

 An Astragalus oligosaccharide (AOS) degraded from Astragalus polysaccharide (APS) and purified by membrane dialysis and silicon gel chromatography is studied in this paper.
An Astragalus oligosaccharide (AOS) degraded from Astragalus polysaccharide (APS) and purified by membrane dialysis and silicon gel chromatography is studied in this paper.
Abstract
An Astragalus oligosaccharide (AOS) degraded from Astragalus polysaccharide (APS) and purified by membrane dialysis and silicon gel chromatography is studied in this paper. The structural features of AOS were investigated by a combination of chemical and instrumental analysis, such as monosaccharide analysis, periodate oxidation-Smith degradation, methylation analysis, electrospray ionization mass spectrometry (ESI-MS), Fourier transform infrared (FT-IR) spectrometry and nuclear magnetic resonance (NMR). The results indicated that AOS is an octasaccharide that consists of (3→)-linked-Rha, (1→3)-linked-Rha, (1→3,4)-linked-Araf, (1→3)-linked-Gal, terminal-linked-Gal and terminal-linked-Glc. The effects of AOS on cyclophosphamide-induced immunosuppression were determined by various studies, such as the proliferation of nucleated marrow, red blood cell (RBC) and white blood cell (WBC) populations, growth of the spleen and thymus, and increases in hemoglobin (HGB) concentration and granulocyte-macrophage colony stimulating factor (GM-CSF) level. The results indicated that AOS can restore cyclophosphamide-induced immunosuppression by stimulating the secretion of GM-CSF, which promoted the differentiation of progenitor cells after proliferation.
1. Introduction
Leukopenia is a disease that involves a decrease in WBC count to fewer than 4 × 109 L–1. It is one of the most common side effects encountered with the use of myelotoxic anticancer agents.1 Almost all patients undergoing chemotherapy are at risk for leukopenia, although the incidence can be influenced by regimen, dose, cancer type, etc.2 For example, with the bleomycin–etoposide–doxorubicin–cyclophosphamide–vincristine–procarbazine–prednisone (BEACOPP) regimen, 98% of subjects who were administered an increased dose for the treatment of advanced-stage Hodgkin's disease developed grade 3 or 4 leukopenia.3,4 The immune system of the host can be impaired and threatened by chemotherapy-induced leukopenia.5 As a result, the occurrence of leukopenia often leads to prolonged hospitalization periods and increased mortality rates.6 Moreover, the occurrence of leukopenia in cancer patients is usually accompanied by delayed chemotherapy treatment and weakened drug dose intensity. Thus, some potentially curable cancers may become incurable.7,8
The most commonly used drugs to restore reduced leukocyte levels resulting from chemotherapy are GM-CSFs.9,10 Most currently administered drugs are molecules with low molecular weights because only small molecules can enter the blood recycling system easily and be recognized by the cell surface. By this method, the secretion of several cytokines can be induced. This finally contributes to the direct activation of leukocytes. Although these hematopoietic colony stimulating factors are efficient, these agents can also lead to severe side effects, such as allergic reactions (including anaphylaxis), musculoskeletal pain, fever, alveolar hemorrhage, respiratory distress syndrome, cutaneous vasculitis and splenic rupture.11,12 The effects of this treatment may endure only for a temporary period. Additionally, because these drugs are usually difficult to prepare, their prices are prohibitive.13 Thus, it is necessary to find alternative drugs to increase leukocyte levels.
Numerous studies have suggested that several polysaccharides (PS) can activate the proliferation of lymphocytes, including natural killer (NK) cells, T cells, and B cells.14–16 However, PS are generally molecules of high molecular weight, structural variation and viscosity. Due to these characteristics, it is difficult for PS to enter the blood circulation system directly. As a result, most of these activities can only be achieved after digestion or metabolism by beneficial bacteria present in the intestinal system. Due to this fact, PS cannot function in the same direct way as most medicines with low molecular weights.
Oligosaccharides (OS) consist of a small number (three to nine) of monosaccharides; they are widely used as sugar supplements. They can be easily incorporated into other foods and are widely applied as prebiotic agents.17,18 Also, research shows that certain OS can enhance immune system performance in rats.19 Because OS are molecules with low molecular weights, they can directly enter the blood circulation system and function directly. In the search for alternative drugs, it is a good strategy to increase leukocyte levels by deriving OS from PS with immune-enhancing abilities.
Research shows that the administration of APS can improve immune function in mice by significantly increasing the spleen and thymus indices, the spleen lymphocyte activity and the WBC population in mouse serum.20 It may be possible to derive an OS from APS that can activate the proliferation of lymphocytes.
There are two main methods to derive OS from PS. One is enzyme catalysis and the other is acid hydrolysis. Our previous studies have demonstrated that APS is mainly composed of (1→3)-linked glucan.21 Although enzyme catalysis has been widely used in the hydrolysis of PS, no enzyme has yet been discovered that can degrade the type of glycosidic bond that exists in APS. Research indicated that OS could be effectively derived from PS by acid degradation. This process is relatively effective, economical and simple for the preparation of certain OS. For example, only a few days were required to derive an OS from Crassostrea gigas.22
In the present study, APS was extracted from Astragalus membranaceus (AM) and acid-hydrolyzed into AOS. To elucidate its structure, AOS was isolated and purified by membrane dialysis and silicon gel chromatography. Then, chemical methods were applied to analyze its monosaccharide composition, molecular weight and glycosidic linkages. Afterward, the ability of AOS to restore cyclophosphamide-induced immunosuppression in mice was assessed.
2. Materials and methods
2.1. Materials
AM (Fisch) Bunge var. mongholicus (Bunge) Hsiao was purchased from Shanghai Medicinal Materials (Shanghai, China). Identification was carried out by the Department of Authentication of Chinese Medicine, Hubei College of Chinese Traditional Medicine (Hubei, China). A voucher specimen (No. AM201003) was deposited in the herbarium of the Medical College, Tianjin University of Science and Technology, China.
