). The reaction was stirred at room temperature for 20 minutes and then quenched with hydrochloric acid (1 M aqueous). The resulting precipitate was suspended in dichloromethane and concentrated in vacuo, the resulting solid was then concentrated from toluene and then again from dichloromethane. The intermediate was dissolved in dichloromethane (250 mL) and 3-chloroperbenzoic acid (75%, 0.624 g, 3.52 mmol) was added. The reaction was stirred for 20 minutes and then concentrated, the crude product was triturated with ethyl acetate and filtered, rinsing with ethyl acetate. 36 was collected as a yellow powder (668 mg, 64% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.79 (s, 1H), 8.09 (br. s., 1H), 8.06–7.97 (m, 2H), 7.35 (s, 1H), 7.22 (s, 2H), 6.74 (s, 1H), 3.50 (dd, J = 13.9, 7.3 Hz, 1H), 3.26 (dd, J = 14.0, 7.4 Hz, 1H), 2.29 (s, 6H), 1.12 (t, J = 7.4 Hz, 3H). MS (E+) m/z: 358.1 (MH+).
To a suspension of 36 (10 mg, 0.028 mmol) in tetrahydrofuran (0.2 mL) was added aniline (0.038 mL, 0.420 mmol) and the vessel was purged of air using a stream of nitrogen. Next sodium hydride (60%, 11.19 mg, 0.280 mmol) was added and the reaction was heated to 75 °C. After 30 minutes the solvent was removed in vacuo and the product was purified using preparative LC to provide 22 as the trifluoroacetate salt (5.5 mg, 38% yield). 1H NMR (400 MHz, CHLOROFORM-d) δ 8.63 (br. s., 1H), 8.20 (s, 1H), 7.50–7.41 (m, 2H), 7.39–7.33 (m, 2H), 6.95 (s, 2H), 6.80 (s, 1H), 6.34 (s, 1H), 6.17 (s, 1H), 5.73 (br. s., 1H), 2.32 (s, 6H). MS (E+) m/z: 373.2 (MH+).
13 6-chloro-8-(cyclopropylamino)imidazo[1,2-b]pyridazine-3-carbonitrile (100 mg, 0.428 mmol), cyclohexanamine (424 mg, 4.28 mmol), iPr2NEt (0.747 mL, 4.28 mmol) and NMP (1 mL) were taken in a 10 mL microwave tube and heated to 150 °C overnight. The reaction was diluted with water and extracted (×3) with ethyl acetate, the combined organics were washed with water and brine, dried over sodium sulfate, filtered, concentrated and purified via automated chromatography providing the cyano precursor to 13. This was dissolved in DMSO (1 mL) and KOH (5 M, 1 mL, 5 mmol) was added followed by the dropwise addition of H2O2 (30% aq., 0.5 mL, 4.9 mmol). The reaction was stirred at room temperature for 2 hours and then diluted with ethyl acetate, washed with water (×2) and brine. The organic layer was dried over sodium sulfate, filtered, concentrated and purified using preparative LC to provide 13 (15 mg, 11% yield over two steps). 1H NMR (400 MHz, DMSO-d6) δ 8.52 (s, 1H), 7.83 (s, 1H), 7.78 (br. s., 1H), 7.36 (br. s., 1H), 6.85 (br. s., 1H), 6.00 (s, 1H), 3.48 (m, 1H), 1.99 (m, 2H), 1.75 (m, 2H), 1.61 (m, 1H), 1.37–1.20 (m, 6 H), 0.79 (m, 2H), 0.62 (m, 2H). MS (E+) m/z: 315.2 (MH+).
TYK2 JH2 SPA binding assay14
Assays were performed in 384-well plates with a final assay volume of 20 μL containing copper-polyvinyltoluene scintillation proximity assay beads (PerkinElmer Life Sciences, catalogue no. RPNQ0095) at 80 g ml–1, [3H] 3 (20 nM), the N-terminal His-tagged TYK2 pseudokinase domain (2.5 nM), and test compounds in assay buffer (50 mM HEPES, pH 7.5, 100 g ml–1 BSA, 5% DMSO). After incubating at room temperature for 30 min, the inhibition was calculated by the displacement of [3H] 3 binding as determined by scintillation counting. Dose-response curves were generated to determine the concentration required to inhibit [3H] 3 binding by 50% (IC50).
