

 This review summarizes recent work directed toward the discovery of selective inhibitors of the protein–protein interactions between RPA and proteins involved in the initiation of DNA damage response pathways.
This review summarizes recent work directed toward the discovery of selective inhibitors of the protein–protein interactions between RPA and proteins involved in the initiation of DNA damage response pathways.
Abstract
Due to the relatively high rate of DNA damage that can occur during cell cycle progression, the DNA damage response (DDR) pathway is critical for the survival of eukaryotic cells. Replication protein A (RPA) is an essential cell cycle checkpoint protein that mediates the initiation of the DDR by binding to single-stranded DNA (ssDNA) and recruiting response partners via protein–protein interactions (PPIs). This important role of RPA in initiating the DDR and cell survival has led to interest within the scientific community to investigate RPA as a potential cancer drug discovery target. To this end, RPA inhibitors have been explored via a variety of methods. This review summarizes the structure and function of RPA and highlights recent efforts to discover inhibitors of RPA–protein interactions.
Introduction
Since its initial purification and characterization in 1988 by Wold and Kelly, replication protein A (RPA) has been extensively studied to elucidate both its structure and function.1 From these studies, the critical role of RPA as both an initiator and regulator of the DNA damage response (DDR) has become increasingly clear. RPA has also been shown to play a role in other crucial processes, including DNA replication, telomere maintenance, and checkpoint activation. The pivotal role of RPA in the response to genomic stress and DNA damage has led many researchers to investigate the role of RPA in cancer. In the past several years, research on RPA has involved the investigation of the role of the protein as a cell checkpoint regulator and initiator of the DDR response as well as the influence of RPA and its effector proteins on cell survival. Specifically, inhibiting RPA from initiating the DDR is an intriguing possible target for drug discovery and a possible new approach for cancer therapy. One strategy for inhibiting RPA-mediated activation of the DDR is to interfere with its ability to interact with other proteins. The discovery and optimization of protein–protein interaction (PPI) inhibitors mediated by RPA has become a rich and interesting field, utilizing in silico methods, high throughput screening (HTS) campaigns, fragment-based drug discovery (FBDD), and structure-based methods.
RPA structure and function
RPA is a heterotrimeric ssDNA-binding protein composed of 70, 32, and 14 kDa subunits, and is essential for eukaryotic DNA replication, damage response and repair (Fig. 1).1,2 The RPA70 subunit is comprised of four domains (A, B, C, and N). Each of these domains contains an oligonucleotide/oligosaccharide binding (OB) fold. The OB fold is formed from a series of beta strands that, in turn, form beta-barrels. RPA70A (amino acids 181–290) and RPA70B (amino acids 301–422) are high affinity DNA binding domains and are responsible for anchoring the binding of RPA to ssDNA.2–5 RPA70C (amino acids 436–616) also contains an OB fold that binds to ssDNA, but with much lower affinity.4–7 RPA70N is the N-terminal domain of the RPA70 subunit (amino acids 1–110)8 and is attached to RPA70A by a flexible 70 amino acid residue linker. This domain also contains an OB fold, but does not bind ssDNA with high affinity. The RPA32 subunit consists of an OB fold (D domain), which helps RPA bind ssDNA, several phosphorylation sites that help regulate DNA metabolism, and a domain that interacts with proteins.4,5,9–20 The RPA14 subunit also contains an OB fold. It does not bind ssDNA, but is instead important to the stability of the heterotrimer.7,21
Fig. 1. Multi-domain structure of RPA. RPA70 (gold) is comprised of OB-fold-containing high affinity ssDNA binding domains and the RPA70N domain that mediates several PPIs. RPA32 (maroon) and RPA14 (orange) also contain OB-folds.