2.2. Preparation of the AOS
APS was extracted with optimized techniques using the previously described method of direct water extraction.21 Briefly, the raw materials were first dried and then ground into fine powder. The powder (100 mg) was suspended in 500 mL of distilled water at 80 °C for 2 h before the supernatants were collected by centrifugation. This process was then repeated two more times. The extracts were gathered in a container, and four volumes of 95% (v/v) ethanol were added; precipitation occurred overnight at 4 °C before centrifugation at 4500 × 1000 rpm for 20 min.
Then, the collected precipitates were further dissolved in 100 mL distilled water, treated with Sevag reagent to remove proteins, and lyophilized to obtain APS. Afterward, APS (0.05 g) was dissolved in 50 mL 1 M H2SO4 and acid-hydrolyzed with boiling water for 2 h. Subsequently, the pH was adjusted to 7 with 1 M NaOH, followed by membrane dialysis with a molecular cutoff size of 100 Da in flowing deionized water for 24 h. Then, the reaction products were concentrated in a rotary evaporator and lyophilized. The lyophilized fractions were dissolved in distilled water to make a solution with a concentration of 2 mg mL–1. Then, 2 mL of solution was filtered through Sephadex-G15 equipped on a column (50 × 1.6 cm) at a flow rate of 1 mL min–1 with distilled water. The phenol-sulfuric acid method23 was used to monitor the purification process. The uniform fraction with a larger proportion was collected, concentrated in a rotary evaporator and lyophilized. The obtained product was named AOS.
2.3. Determination of the total carbohydrate content and the purity of AOS
The total carbohydrate content was identified using the phenol-sulfuric acid method.23 The purity of the AOS was further determined using a HPLC (Agilent-1200) equipped with an APS-2 HYPERSIL column (Thermo Fisher Scientific, USA) (4.5 mm × 300 mm, column temperature 30 °C) and a refractive index detector (RID, detecting temperature 35 °C). 20 μL sample solution (1 mg mL–1) was injected and eluted with a mobile phase consisting of acetonitrile and water (5 : 1) at 1.0 mL min–1.
2.4. Monosaccharide composition analysis
The AOS was acid-hydrolyzed with 2 M TFA at 110 °C in a sealed tube for 3 h. After TFA was removed, the product was acetylated and analyzed by GC.24 Acetylated d-glucose (d-Glc), d-galactose (d-Gal), l-rhamnose (l-Rha), d-xylose (d-Xyl), d-mannose (d-Man), and d-arabinose (d-Araf) were used as standards.
2.5. FT-IR and NMR analyses
Before the FT-IR analysis, a 1 mm thick disk was pressed with a mixture of 1 mg of sample and 150 mg of dried KBr. Afterward, the analysis was performed in the range of 4000 to 400 cm–1 on a Fourier transform IR spectrophotometer (VECTOR-22).251H NMR and 13C NMR spectra were recorded on a Bruker spectrometer (400 MHz) at a probe temperature of 298 K after the freeze-dried sample was exchanged twice with D2O.26
2.6. Periodate oxidation and Smith degradation analysis
Periodate oxidation and Smith degradation analysis were performed based on literature methods.25
2.7. Methylation of AOS
The AOS (0.0263 g) was methylated initially by the Hakomori method27 using sodium hydride–dimethyl sulfoxide. The methylated oligosaccharide was recovered by chloroform extraction. After evaporation of the chloroform extracts to dryness, the partially methylated AOS was subjected to the Purdie and Irvine method (1904)28 for complete methylation. Briefly, partially methylated AOS was dissolved in methyl iodide (5 mL) with stirring under inert atmosphere. Silver oxide (0.50 g) was then added periodically. Then, the reaction was continued for 6 h. This process was repeated two times on successive days. The fully methylated AOS was extracted further, as above. The final product obtained was a dark brown syrup with a yield of 0.0057 g.
2.8. Alditol acetates
The pre-methylated AOS was degraded by 2 mL 2 M TFA under 110 °C for 3 h. Then, the TAF residues were removed by reduced pressure distillation 3 to 5 times after the addition of 2 mL methanol. Then, the degraded AOS was acetylated by the method described in 2.4. The final products were dissolved in 4 mL of trichloromethane and washed with deionized water twice before GC-MS analysis.
2.9. ESI-MS
The mass spectrometer (model 9.4 T Solarix; Bruker Daltonics, Bremen, Germany) was set to negative ion mode, ESI(–), over a mass range of m/z 150 to 1500. The ESI source conditions were as follows: a nebulizer gas pressure of 1.4 bar, a capillary voltage of 3.8 kV, and a capillary transfer temperature of 200 °C. Ion time accumulation was 0.010 s. ESI(–)FT-ICR mass spectra were acquired by accumulating 32 scans of time-domain transient signals in 16 mega-point time-domain data sets. All mass spectra were externally calibrated using NaTFA (m/z from 0 to 1400). The mass spectra were acquired and processed using Data Analysis software (Bruker Daltonics, Bremen, Germany). The elemental compositions of the compounds were determined by measuring the m/z values. The proposed structures for each formula were assigned using the ChemSpider (; www.chemspider.com) database. The degree of unsaturation for each molecule can be deduced directly from its DBE value according to the equation DBE = c – h/2 + n/2 + 1, where c, h, and n are the numbers of carbon, hydrogen, and nitrogen atoms, respectively, in the molecular formula.
Tandem mass spectrometry (MS2) experiments were performed on a quadrupole analyser coupled to the FT-ICR mass spectrometer. The ESI(–)-MS/MS spectra were acquired under the following conditions: infusion flow rate of 5 μL min–1, capillary voltage of 3.0 kV, nebulising temperature of 250 °C, argon as collision gas, ion accumulation time of 1 s, isolation window of 1.0 (m/z units), and 25% to 45% eV of collision energy.