Radiometric [3H]cAMP SPA binding assay with PDE4
Binding of compounds to PDE4 enzyme was assessed by a radiometric competition assay measuring their inhibitory activity on [3H]cAMP binding. The PDE4 construct encoding His-Tb-hPDE4D2(86-413) was expressed in E. coli, and purified by nickel affinity and gel filtration chromatography. Assays were performed in 384-well Proxiplates (Perkin-Elmer). The purified His-Tb-hPDE4D2(86-413) protein was diluted to 0.5 ng mL–1 in assay buffer (25 mM HEPES, pH 7.4, 2.5 mM MgCl2, 0.1% BSA). Test compounds were pre-incubated for 10 minutes with the diluted His-Tb-hPDE4D2(86-413) protein in a concentration series from 0.5 nM to 30 μM, in 3-fold serial dilutions for 11 points. An equal volume of [3H]cAMP (isotopically diluted to 0.6 μM) was added and further incubated for 1 hour. Subsequently, a mixture containing YtO beads (GE Healthcare, RPNQ1280, 10 mg mL–1), IBMX (2 mM), glycerol (20%) was added as twice the original volume. The final reaction mixtures were incubated for another 3 hours. Radioactivity was measured on the Leadseeker (GE Healthcare). Competition data was fitted to a 4-parameter logistic equation. IC50 was determined as the concentration of compound that yielded 50% inhibition to the [3H]cAMP binding.
Homogeneous time resolved fluorescence (HTRF) assay (Fig. 6)
Binding assays contained protein construct, 0.2 nM terbium-anti-His antibody, fluorescein-labeled kinase tracer at the respective kinase Kd, and test compounds in assay buffer consisting of 20 mM Hepes pH 7.5, 10 mM MgCl2, 0.015% Brij-35, 2 mM DTT, and 50 μg ml–1 BSA. Assays were performed in black, flat-bottom, 1536-well plates. The reactions were incubated at room temperature for 90 minutes, following which the HTRF signal, ratio of fluorescence intensities at emission wavelengths for fluorescein acceptor (520 nm) and terbium donor (495 nm), the 520/495 ratio, generated were then measured on the Envision Plate reader. Inhibition data were calculated from the 520/495 ratio generated by the no protein control reactions for 100% inhibition and vehicle-only reactions for 0% inhibition. Dose response curves were generated to determine the concentration required for inhibiting 50% of the HTRF signal (IC50). Compounds were dissolved at 10 mM in dimethylsulfoxide (DMSO) and evaluated at eleven concentrations (3-fold dilutions).
IKKβ assays
The IKKβ activity of compound 3 was determined using a previously reported fluorescence polarization assay.23 The IKKβ activity of compound 7 was determined using a mobility shift assay (384-well plates): to a plate containing 11 serial dilutions of the ligand in 0.5 μL in DMSO was added 15 μL of a substrate solution containing IKKβ peptide substrate (3 μM) and ATP (28 μM) in buffer (100 mM HEPES, 10 mM MgCl2, pH 7.5, 10 mM MgCl2, 0.015% Brij-35, 4 mM DTT) followed by 15 μL of IKKβ (3.2 nM) in buffer. After incubating at room temperature for 60 min, 40 μL of and EDTA buffer was added to the wells. Assay plates were read with the Caliper LabChip® 3000 and IC50 values were determined by comparison to background.
IL-23/IFNα cell assay
Kit225 T cells with a stably-integrated STAT-dependent luciferase reporter were plated in RPMI (GIBCO) containing 10% heat-inactivated FBS (GIBCO) and 100 U mL–1 PenStrep (GIBCO). The cells were then stimulated with either 20 ng mL–1 human recombinant IL-23 or 200 U mL–1 human recombinant IFN-α (PBL InterferonSource) for 5–6 hours. Luciferase expression was measured using the STEADY-GLO® Luciferase Assay System (PROMEGA®) according to the manufacturer's instructions. Inhibition data were calculated by comparison to no inhibitor control wells for 0% inhibition and non-stimulated control wells for 100% inhibition. Dose response curves were generated to determine the concentration required to inhibit 50% of cellular response (IC50) as derived by non-linear regression analysis.