Through the interactions of the A, B, C, and D domains of RPA with DNA, RPA serves a protective function, preventing formation of aberrant DNA structures at replication foci.9,12,22–24 RPA also interacts with a wide range of DNA processing proteins through its 70N and 32C domains.2,9,12,22,23,25–27 RPA70N utilizes a shallow basic cleft that binds the acidic helices of various protein binding partners, such as p53, Rad9, ATRIP, and Mre11.28 Thus, RPA functions as a scaffold upon which DNA processing proteins assemble and initiate the DDR pathway in both the G1/S and G2/M cell cycle checkpoints (Fig. 1).4,5,23,24,29,30
Disruption of the protein–protein interactions of RPA70N by mutation of either RPA or its binding partners leads to decreased signalling through ATR and increased sensitivity to DNA damage and replication stress.24,31 Removal of the entire RPA protein through the use of siRNA, however, is cytotoxic to cells, as would be expected given the essential role of this protein in DNA metabolism.32 Blocking the function of RPA might be accomplished by either disrupting the ssDNA interactions of RPA70A–D or by blocking the ability of the 70N subunit to recruit DNA processing proteins. Affecting the ability of RPA to bind ssDNA may be expected to present a cytotoxic phenotype similar to the results of siRNA knockouts and may not allow a suitable therapeutic window. Despite this possible liability, Turchi and colleagues have identified several interesting inhibitors of RPA–ssDNA binding thus far.32–34
Alternatively, one can envision an inhibitor that would bind only the RPA70N protein-binding domain and block its interaction with DNA processing proteins. Such an inhibitor would allow RPA to bind ssDNA while selectively and simultaneously inhibiting several critical DNA damage response pathways, such as those involving ATR, p53, and Rad9. This should present a different phenotype from the full protein knockout, since, ideally, the protective ssDNA binding and other functions of RPA would not be affected. Indeed, it has become well-known that RNA-mediated silencing of a protein and genetic loss-of-function experiments can exhibit different effects than pharmacologically inhibiting the function of a protein with a small molecule.35 In this scenario, one may be able to achieve a therapeutic window, as initiation of the DDR in cancer cells would be inhibited. As cancer cells are undergoing higher replication stress than normal cells, this inhibition would thus be cytotoxic.
A molecule that binds to the basic cleft of RPA70N should interrupt pathways mediated by the interaction of this domain, but not affect those mediated by RPA32C.28 An important point to also consider is that a putative RPA inhibitor might exert differential effects on the different pathways downstream of RPA70N, due to the possibility for different potencies or kinetics of interrupting the interaction of RPA and its various effectors. Thus, a selective RPA PPI inhibitor may be useful for dissecting the specific contributions of individual RPA binding proteins to the biological function of the protein, or an analysis of specific toxicities that may be associated with the various downstream pathways.
One also has to consider the fact that a small molecule that blocks the interaction of RPA70N with all of its effector molecules may display a significant effect on several diverse biological processes. This may lead to expansion of the possible therapeutic utility of an RPA70N binder, or may lead to unacceptable toxicities. Ultimately, then, the discovery of RPA70N inhibitors would enable significant advancements in the field, while also presenting a possible therapeutic opportunity. The hypothesis of selective RPA-mediated PPI inhibition as a means to a therapeutic window has driven recent research on RPA inhibitors.
It should be noted that, while the discovery of PPI inhibitors of RPA70N might be a very attractive approach for potential therapeutic agents, it is potentially a difficult endeavour, since the basic cleft of RPA70N is rather shallow and exhibits significant flexibility.23 Further, the binding cleft of RPA contains three arginine residues (R31, R41, and R43) that all play a role in binding to the relatively conserved acidic helices present in all of RPA70N's binding partners. Thus, the identification of a small molecule that is capable of binding in a potent and selective manner to the fluid basic binding cleft of RPA may not be a trivial task.
High throughput screening
Fumaroprimaric acid
In 2011, Oakley and colleagues described the first inhibitor of the PPI between RPA70N and RAD9.31 In the study, a plate-based ELISA-like HTS assay was developed to assess the disruption of the interaction between a RPA70/ssDNA-biotin complex and GST-Rad9. This optimized assay was used to screen the 1500 compound NCI Diversity Set II at a single concentration. 44 molecules were identified that inhibited at least 50% of the RPA/Rad9 interaction at 200 μM. These molecules were then screened in an electrophoretic mobility shift assay (EMSA) for the unwanted ability to disrupt RPA binding to ssDNA. In the EMSA, 14 compounds were triaged due to their ability to displace >20% of RPA from labeled ssDNA at 200 μM. The 30 selective inhibitors were retested in two separate assays to reconfirm their activity. Four compounds, including NSC15520, fumaropimaric acid (1, Fig. 2), were confirmed as strong inhibitors of the RPA-Rad9 PPI.