2.10. Animal experiments
2.10.1. Animals
70 female Kunming mice weighing 20 ± 2 g were provided by the Experimental Animal Center of the China Academy of Chinese Medical Sciences. They were maintained in a pathogen-free environment (23 ± 2 °C, 55 ± 5% humidity) on a 12 h light/12 h dark cycle with a standard pellet diet and water supplied ad libitum throughout the experimental period. Animal procedures were performed in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals procedures, which were approved by the Animal Ethics Committee of the China Academy of Chinese Medical Sciences.
2.10.2. AOS dosage
The AOS dosage was set previously by applying it at several high levels of 2000 mg kg–1, 1000 mg kg–1, 500 mg kg–1 and 300 mg kg–1 to a group of normal mice under the protocol described in 2.10.3.
2.10.3. Animal experiments and treatment protocol
Seventy female Kunming mice were grouped (n = 10) as follows: model group, normal group, lithium carbonate group with a dose of 200 mg kg–1 (as positive control), a medium dose APS group with a dosage of 200 mg kg–1, a high dosage AOS group, a medium dosage AOS group and a low dosage AOS group; the dosages were 100 mg kg–1, 200 mg kg–1 and 300 mg kg–1, respectively. The mice in the model and normal groups were given the same volumes of physiologic saline instead of the test solution. All the groups were processed through intragastric administration by the described method for 15 days. During this period, all the groups except for the normal group were injected with 100 mg kg–1 of cyclophosphamide (CY) at the armpit of the right hind limb subcutaneously under aseptic conditions for 3 consecutive days by the ninth day. Four days after this period, all the mice were executed with the following procedure.29 Firstly, 100 μL of blood were taken from the eyes of each mouse using metal plumbing through the eyes. Then, all the mice were processed by cervical dislocation. The spleen and thymus of the mice were excised and weighed on a balance after being rinsed with Hank's solution. The nucleate marrow cells (NMCs) were then extracted and resuspended with 5 mL of complete RPMI-1640 intermediate to calculate the population of NMC under a microscope. Successively, the levels of HGB and the WBC and RBC populations in the serum were measured by a globulimeter (BC-5500, Shanghai, China).
Finally, the GM-CSF content in the mouse serum was assessed. Briefly, the supernatants of the serum of the mice were collected by centrifugation at 2000 rpm for 10 min at 4 °C or below. The subsequent detections were conducted according to the procedures recommended in the manual of the commercial GM-CSF (CSF-2) Mouse ELISA kit provided by Abcam Company. Briefly, 50 μL of all samples or standards were added to a 96-well microtitre plate. Next, 50 μL of antibody cocktail were added to each well. Then, the plates were incubated for 1 h at room temperature on a plate shaker set to 400 rpm before each well was washed with 350 μL 1× Wash Buffer PT three times. Afterward, 100 μL of TMB substrate was added to each well and incubated for 10 min in the dark on a plate shaker set to 400 rpm. Finally, 100 μL of Stop Solution was added to each well. The plates were shaken on a plate shaker for 1 min to fully mix the solutions. The absorbance was then measured at 450 nm on an automatic ELISA plate reader. Finally, the GM-CSF level was calculated according to the standard curve equation of GM-CSF.
2.11. Statistical analysis
Data are expressed as mean ± S.D. Statistical analysis was performed by one-way analysis of variance followed by Sidak's multiple comparison test using GraphPad software (GraphPad Prism 5.04; San Diego, CA); p values < 0.05 were considered significant, and p values < 0.01 were considered extremely significant.
3. Results and discussion
3.1. Chemical content and monosaccharide composition
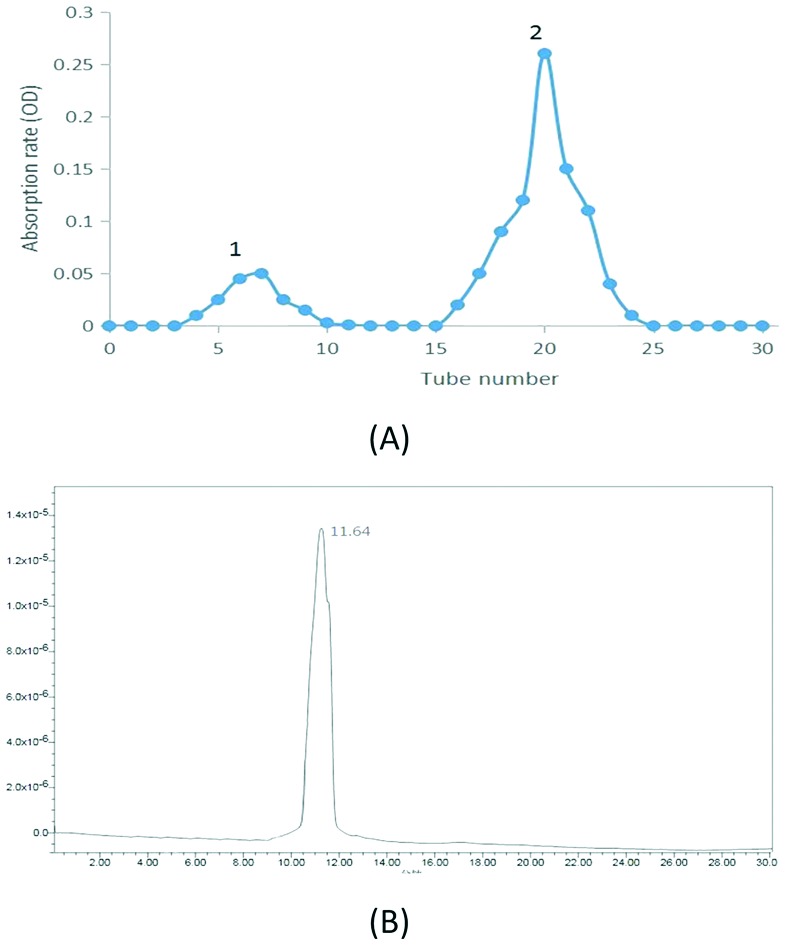
As shown in Fig. 1A, the hydrolyzed APS were mainly composed of two fractions. The second fraction with a higher proportion and lower molecular weight was collected and named AOS. According to the results, 0.036 g AOS were obtained by the process. The yield was calculated as 72.0% (0.036/0.05). The total sugar content was calculated as 98.5% by the phenol-sulfuric acid method; the standard curve was measured to be y = 0.001x + 0.01273, R2 = 0.9994.