Protein binding assay
The protein binding assay was performed with equilibrium dialysis in a 96-well plate format using HTDialysis (HTD) block on a custom fully-automated platform. The assay incubation was done in triplicate at 37 °C for 6.5 hours, at a compound incubation concentration of 10 μM. At the end of the incubation, all samples generated (dialysate, serum and T0), were diluted with blank serum or buffer to achieve a balanced matrix of 50 : 50 serum/buffer, followed by protein precipitation using acetonitrile containing internal standards, at a ratio of 3 : 1 (ACN/sample). The resulted samples in 96-well plates were then vortexed and centrifuged, before the supernatant was pipetted to clean 96-well plates for LC-MS/MS analysis. The LC-MS/MS analysis was performed with a modified Aria LX-2 multiplexed system (ThermoFisher), where two sets of independent LC gradient pumps and auto-samplers were configured to a single TSQ Quantum Ultra triple quadrupole mass spectrometer equipped with a heated electrospray (H-ESI) source. Time staggered injections were enabled on the multiplexed system, so that at any given moment only the LC channel with an active data window was switched inline with the mass spectrometer for data collection, to reduce the mass spectrometer idle time and increase throughput. The column used was fused-core Ascentis® Express C18, 2.7 μm, 2.1 × 30 mm (Supelco, Bellefonte, PA) at a flow rate of 0.7 mL min–1 and the injection volume was 15 μL onto a 5 μL sample loop. The total LC runtime for each injection was 1.48 min, and the active data window for mass spectrometric acquisition for each channel was from 0.4–0.9 min after injection, for a total of 30 seconds. Post data acquisition, the chromatographic peak review for all the samples were performed and the integration results were exported to an Excel report. A custom VBA macro was used to automatically perform regression for the analytical standard curves, as well as calculation of sample concentration and biological end results. The formula used to calculate the biological end results are as following: % free = [dialysate]/[serum]; % recovery = {[dialysate] + [serum]}/[T0].
Protein production and X-ray crystallography
Protein production, purification, crystallization and structure determination of TYK2 JH2 (575-869) in complex with 18 was carried out as previously reported (14) and deposited to RCSB with PDBID 5TKD. The structure of PDE4 in complex with 30 was determined by Beryllium Discovery Corp. (; http://be4.com) and deposited to RCSB with PDBID 5TKB. Refinement tables included in the ESI.‡
Supplementary Material
Footnotes
†The authors declare no competing interests.
‡Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00560h
References
- Clark J. D., Flanagan M. E., Telliez J.-B. J. Med. Chem. 2014;57:5023–5038. doi: 10.1021/jm401490p. [DOI] [PubMed] [Google Scholar]
- For a review of progress towards JAK1-selective inhibitors see: Menet C. J., Mammoliti O., López-Ramos M., Future Med. Chem., 2015, 7 , 203 –235 . [DOI] [PubMed] [Google Scholar]
- Inhibition of JAK2 could result in anemia and other undesired hematopoiteic defects: Vincenti F., Tedesco Silva H., Busque S., O'Connell P., Friedewald J., Cibrik D., Budde K., Yoshida A., Cohney S., Weimar W., Kim Y. S., Lawendy N., Lan S.-P., Kudlacz E., Krishnaswami S., Chan G., Am. J. Transplant., 2012, 12 , 2446 –2456 . [DOI] [PubMed] [Google Scholar]
- For a review of the use of Ustekinumab in psoriasis and psoriatic arthritis see: Croxtall J. D., Drugs, 2011, 71 , 1733 –1753 .21902296 [Google Scholar]