Fig. 2. Structure of fumaropimaric acid and its predicted binding mode to RPA70N.

To further characterize fumaropimaric acid, the GST-Rad9 used in the HTS assay was replaced with a GST-fused p53 18mer (GST-p53). Compound 1 was shown to also inhibit the RPA-p53 PPI with an IC50 of 10 μM. It was also shown that 1 inhibits the ability of RPA to bind to double stranded DNA (dsDNA) at concentrations below 100 μM. Since the RPA70N domain is essential for RPA binding to dsDNA, this finding further supports tri-acid 1 as a selective inhibitor of RPA70N.36,37 In silico docking was used to gain structural insight into the binding mode of fumaropimaric acid. Using the Autodock Vina software suite, compound 1 and a virtually constructed Rad9 peptide, containing its acidic helix, were docked into a large section of RPA70N containing the basic cleft.38 From this model, it was evident that inhibitor 1 binds to the basic cleft and interacts with R41 in a similar fashion as the Rad9 peptide, which is to be expected, based upon the essential role of R41 in Rad9 binding to RPA70N (Fig. 2).24
In later studies, it was shown that tri-acid 1 inhibits the binding of GST-p53 to RPA70N with an affinity of 9.0 μM.39 Seven structurally related analogs were also tested; only two analogs showed any ability to inhibit GST-p53 binding to RPA70N. The analogs were not selective for inhibiting the PPIs of RPA versus its ability to bind ssDNA and thus were not studied further. By measuring the tryptophan quenching of RPA residues, based upon the conformational changes induced upon compound 1 binding to RPA, the affinity of inhibitor 1 was determined to be 29 μM using full-length heterotrimeric RPA. RPA70N mutational studies revealed that an R41E, R43E double mutant impaired the ability of the compound to cause a conformational change, and thus quench tryptophan fluorescence, by approximately 10-fold (Kd = 230 μM), further supporting the importance of these two basic residues in the binding of molecules to RPA70N.
The identification of fumaropimaric acid was significant, as it was the first PPI inhibitor of the RPA70N-Rad9 interaction, demonstrating that, despite the challenging nature of this protein, the discovery of small molecule inhibitors of the RPA PPIs was indeed possible.
HAMNO
Similarly, using the previously described HTS assay, the Oakley laboratory identified HAMNO (Fig. 3) as a RPA PPI inhibitor.40 HAMNO is unique in that it is neutral at pH 7, unlike compound 1, which has three carboxylic acids and is therefore trianionic at physiological pH. After the initial discovery, HAMNO was investigated using in silico methods to predict its binding mode (Fig. 3).38 From this study, the site of highest predicted affinity was at the end of the basic cleft, interacting with R43, a residue previously shown to be important in the PPIs of RPA.24 HAMNO was shown to disrupt the binding of RPA to dsDNA at 100 μM, but not to interfere with RPA–ssDNA binding at 200 μM. These data show HAMNO is selective for RPA70N over RPA domains A–D at the micromolar levels and is thus most likely a selective PPI inhibitor.
Fig. 3. Structure of HAMNO and its predicted binding mode to RPA70N.

A molecule that inhibits the PPIs of RPA with ATRIP and Rad9 should inhibit the recruitment of the proteins ultimately responsible for ATR activation, leading to impaired ATR signalling and increased replication stress.41–43 To assess whether HAMNO was inducing replication stress by inhibiting the RPA PPIs, UMSCC38 cells were exposed to HAMNO. A dose dependent increase in pan-nuclear γ-H2AX staining was observed, and it was further shown that 20 μM HAMNO caused an increase in H2AX phosphorylation in both UMSCC38 and UMSCC11B cancer cell lines, but not in non-cancerous OK4 cells. A comparison of the pan-nuclear γ-H2AX staining between the effects of 20 μM and 50 μM HAMNO showed the ratio of pan-nuclear γ-H2AX positive cells to negative cells was 6–8 fold larger in UMSCC38 cells versus OK4 cells. Taken together, these experiments show HAMNO causes increased replication stress that leads to aberrant signalling in the ATR pathway.