Fig. 1. (A) Elution profile of hydrolyzed APS, (B) HPLC profile of AOS.

Its specific rotation was found to be [α]25°CAOS = –35° (25 °C, 0.001 g mL–1, H2O). The single and symmetric peak that appeared in the HPLC profile of AOS (Fig. 1B) indicated the effectiveness of the purification method and that AOS was an OS with a uniform molecular weight.
The monosaccharide compositions of AOS are shown in Fig. 2. The results showed that AOS was primarily composed of Rha, Araf, Glc and Gal in a mole ratio of 1 : 0.95 : 1.24 : 1.22.
Fig. 2. (A) Standard monosaccharide. (B) GC spectra of derivatives from the monosaccharide composition of AOS. Peaks: (1) l-Rhamnose, (2) d-arabinose, (3) d-xylose, (4) d-mannose, (5) d-glucose, (6) d-galactose, (7) internal standard.

3.2. FT-IR analysis
As shown in Fig. 3, the broad stretching peaks at 3405.06 cm–1 were ascribed to the stretching vibration of hydroxyl groups. The weak absorption peaks at 2929.15 cm–1 were characteristic of C–H stretching vibrations. In addition, the strong absorption peak at 1636.88 cm–1 was indicative of the C O stretching vibration. The band at 1412.45 cm–1 was assigned to the C–H bending vibration.
Fig. 3. FT-IR spectrum of AOS.

3.3. NMR analysis
Normally, the configuration of anomeric carbon atoms is distinguished by the coupling constant (J) in the 1H NMR spectrum. Due to the fact that some peaks appeared as broad singlets, the proton–proton coupling constants could not be measured. Therefore, the anomeric configuration was determined based on the chemical shifts of the anomeric protons (typically δ 5.2 to 5.0 for α-sugars and δ 4.6 to 4.3 for β-sugars).30 Thus, the anomeric signals at 5.338 (5.328), 5.034, 5.158 (5.148), 5.120 (5.111), 5.034 and 5.034 were determined to be the peaks of α-Glc, Gal and Araf, respectively, while the peaks at 4.788 and 4.587 (4.507) were assigned to β-Gal and Rha, respectively (Fig. 4A). The signals at 1.190, 1.205 and 1.223 ppm were attributed to the methyl group of Rha and the methylene group of Araf.31 The signal at 4.70 ppm was solvent resonance (Fig. 4A). The identification of peaks in the 13C NMR spectrum of AOS (Fig. 4B) was based on the analysis of the results of the 1H NMR spectrum. The signals at 99.633, 99.550 (96.626), 95.771 (93.984), 93.505, and 92.234 (91.892) were assigned to the anomeric carbons (C-1) of β-Gal, β-Rha, α-Glc, α-Gal and α-Araf, respectively.32 The peaks at 16.81 and 16.77 ppm corresponded to the methyl group of Rha and the methylene group of Araf.
Fig. 4. (A) 1H NMR and (B) 13C NMR spectra of AOS.

3.4. Linkage analysis of AOS
The periodate oxidation results are shown in Table 1.
Table 1. Results of the periodate oxidation of AOS.
| Glycosyl group (mmol) | Consumption of periodate (mmol) | Formic acid generation (mmol) | |
| AOS | 0.139 | 0.125 | 0.061 |
After Smith degradation of the oxidized product of AOS, glycerol (Gly), Gal, Rha and Araf were found in the degradation products. The molar ratio of Gal, Gly, Rha and Araf was 1 : 2.58 : 1.86 : 1.51 in AOS.
The individual peaks of the degradated premethylated AOS from the GC-MS analysis were identified by their retention times and by comparison with the mass spectrum. As listed in Table 2, the molar ratio of each sugar residue was calculated from the peak area in the GC-MS chromatogram. As shown in Table 2, the results showed the presence of six derivatives, namely 2,3,4,6-tetra-O-Me-d-Gal, 2,3,4,6-tetra-O-Me-d-Glc, 2,4,6-tri-O-Me-Gal, 1,2,4-tri-O-Me-d-Rha, 2,4-d-O-Me-d-Rha and 2-O-Me-l-Araf, at a molar ratio of 1 : 2.07 : 0.86 : 1.02 : 1.21 : 1.87.
Table 2. Results of the methylation analysis of AOS.
| T R/min | Methylation sugar residue | Linkage type | Relative molar ratio | Major mass fragments |
| 16.415 | 2,3,4,6-tetra-O-Me-d-Gal | Gal(1→ | 1 | 43.0, 45.0, 71.9, 101.0, 117.13, 145, 161, 205 |
| 16.949 | 2,3,4,6-tetra-O-Me-d-Glc | Glc(1→ | 2.07 | 43.0, 45.0, 100.0, 117.2, 161.0, 205.0 |
| 17.168 | 2,4,6-tri-O- Me-d-Gal | 3→)Gal(1→ | 0.86 | 43.0, 86.9, 129.0, 189.0 |
| 17.212 | 1,2,4-tri-O-Me-d-Rha | Rha(3→ | 1.02 | 43.0, 88.0 |
| 18.247 | 2,4-d-O-Me-d-Rha | 3→)Rha(1→ | 1.21 | 43.0, 88.0 |
| 18.924 | 2-O-Me-l-Araf | →3,4)Araf(1→ | 1.87 | 43.0, 85.0, 101.0, 117.0 |
3.5. Mass spectral analysis
ESI(–)FT-ICR mass spectra were obtained to evaluate the chemical profile of AOS (Fig. 5A). The acquisition conditions of the ESI source were kept constant in all analyses. The relevant ion fragments were listed as follows. Briefly, the value of m/z at 178.99 was projected to [C12H20O10 – H+ + Cl–]2– ion (calculated as (324 + 35.5–1)/2). The m/z values of 260.86 and 331.76 were attributed to [C17H26O14 + 2Cl–]2– and [C22H36O18 + 2Cl–]2– ions, respectively (calculated as (456 + 35.5 × 2)/2 and (592 + 35.5 × 2)/2). The values of m/z at 473.80, 899.74, and 1112.36 were assigned to the ions of [C17H30O15 – H+]–, [C34H58O27 – H+]– and [C40H70O33 + Cl–]–, respectively. Finally, the m/z value of 1255.03 was projected to [C46H78O37 + Cl–1]–. The structure of AOS can be verified as Fig. 5B according to the relevant analysis in the discussion.