- For a review of the use of Ustekinumab in Crohn's disease see: Tuskey A., Behm B. W., Clin. Exp. Gastroenterol., 2014, 7 , 173 –179 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furie R., Merrill J., Werth V., Khamashta M., Kalunian K., Brohawn P., Illei G., Drappa J., Wang L., Yoo S. Arthritis Rheumatol. 2015;67(suppl 10) doi: 10.1002/art.39962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The P1104A SNP, which shows a 50% reduction in TYK2 activation has been shown to be protective against multiple sclerosis: ; (a) Ban M., Goris A., Lorentzen A. R., Baker A., Mihalova T., Ingram G., Booth D. R., Heard R. N., Stewart G. J., Bogaert E., Dubois B., Harbo H. F., Celius E. G., Spurkland A., Strange R., Hawkins C., Robertson N. P., Dudbridge F., Wason J., De Jager P. L., Hafler D., Rioux J. D., Ivinson A. J., McCauley J. L., Pericak-Vance M., Oksenberg J. R., Hauser S. L., Sexton D., Haines J., Sawcer S. Eur. J. Hum. Genet. 2009;17:1309–1313. doi: 10.1038/ejhg.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Couturier N., Bucciarelli F., Nurtdinov R. N., Debouverie M., Lebrun-Frenay C., Defer G., Moreau T., Confavreux C., Vukusic S., Cournu-Rebeix I., Goertsches R. H., Zettl U. K., Comabella M., Montalban X., Rieckmann P., Weber F., Müller-Myhsok B., Edan G., Fontaine B., Mars L. T., Saoudi A., Oksenberg J. R., Clanet M., Liblau R. S., Brassat D. Brain. 2011;134:693–703. doi: 10.1093/brain/awr010. [DOI] [PubMed] [Google Scholar]; (c) Dendrou C. A., Cortes A., Shipman L., Evans H. G., Attfield K. E., Jostins L., Barber T., Kaur G., Kuttikkatte S. B., Leach O. A., Desel C., Faergeman S. L., Cheeseman J., Neville M. J., Sawcer S., Compston A., Johnson A. R., Everett C., Bell J. I., Karpe F., Ultsch M., Eigenbrot C., McVean G., Fugger L. Sci. Transl. Med. 2016;363:363ra149. doi: 10.1126/scitranslmed.aag1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAK1 deficient mice die perinatally and suffer from neurological deficits: Rodig S. J., Meraz M. A., White J. M., Lampe P. A., Riley J. K., Arthur C. D., King K. L., Sheehan K. C. F., Yin L., Pennica D., Johnson E. M., Schreiber R. D., Cell, 1998, 93 , 373 –383 . [DOI] [PubMed] [Google Scholar]
- Oyamada A., Ikebe H., Itsumi M., Saiwai H., Okada S., Shimoda K., Iwakura Y., Nakayama K. I., Iwamoto Y., Yoshikai Y., Yamada H. J. Immunol. 2009;183:7539–7546. doi: 10.4049/jimmunol.0902740. [DOI] [PubMed] [Google Scholar]
- Ortmann R., Smeltz R., Yap G., Sher S., Shevach E. M. J. Immunol. 2001;166:5712–5719. doi: 10.4049/jimmunol.166.9.5712. [DOI] [PubMed] [Google Scholar]
- There are eight examples of human patients with a TYK2 deficiency, susceptibility to mycobacterial and/or viral infections, consistent with contributions of TYK2 to normal immune homeostasis: ; (a) Minegishi Y., Saito M., Morio T., Watanabe K., Agematsu K., Tsuchiya S., Takada H., Hara T., Kawamura N., Ariga T., Kaneko H., Kondo N., Tsuge I., Yachie A., Sakiyama Y., Iwata T., Bessho F., Ohishi T., Joh K., Imai K., Kogawa K., Shinohara M., Fujieda M., Wakiguchi H., Pasic S., Abinun M., Ochs H. D. Immunity. 2006;25:745–755. doi: 10.1016/j.immuni.2006.09.009. [DOI] [PubMed] [Google Scholar]; (b) Kreins A. Y., Ciancanelli M. J., Okada S., Kong X.-F., Ramírez-Alejo N., Kilic S. S., El Baghdadi J., Nonoyama S., Mahdaviani S. A., Ailal F., Bousfiha A., Mansouri D., Nievas E., Ma C. S., Rao G., Bernasconi A., Kuehn H. S., Niemela J., Stoddard J., Deveau P., Cobat A., El Azbaoui S., Sabri A., Lim C. K., Sundin M., Avery D. T., Halwani R., Grant A. V., Boisson B., Bogunovic D., Itan Y., Moncada-Velez M., Martinez-Barricarte R., Migaud M., Deswarte C., Alsina L., Kotlarz D., Klein C., Muller-Fleckenstein I., Fleckenstein B., Cormier-Daire V., Rose-John S., Picard C., Hammarstrom L., Puel A., Al-Muhsen S., Abel L., Chaussabel D., Rosenzweig S. D., Minegishi Y., Tangye S. G., Bustamante J., Casanova J.-L., Boisson-Dupuis S. J. Exp. Med. 2015;212:1641–1662. doi: 10.1084/jem.20140280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J., Abbema A. V., Balazs M., Barrett K., Berezhkovsky L., Blair W., Chang C., Delarosa D., DeVoss J., Driscoll J., Eigenbrot C., Ghilardi N., Gibbons P., Halladay J., Johnson A., Kohli P. B., Lai Y., Liu Y., Lyssikatos J., Mantik P., Menghrajani K., Murray J., Peng I., Sambrone A., Shia S., Shin Y., Smith J., Sohn S., Tsui V., Ultsch M., Wu L. C., Xiao Y., Yang W., Young J., Zhang B., Zhu B.-Y., Magnuson S. J. Med. Chem. 2013;56:4521–4536. doi: 10.1021/jm400266t. [DOI] [PubMed] [Google Scholar]
- Nagamiya H., Yoshida M., Seto M., Marui S., Oda T., Ishichi Y., Suzuki H., Kusumoto T., Yogo T., Rhim C. Y., Yoon C., Lee G. N., Kang H. B., Kim K. O. and Jeoh H. S., PCT Int. Appl., WO2013/125543A1 20130829, 2013.