Exposure to HAMNO alone at low micromolar concentrations inhibited both UMSCC38 and UMSCC11B cells from forming colonies. UMSCC38 cells were also exposed to 20 μM etoposide, a topoisomerase 2 inhibitor, and HAMNO in combination, resulting in greater inhibition of colony formation versus treatment with HAMNO alone. The molecule was further evaluated in a xenograft study, which showed that treatment of tumor-bearing mice with 1 mg kg–1 of HAMNO slowed UMSCC11B tumor progression. Treatment of either UMSCC38 or UMSCC11B tumors in mice with a combination of etoposide (10 mg kg–1) and HAMNO (2 mg kg–1) also showed a slowing of tumor progression.
HAMNO represents the first PPI inhibitor of RPA70N that has shown an effect both in cells and an in vivo tumor model, as a single agent and in a synergistic combination with etoposide. Based upon these results, and the fact HAMNO is well tolerated in mice at concentrations that affect tumor progression, Oakley and colleagues are currently researching various derivatives of HAMNO to identify a more potent RPA70N inhibitor.
Anthranilic acid based inhibitors
Fesik and colleagues also conducted a HTS screen to identify compounds that bound to RPA, using 90 000 compounds from the Vanderbilt HTS collection and a competition-based fluorescence polarization anisotropy (FPA) assay.44,45 The initial screen was conducted at a single concentration of 30 μM. From this screen, 674 compounds were shown to displace >10% of the fluorescently labelled, peptide-based probe. These initial hit molecules were filtered for fluorescence interference, chemical reactivity, and drug-likeness.45 Of the remaining 90 compounds, 52 compounds were determined to possess a Kd value <100 μM. Based on its combination of modular synthesis, ligand efficiency, and potency, anthranilic acid 3 was chosen for further optimization (Fig. 4).
Fig. 4. Identification and optimization of anthranilic acid inhibitors.
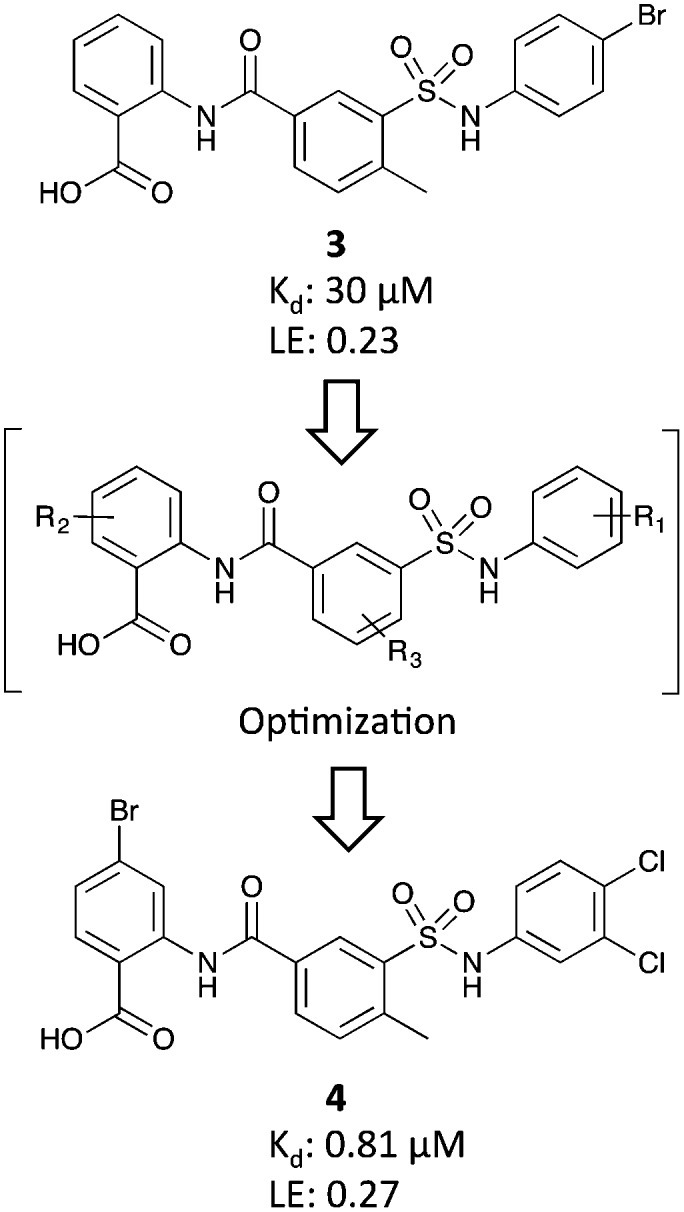
In an effort to optimize inhibitor 3, a multipronged SAR campaign was undertaken using the crystal structure of 3 in complex with RPA70N for guidance (Fig. 5). Initially, the R1 substituents on the right phenyl ring were varied and replacement of the phenyl with other aromatic and nonaromatic ring systems was explored. It was determined the best substituents were 4-Br, 3-Cl, and 3,4-diCl. Holding the R1 substituent as a 3,4-diCl, the R2 and R3 substituents were varied independently. The culmination of this work was compound 4 (Kd = 0.81 μM) containing a 3,4-diCl substitution for R1, a 4-Br substitution for R2, and a 3-methyl substitution on R3 (Fig. 4).