Fig. 5. (A) Mass spectrum of AOS. (B) The structure of AOS.

3.6. Animal experiments
3.6.1. AOS dosage
The dosages were set according to the maximum human dietary intake of AM, the average PS content and the dose translation standard from human to animals. Death was observed in groups who were administered 1000 mg kg–1 and 2000 mg kg–1 after 3 days. Toxic effects such as weight loss and decreased food intake and activity were observed in the group administered with 500 mg kg–1. These phenomena suggested that these dosages were too high for the mice. None of these phenomena were observed in the group administered with 300 mg kg–1. Again, this dosage was translated to a human dietary intake of 2 g kg–1, which is within the average human daily intake of AM. Thus, this dosage was set as the highest level for the following experiment. This dosage was consistent with past studies of the immune-enhancing activity of APS.33
3.6.2. Effects on the indices of spleen and thymus
The spleen and thymus are crucial organs in the regulation of the immune system. The weights of these two organs were thus measured as a way to assess the physiological effects of AOS. According to the results (Table 3), compared with the normal group, the thymus figure for the model group was significantly lower than that of the normal group (P < 0.01), indicating that the immunosuppressed model was successfully established. The spleen and thymus figures of the AOS group were both significantly higher than those of the model group (P < 0.05) in a dose-dependent manner. This suggested that AOS can protect these two organs against the toxicity of CY. Similarly, the figures for the APS group were significantly higher (P < 0.05) than those of the model group and the AOS group (medium-dose). This saccharide may protect various organs from damage by stimulating immunosuppression inducers.
Table 3. Results of the immunity experiment.
| Normal | Model | Low-dose | Medium-dose | High-dose | Lithium carbonate | APS | |
| Dosage (mg kg–1) | 0 | 0 | 100 | 200 | 300 | 200 | 200 |
| Thymus weight (mg) | 48.77 ± 5.36 | 14.66 ± 2.57†† | 18.07 ± 4.38* | 19.44 ± 6.32* | 25.44 ± 4.28** | 28.57 ± 9.62** | 19.59 ± 3.73* |
| Spleen weight (mg) | 166.30 ± 21.36 | 119.50 ± 16.30†† | 147.47 ± 21.24** | 154.47 ± 14.73** | 160.95 ± 23.58** | 167.73 ± 31.67** | 151.29 ± 17.16** |
| WBC population (109/L) | 9.33 ± 2.32 | 5.42 ± 1.42†† | 7.74 ± 2.01** | 8.54 ± 3.25** | 8.73 ± 2.14** | 8.97 ± 2.35** | 8.41 ± 2.51** |
| RBC population (1012/L) | 9.26 ± 1.26 | 7.51 ± 1.59† | 8.04 ± 2.14* | 8.34 ± 2.73* | 8.79 ± 3.01* | 9.30 ± 2.82* | 9.14 ± 2.78* |
| Hemoglobin concentration (g L–1) | 174.26 ± 23.85 | 146.33 ± 32.64† | 142.50 ± 21.52* | 157.84 ± 42.85* | 169.14 ± 51.45** | 180.17 ± 23.64* | 165.65 ± 48.19* |
| Nucleated marrow cell population (106/L) | 119.26 ± 29.21 | 34.52 ± 11.36† | 65.18 ± 10.21* | 71.26 ± 14.58* | 88.91 ± 26.35** | 119.01 ± 29.34** | 82.61 ± 17.48* |
| GM-CSF level in peripheral blood (ng L–1) | 228.52 ± 56.32 | 125.32 ± 41.24†† | 190.31 ± 32.97** | 223.63 ± 25.73* | 256.71 ± 64.37** | 265.32 ± 24.26* | 207.39 ± 30.34* |
3.6.3. Effects of AOS on serum HGB levels and populations of WBC and RBC
As shown in Table 3, compared with the normal group, the levels of these three factors were remarkably down-regulated in the model group (p < 0.01), indicating that the immunosuppressed model was successfully established. All the AOS groups showed significant increases (P < 0.05) in these factors in a dose-dependent manner. The APS group was also higher (P < 0.05) than the module group. In addition, in the AOS group (medium-dose), the serum HGB levels and the population of WBC were higher than in the APS group. One difference was that in the AOS group (medium-dose), the population of RBC was lower than in the APS group. That is, AOS and APS demonstrated certain immune characteristics.
3.6.4. Effects on the NMC population
A change in the number of bone marrow nucleated cells can reflect the hematopoietic ability of bone marrow and is directly related to the number of peripheral blood leukocytes. After the injection of CY, a dramatic decline in the NMC population was shown in the model group compared with the normal group (P < 0.05); this suggested that the immunosuppressed model was established successfully (see Table 3). Compared with the model control group, the NMC of the AOS group was significantly increased (P < 0.05), and there were no obvious differences with lithium carbonate administration (P > 0.05) for the intermediate-dose and high-dose groups; this suggests that AOS has a positive effect on the nucleated marrow cell population at the tested level. Compared with the APS group, the NMC population of the AOS group was slightly higher at the same dosage. Our result is consistent with some other studies. Yang et al. found that APS could reconstitute bone marrow in a myelosuppressed animal model.29
3.6.5. Effects on GM-CSF levels in peripheral blood
To further understand the mechanism of the performance-enhancing effects of AOS on the immune system, the GM-CSF levels in the peripheral blood of the mice were tested. The standard curve equation of GM-CSF was found to be y = 0.0004x – 0.0018, where y represents the absorbance rate of each sample at the wavelength of 450 nm and x represents the l g value of the GM-CSF content in each sample. The standard curve was adjusted to the correlation coefficient of 0.9964.