- Tokarski J. S., Zupa-Fernandez A., Tredup J. A., Pike K., Chang C., Xie D., Cheng L., Pedicord D., Muckelbauer J., Johnson S. R., Wu S., Edavettal S. C., Hong Y., Witmer M. R., Elkin L. L., Blat Y., Pitts W. J., Weinstein D. S., Burke J. R. J. Biol. Chem. 2015;290:11061–11074. doi: 10.1074/jbc.M114.619502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Luo H., Rose P., Barber D., Hanratty W., Lee S., Roberts T. M., D'Andrea A. D., Dearolf C. R. Mol. Cell. Biol. 1997;17:1562–1571. doi: 10.1128/mcb.17.3.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Saharinen P., Silvennoinen O. J. Biol. Chem. 2002;277:47954–47963. doi: 10.1074/jbc.M205156200. [DOI] [PubMed] [Google Scholar]; (c) Chen M., Cheng A., Candotti F., Zhou Y.-J., Hymel A., Fasth A., Notarangelo L. D., O'Shea J. J. Mol. Cell. Biol. 2000;20:947–956. doi: 10.1128/mcb.20.3.947-956.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min X., Ungureanu D., Maxwell S., Hammarén H., Thibault S., Hillert E.-K., Ayres M., Greenfield B., Eksterowicz J., Gabel C., Walker N., Silvennoinen O., Wang Z. J. Biol. Chem. 2015;290:27261–27270. doi: 10.1074/jbc.M115.672048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupardus P. J., Ultsch M., Wallweber H., Kohli P. B., Johnson A. R., Eigenbrot C. Proc. Natl. Acad. Sci. U. S. A. 2014;111:8025–8030. doi: 10.1073/pnas.1401180111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallweber H. J. A., Tam C., Franke Y., Starovasnik M. A., Lupardus P. J. Nat. Struct. Mol. Biol. 2014;21:443–448. doi: 10.1038/nsmb.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a discussion of pseudokinases see: Boudeau J., Miranda-Saavedra D., Barton G. J., Alessi D. R., Trends Cell Biol., 2006, 16 , 443 –452 . [DOI] [PubMed] [Google Scholar]
- Kieltyka K., Zhang J., Li S., Vath M., Baglieri C., Ferraro C., Zvyaga T. A., Drexler D. M., Weller H. N., Shou W. Z. Rapid Commun. Mass Spectrom. 2009;23:1579–1591. doi: 10.1002/rcm.4037. [DOI] [PubMed] [Google Scholar]
- All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee and conformed to the “Guide for the Care and Use of Laboratory Animals” published by the National Institutes of Health (NIH Publication No. 85-23, revised 2011).
- Fink B., Chen L., Gavai A., He L., Kim S.-H., Nation A., Zhao Y. and Zhang L., PCT Int. Appl., WO2010/042699A1, 2010.
- Dyckman A. J., Langevine C. M., Quesnelle C., Kempson J., Guo J., Gill P., Spergel S. H., Watterson S. H., Li T., Nirschl D. S., Gillooly K. M., Pattoli M. A., McIntyre K. W., Chen L., McKinnon M., Dodd J. H., Barrish J. C., Burke J. R., Pitts W. J. Bioorg. Med. Chem. Lett. 2011;21:383–386. doi: 10.1016/j.bmcl.2010.10.133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.