Fig. 5. Co-crystal structures of fragment-based inhibitors and HTS inhibitors in complex with RPA70N. A: Compound 4 in complex with RPA70N (5E7N). B: An analog of compound 9 in complex with RPA70N (4IJH). C: Compound 14 in complex with RPA70N (4O0A). D: Overlay of co-crystal structures of compound 4, an analog of compound 9, and compound 14 in complex with RPA70N.

Compound 4 was progressed further to assess its selectivity and potential for cellular studies. A fluorescence-based DNA binding assay showed that the ability of neither RPA70AB nor RPA70NAB to bind to ssDNA was affected by the presence of 4. To determine if inhibitor 4 was appropriate to take further into cellular studies both its protein binding and cellular permeability were determined. While the protein binding of 4 was high at 99.8% bound, the compound was cell penetrant, with a Papp of 29.2 × 10–6 cms–1 (Caco-2). Due to its potency and permeability, compound 4 is currently being evaluated in cellular studies.
Fragment-based screening
Chlorobenzothiophene inhibitors
The Fesik laboratory also identified compounds that bind to RPA70N by screening a 14 976 membered fragment library against 15N labeled RPA70N using HSQC NMR.46,47 149 confirmed fragment hits (1% hit rate) were identified from the screen by monitoring compound-induced shifts in serine 55 (S55) and threonine 60 (T60), two amino acids on either end of the binding cleft. These 149 hits were subdivided into two categories, based upon the HSQC shifts of either S55 or T60, and the presumption that independent shifts in these residues were indicative of two distinct binding sites, site-1 and site-2, respectively. The fragment hits displayed affinities ranging from 600 μM to 2 mM, based on titration of the NMR shifts, and corresponding ligand efficiencies ranging from 0.15 to 0.26.
Without available guidance from a co-crystal structure, an initial merging strategy led to the identification of two lead molecules 7 and 8 (Fig. 6) with improved affinities of 130 and 135 μM, respectively, but with reduced ligand efficiencies. An SAR campaign based on these two leads was undertaken to further improve the binding affinities. Analogs were assessed for their ability to inhibit the interaction of RPA70N and a FITC-labeled ATRIP-derived peptide (FITC-ATRIP) in the previously described FPA assay. This campaign resulted in the identification of compounds 9 and 10 (Fig. 6), which had affinities of 10 μM and 18 μM, respectively. To assess the domain selectivity of 9 and 10, the compounds were analyzed for their ability to inhibit binding of RPA to ssDNA using EMSA. Neither compound showed evidence of disruption of RPA–ssDNA interactions at concentrations up to 100 μM, indicating that these compounds are specific for inhibition of the RPA70N–ATRIP interaction.
Fig. 6. Identification and optimization of chlorobenzothiophene inhibitors.

The binding modes of the two different lead series were characterized using X-ray crystallography. Co-crystal structures of compound 8 and an analog of 9 bound to RPA70N were obtained from compound soaks into crystals of E7R RPA70N (Fig. 5).48 The E7R RPA70N was designed to eliminate E7 on the N terminus of WT RPA70N from binding to and occluding the basic cleft of an adjacent RPA70N in the crystal lattice, thus allowing for the molecules to bind to the exposed binding cleft and the structures of RPA70N/ligand complexes to be obtained.48 The co-crystal structures show several common features of the binding modes of these molecules. A carboxylic acid in each molecule has an interaction with R31 and a 3,4 di-chloro-substituted phenyl ring of each inhibitor is oriented into the hydrophobic site-1 that also accommodates the phenylalanine of p53.23 The aromatic ring of the inhibitors also appears to make a cation–pi interaction with R43 of RPA70N.