The GM-CSF level of the model group was significantly lower than that of the experimental group (P < 0.01), which suggested that the immunosuppressed model was established successfully. This figure was significantly higher in all AOS groups than in the model group (P < 0.05) in a dose-dependent manner. The highest GM-CSF level of 256.7 ng L–1 was observed at the highest dosage; this value is almost the same as that of the positive control. At the same time, the GM-CSF level of AOS was higher than that of APS. This is consistent with several previous studies. Min et al. suggested that APS can enhance the expression of various cytokines, including GM-CSF.34–36 Shao et al. also suggested that the extractions from the roots of AM PS could stimulate the secretion of several cytokines.37
4. Discussion
AOS was obtained from AM by acid hydrolysis and was purified by membrane dialysis and silicon gel chromatography. The purified and unified AOS, with slight impurities, contained about 98.5% carbohydrate content and was mainly composed of Rha, Araf, Glc and Gal in a mole ratio of 1 : 0.95 : 1.24 : 1.22. This is consistent with the experimental result of PS from Pu et al.,38 who reported that the major sugar constituents of a PS extracted from Astragalus were Rha, Gal, Araf and Glc.
The characteristic peaks of the OS were found in the FT-IR and NMR spectra. β-d-Glc, α-d-Rha, α-d-Araf and α-d-Gal are contained in AOS, judging from the results of 1H and 13C NMR spectroscopy. The molar ratios of each permethylation derivative were consistent with the results of GC analysis.
The periodate oxidation of AOS produced 0.061 mmol formic acid, which is indicative of 1→ or 1→6 linkages. The fact that 0.122 mol (about 0.125 mmol, two times the production of formic acid) periodate was consumed indicated the absence of 1→2, 1→2,6, 1→4 and 1→4,6 linkages, which consume periodate without producing formic acid. The periodate oxidation of AOS consumed 0.125 mol of periodate (less than 0.139 mmol, the content of glycosyl groups). This indicated the presence of a 1→3 linkage in AOS because less than 1 mol of periodate was consumed per mole of Glc residue; the 1→3 linkage does not consume periodate during the oxidation.39
According to the Smith degradation results, the presence of Araf and Rha indicated the existence of several types of linkages, such as 1→3, 1→3,6, 1→2,3, 1→2,4, 1→3,4, and 1→2,3,4 linkages, or non-reducing terminals which cannot be oxidized.40
The absence of erythritol, which is a chemical indicative of 1→4 and 1→4,6 linkages, suggested that these linkages were not present. The presence of Gly suggested that 1→, 1→6, 1→2 or 1→2,6 linkages, which produce Gly, might exist. The periodate oxidation-Smith degradation results showed that 1→, 1→3, 1→3,6, 1→2,3, 1→2,4, 1→3,4 or 1→2,3,4 linkages or non-reducing terminals might exist in AOS.
Specifically, the GC-MS analysis results indicated that AOS consisted of (1→)-linked-Gal, (1→)-linked-Glc, (1→3)-linked-Gal, (3→)-linked-Rha, (1→3)-linked-Rha and (1→3,4)-linked-Araf in a molar ratio of 1 : 2.07 : 0.86 : 1.02 : 1.21 : 1.87. This was in accordance with the monosaccharide analysis results as well as with the results of periodate oxidation and Smith degradation, which showed that 1→3, 1→4, 1→2,3, 1→6 and 1→3,6 linkages or non-reducing terminals might exist in the structure of AOS. Additionally, this finding was found to be in agreement with references which studied the linkages existing in the PS of AM.38,41 The similarity of linkage types between AOS and previous studies of APS is due to the fact that AOS is obtained from APS and may share similar structures.
Several molecular formulas were deduced from the mass spectra analysis. Because they were fractions of AOS, the structure of AOS could be verified by identification of these fractions. Specifically, C12H20O10, C17H26O14, C17H30O15, C34H58O27, and C46H78O37 consisted of a molecule of Rha and Gal; a molecule of Glc, Rha and Araf; a molecule of Gal, Araf and Glc; two molecules of Gal, Glc, Araf and one molecular of Rha; and two molecules of Gal, Glc, Araf and Rha, respectively. The specific structure of AOS could thus be deduced in relation to the results of GC-MS and NMR analysis.
Furthermore, the efficacy profiles of low-, intermediate- and high-dose AOS treatment for enhancing immune function in immunosuppressed Kunming mice were determined. Compared with immunosuppressive drugs, AOS has the advantages of safety, efficiency, low toxicity, low residue and low cost. This natural product has excellent development potential and good application prospects.42 The major findings were that in CY-induced immunosuppressed mice, chronic treatment with AOS resulted in (1) initiation of the restoration of WBC, NMC, and RBC populations and HGB levels; (2) acceleration of the recovery of spleen and thymus indices; (3) recovery of the GM-CSF levels in serum. None of the mice treated with AOS died, nor did their body weights change significantly compared with the normal group (P > 0.05, data not shown) during the experiment period. The results were consistent with the fact that AOS was derived from AM and might share some immunomodulating activities with AM.37,43 According to the results of the study, the mechanism of these effects was enhancement of the effects of terminal β-glucans existing in the structure of AOS toward the secretion of GM-CSF in serum. These effects of β-glucans were reported in in vitro experiments, which showed that β-glucans could generate the secretion of proinflammatory mediators such as cytokines and chemokines, leading to the direct activation of leukocytes.44 GM-CSF is a cytokine capable of stimulating the formation of neutrophil colonies and macrophage colonies in bone marrow cells; it plays an important role in the differentiation of progenitor cells, including nucleus pulposus nucleated cells, after proliferation. AOS exhibited significant immunomodulating effects by stimulating the proliferation of human peripheral blood mononuclear cells and enhancing interleukin production.45 Because the GM-CSF level was associated with promotion of the differentiation of progenitor cells, including marrow nucleated cells, after proliferation, the increasing level of GM-CSF eventually led to increases in the WBC and RBC populations.