These molecules were the first reported PPI inhibitors of RPA70N and ATRIP and the first PPI inhibitors of RPA70N discovered using fragment-based techniques.
Pyrazole inhibitors
In a related effort, Fesik and colleagues used an optimized diphenyl pyrazole 11, derived from a phenyl pyrazole fragment hit, and a phenyl furan acid fragment hit 12 in a fragment-linking approach (Fig. 7).47,49 A ternary structure of E7R RPA70N bound to two fragments (11 and 12) revealed that the two fragments bound in site-1 and site-2, respectively, with a distance of 4.9 Å between them. Based upon this information, compounds 11 and 12 were linked using a short flexible linker to create 13, which had an affinity of 20 μM. While this linked compound showed marked improvement over the individual fragments, its affinity was far less than the predicted 1 μM (580 μM for 11 × 1870 μM for 12 = 1.0 μM).
Fig. 7. Identification and optimization of pyrazole inhiitors.

Theorizing that a sub-optimal linker was at least partially responsible for the reduced affinity, a structure-based optimization campaign was undertaken to improve 13. A library of linked compounds was generated with the aim of exploiting unused hydrophobic space in the basic cleft, particularly the site-1 pocket, maintaining the key electrostatic interactions, and optimizing the geometry and length of the linker. This study produced 14 with an FPA-derived affinity of 0.19 μM. A co-crystal structure of 14 bound to RPA70N was obtained and used to rationalize the gains in potency (Fig. 5). The 3,4-dichlorine substitutions on the N-phenyl ring of the pyrazole bound in site-1 recapitulated previous SAR observations. The chlorine atom from the original fragment hit was incorporated onto the phenyl ring adjacent to the furan. An important improvement to the linker region was the sulfur atom in the thioamide, which was found to occupy a small lipophilic space under L87, providing a 15-fold improvement in affinity compared to the corresponding amide analog.
The binding affinity of inhibitor 14 to other RPA constructs was measured to determine the effects of additional RPA domains on binding to RPA70N and to assess relative selectivity other OB-folds of RPA. An FP competition assay using RPA70NAB demonstrated that the affinity of compound 14 to the N-terminal domain of RPA was minimally affected by the presence of the high affinity ssDNA binding OB-folds of RPA70 A and B (affinity 0.29 μM versus 0.19 μM to RPA70N alone). The selectivity of 14 was established based on an FP displacement assay using RPA70NAB and a FITC-labeled ssDNA probe. Under these conditions, inhibitor 14 exhibited 15-fold selectivity for disrupting the ATRIP–RPA70N interaction over ssDNA displacement.
Linked inhibitor 14 shows the power of FBDD for generating potent molecules from relatively weak starting points for difficult target proteins. This compound represents the first submicromolar in vitro inhibitor of RPA70N and the most potent RPA70N inhibitor identified to date. Further optimization of the physiochemical properties of the molecule, specifically its dianionic nature, will have to be addressed moving forward to improve the utility of the molecule in a cellular context.
Stapled helix peptides
The Fesik laboratory also developed a PPI inhibitor using a stapled helix peptide approach based on a sequence from ATRIP, an endogenous RPA binding partner.50 Starting with the 15 amino acid ATRIP-derived peptide (Fig. 8) that had been the basis of the fluorescent probe employed in the FP assay used to evaluate small molecule inhibitors, an alanine scan was conducted to identify key residues mediating the binding of the probe to RPA70N. The alanine scan revealed that D1, F2, T3, D6, L7, D11, and L13 were important residues for binding to RPA70N, with the peptide losing 2–5 fold affinity when any of these were replaced with an alanine. Residue F2 proved to be critical for binding, as replacement of this residue led to a 50-fold reduction in affinity. Interestingly, two of the mutations to the ATRIP peptide, E8A and T12A, led to minor improvements in binding. Surprisingly, replacement of D5 and E8, both of which are conserved in multiple endogenous binding partners, resulted in no loss of affinity.