5. Conclusions
In conclusion, an oligosaccharide was derived from APS by acid hydrolysis. Its structure has been elucidated. The immunological assay results showed that AOS can increase RBC, WBC and NMC populations and GM-CSF levels as well as other characteristics, such as the weight of immune organs in mice and HGB concentration in CY-induced immune-suppressed mice. The mechanism of the ability of AOS to proliferate the secretion of GM-CSF and thus restore immunosuppression was related to the two terminal β-glucans present in the structure. It is well known that cancer patients generally experience immunosuppression. Therefore, AOS could be considered and recommended for use as a useful adjuvant in the clinical treatment of cancer patients, especially when chemotherapy (CY or other drugs) is used, because of its elucidated structure and ability to restore CY-induced immunosuppression.
Conflict of interest
The authors declare no competing interests.
Acknowledgments
This work was financially supported by the key program of the Natural Science Foundation of Tianjin (16JCZDJC34100) and the National Spark Key Program of China (2015GA610001).
References
- Lyman G. H., Lyman C. H., Agboola O. Oncologist. 2005;10(2):427–437. doi: 10.1634/theoncologist.10-6-427. [DOI] [PubMed] [Google Scholar]
- Crawford J., Dale D. C., Lyman G. H. Cancer. 2004;100(2):228–237. doi: 10.1002/cncr.11882. [DOI] [PubMed] [Google Scholar]
- Franklin J., Diehl V. Ann Oncol. 2002;13(suppl_1):98–101. doi: 10.1093/annonc/13.s1.98. [DOI] [PubMed] [Google Scholar]
- Diehl V., Franklin J., Pfreundschuh M., Lathan B., Paulus U., Hasenclever D., Tesch H., Herrmann R., Dörken B., Müller-Hermelink H. K., Dühmke E., Loeffler M. N. Engl. J. Med. 2003;348:2386–2395. doi: 10.1056/NEJMoa022473. [DOI] [PubMed] [Google Scholar]
- Li H., Ma Q., Ai P., Ma L. X. Chin. J. Integr. Med. 2015;35(2):157–166. [Google Scholar]
- Kuderer N. M., Dale D. C., Crawford J., Cosler L. E., Lyman G. H. Cancer. 2006;106(10):2258–2266. doi: 10.1002/cncr.21847. [DOI] [PubMed] [Google Scholar]
- Lyman G. H., Dale D. C., Friedberg J., Crawford J., Fisher R. I. J. Clin. Oncol. 2004;22(21):4302–4311. doi: 10.1200/JCO.2004.03.213. [DOI] [PubMed] [Google Scholar]
- Crawford J. Clin. Adv. Hematol. Oncol. 2013;11(8):514–517. [PubMed] [Google Scholar]
- Crawford J., Althaus B., Armitage J., Balducci L., Bennett C., Blayney D. W. J. Natl. Compr. Cancer Network. 2007;5(2):188–202. doi: 10.6004/jnccn.2007.0019. [DOI] [PubMed] [Google Scholar]
- Aapro M. S., Bohlius J., Cameron D. A., Lago L. D., Donnelly J. P., Kearney N., Lyman G. H., Pettengellt R., Tjan-Heijnenu V. C., Walewskiv J., Weberw D. C., Zielinski C. Eur. J. Cancer. 2011;47(1):8–32. doi: 10.1016/j.ejca.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Papaldo P., Lopez M., Marolla P., Cortesi E., Antimi M., Terzoli E., Vici P., Barone C., Ferretti G., Cosimo S. D., Carlini P., Nisticò C., Conti F., Lauro L. D., Botti C., Filippo F. D., Fabi A., Giannarelli D., Calabresiand F. J. Clin. Oncol. 2005;23(28):6908–6918. doi: 10.1200/JCO.2005.03.099. [DOI] [PubMed] [Google Scholar]
- Hershman D., Neugut A., Jacobson J. S., Wang J., Tsai W. Y., McBride R., Bennett C. L., Grann V. R. J. Natl. Cancer Inst. 2007;99(3):196–205. doi: 10.1093/jnci/djk028. [DOI] [PubMed] [Google Scholar]
- Clavarezza W. J. M., Retsky M., Baum M., Demicheli R. J. Natl. Cancer Inst. 2007;99(13):1050–1051. doi: 10.1093/jnci/djm014. [DOI] [PubMed] [Google Scholar]
- Feng H., Du X., Liu J., Han X., Cao X., Zeng X. Int. J. Biol. Macromol. 2014;65:121–128. doi: 10.1016/j.ijbiomac.2014.01.017. [DOI] [PubMed] [Google Scholar]
- Gao Z. Z., Liu K. H., Tian W. J., Wang H. C., Liu Z. G., Li Y. Y., Li E. T., Liu C., Li X. P., Hou R. R., Yue C. J., Wang D. Y. Int. Immunopharmacol. 2015;27(1):104–109. doi: 10.1016/j.intimp.2015.04.052. [DOI] [PubMed] [Google Scholar]
- Ren Z., He C. H., Fan Y. H., Si H. M., Wang Y. W., Shi Z. Y., Zhao X. M., Zheng Y. T., Liu Q. X., Zhang H. B. Int. J. Biol. Macromol. 2014;70:590–595. doi: 10.1016/j.ijbiomac.2014.07.044. [DOI] [PubMed] [Google Scholar]
- Swennen K., Courtin C. M., Delcour J. A. Crit. Rev. Food Sci. Nutr. 2006;46(6):459–471. doi: 10.1080/10408390500215746. [DOI] [PubMed] [Google Scholar]
- Roberfroid M., Gibson G. R., Hoyles L., McCartney A. L., Rastall R., Rowland I., Wolvers D., Watzl B., Szajewska H., Stahl B., Guarner F., Respondek F., Whelan K., Coxam V., Davicco M., Léotoing L., Wittrant Y., Delzenne N. M., Cani P. D., Neyrinck A. M., Hoyles A. L. Br. J. Nutr. 2010;104(S2):S1–S63. doi: 10.1017/S0007114510003363. [DOI] [PubMed] [Google Scholar]
- Nauta A. J., Garssen J. Carbohydr. Polym. 2013;93(1):263–265. doi: 10.1016/j.carbpol.2012.02.021. [DOI] [PubMed] [Google Scholar]
- Zhong R. Z., Yu M., Liu H. W., Sun H. X., Cao Y., Zhou D. W. Anim. Feed Sci. Technol. 2012;174(1–2):60–67. [Google Scholar]
- Li J. J., Sun H., Sun D., Yao Y. L., Yao F. L., Yao K. J. Carbohydr. Polym. 2011;85(4):895–902. [Google Scholar]
- Wu S. J., Yu L. Food Chem. 2015;181:15–18. doi: 10.1016/j.foodchem.2015.02.066. [DOI] [PubMed] [Google Scholar]
- Zhang D. Y., Li S. J., Xiong Q. P., Jiang C. P., Lai X. P. Carbohydr. Polym. 2013;95(1):114–122. doi: 10.1016/j.carbpol.2013.03.015. [DOI] [PubMed] [Google Scholar]
- Chen G. H., Kan J. Q., Li Z. X., Chen Z. D. Carbohydr. Polym. 2005;61(2):125–131. [Google Scholar]
- Dou J., Meng Y. H., Liu L., Li J., Ren D. Y., Guo Y. R. Int. J. Biol. Macromol. 2015;72:31–40. doi: 10.1016/j.ijbiomac.2014.07.053. [DOI] [PubMed] [Google Scholar]
- Leeuwen S. S. V., Leeflang B. R., Gerwig G. J., Kamerling J. P. Carbohydr. Res. 2008;343:1114–1119. doi: 10.1016/j.carres.2008.01.043. [DOI] [PubMed] [Google Scholar]
- Hakomori S. I. J. Biochem. 1964;55(2):205–208. [PubMed] [Google Scholar]
- Purdie T., Irvine J. C. J. Am. Chem. Soc. 1904;85:1049–1070. [Google Scholar]
- Yang B., Xiao B., Sun T. Y. Int. J. Biol. Macromol. 2013;62:287–290. doi: 10.1016/j.ijbiomac.2013.09.016. [DOI] [PubMed] [Google Scholar]
- Synytsya A., Novak M. Ann. Transl. Med. 2014;2(2):17. doi: 10.3978/j.issn.2305-5839.2014.02.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone S., Molinaro A., Dubery I., Lanzetta R., Parrilli M. Carbohydr. Res. 2007;342(11):1514–1518. doi: 10.1016/j.carres.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Zou C., Du Y. M., Li Y., Yang J. H., Tao F., Zhang L., Kennedy J. F. Carbohydr. Polym. 2008;73(2):322–331. [Google Scholar]
- Li R., Chen W. C., Wang W. P., Tian W. Y., Zhang X. G. Carbohydr. Polym. 2009;78(4):738–742. [Google Scholar]
- Yang M., Lin H. B., Gong S. T., Chen P. Y., Geng L. L., Zeng Y. M., Li D. Y. Cytokines. 2014;70(2):81–86. doi: 10.1016/j.cyto.2014.07.250. [DOI] [PubMed] [Google Scholar]
- Liu Q. Y., Yao Y. M., Zhang S. W., Sheng Z. Y. J. Ethnopharmacol. 2011;136(3):457–464. doi: 10.1016/j.jep.2010.06.041. [DOI] [PubMed] [Google Scholar]
- Yuan C. T., Pan X. P., Gong Y., Xia A. X., Wu G. H., Tang J. Q., Han X. D. Int. Immunopharmacol. 2008;8(1):51–58. doi: 10.1016/j.intimp.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Shao B. M., Xu W., Dai H., Tu P. F., Li Z. J., Gao X. M. Biochem. Biophys. Res. Commun. 2004;320(4):1103–1111. doi: 10.1016/j.bbrc.2004.06.065. [DOI] [PubMed] [Google Scholar]
- Pu X. Y., Ma X. L., Liu L., Ren J., Li H. B., Li X. Y., Yu S., Zhang W. J., Fan W. B. Carbohydr. Polym. 2016;137:154–164. doi: 10.1016/j.carbpol.2015.10.053. [DOI] [PubMed] [Google Scholar]
- Wang Z. M., Peng X., Lee K. L. D., Tang J. C., Cheung P. C. K., Wu J. Y. Food Chem. 2011;125(2):637–643. [Google Scholar]
- Zhang Y., Gu M., Wang K. P., Chen Z. X., Dai L. Q., Liu J. Y., Zeng F. Fitoterapia. 2010;81(8):1163–1170. [Google Scholar]
- Yang F., Xiao C. Y., Qu J., Wang G. Y. Carbohydr. Polym. 2016;145:48–55. doi: 10.1016/j.carbpol.2016.03.024. [DOI] [PubMed] [Google Scholar]
- Jiang C. L., Tang C., Qian Y., Li L. Food Sci. 2013;11:327–332. [Google Scholar]
- Cho W. C. S., Leung K. N. J. Ethnopharmacol. 2007;113(1):132–141. doi: 10.1016/j.jep.2007.05.020. [DOI] [PubMed] [Google Scholar]
- Schepetkin I. A., Quinn M. T. Int. Immunopharmacol. 2006;6(3):317–333. doi: 10.1016/j.intimp.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Jun J. Y., Chan B. C. L., Yu H., Lau I. Y. K., Han X. Q., Cheng S. W., Lau C. B. S., Xie M. Y., Fung K. P., Leung P. C. Carbohydr. Polym. 2012;87(1):667–675. doi: 10.1016/j.carbpol.2011.08.045. [DOI] [PubMed] [Google Scholar]