Fig. 8. Optimization of stapled helix peptides.

To improve the affinity of the peptide, the researchers turned to the sequence of another RPA binding partner, p53. The p53 sequence features a hydrophobic cluster (WF) not found in ATRIP. The single point mutated peptides 15 and 16 (L10W and D11F), each containing only one of these hydrophobic residues, did not provide added affinity. However, inserting both hydrophobic residues simultaneously (peptide 17) resulted in a 10-fold improvement in potency. Based upon the SAR from the alanine scan, residue 12 was mutated to alanine; peptide 18, with this WFA motif, showed a further 2-fold improvement. A FITC moiety was attached to this sequence (peptide 19) to enable direct binding measurements to RPA70N. The fluorophore produced a further 3-fold increase in binding affinity to RPA70N (Kd = 0.48 μM). The rationale for the improvement in binding affinity from the FITC is not well understood, but had been documented previously.50
Optimization of the physiochemical properties of the peptide was undertaken due to its highly charged state (overall charge of –6) being a liability for cell penetration. Strategic replacement of the glutamic and aspartic acid residues was investigated to mitigate the overall negative charge. The study resulted in FITC-labeled peptide 20 with a –2 charge, but with a 44-fold reduction in affinity relative to 19 (Kd = 21 μM). While the N-terminal residues had been shown to be necessary from the alanine scan, deletion of two residues from the C-terminus had no effect on the potency (peptide 21).
To further improve the affinity of the peptide to RPA70N, a new and unique strategy, incorporating both an unnatural amino acid and a hydrocarbon staple, was employed. Based upon previous SAR from fragment-based small molecule campaigns, substitution of a phenyl ring with a 3,4-dichlorophenyl resulted in an improved binding affinity. Based on these observations, an unnatural amino acid containing a phenylalanine with a dichloro substitution (Z) was incorporated into the peptide and led to a >100-fold improvement in affinity.46,49,51 A co-crystal structure of peptide 22 bound to RPA70N revealed that the 3,4-dichlorophenyl ring of peptide 22 bound in a similar fashion to the 3,4-dichlorophenyl moiety of the previously characterized small molecule inhibitors of RPA (Fig. 9). A hydrocarbon staple is a known method for improving properties of helical peptides, including increased potency, protease resistance, and cell penetration.52,53 Incorporating a hydrocarbon staple and a FITC label into an optimized peptide containing the F11Z substitution resulted in peptide 26, which displayed a 500-fold boost in affinity (Kd = 0.042 μM, with a net charge of –2).
Fig. 9. Co-crystal structure of peptide 22 bound to RPA70N (4NB3).

The domain specificity and selectivity of stapled helix peptide 26 was further evaluated. An EMSA experiment demonstrated that increasing concentrations of peptide showed no change in the percent of full-length RPA bound to ssDNA. To assess cell penetration, U2OS cells were incubated with the peptide for 24 hours, fixed, and visualized using confocal microscopy for direct observation of the FITC.
The labeled peptide was observed in the cell in a pattern consistent with entry through endocytosis, along with diffuse cytosolic staining and fluorescence in the nucleus.
The selective and potent peptide 26 was the end result of a combination of traditional peptide optimization, a new method for incorporating unnatural amino acids based upon small molecule SAR, and stapled helix peptide technology. This molecule demonstrates a non-traditional route to the discovery of cellular probes and a promising and distinctive starting point for further studies.
Conclusions
RPA is the essential ssDNA binding protein in eukaryotes that is responsible for binding to ssDNA and recruiting a multitude of binding partners to initiate the DDR pathway in response to DNA damage. This crucial role as a hub for the DDR has garnered the attention of the scientific community, which has led to research focusing on the discovery and optimization of inhibitors of RPA PPIs. Despite the difficulty of targeting the PPIs of RPA, due to its promiscuity in binding partners and the nature of its binding cleft, the Oakley and Fesik laboratories have pioneered the research in this field.
The Oakley Laboratory was responsible for identifying the first RPA PPI inhibitor and the first RPA PPI inhibitor to exhibit activity in a mouse model. Importantly, this molecule did not display broad spectrum toxicities, suggesting the possibility for the existence of a therapeutic window, despite the possible effects from the inhibition of the interaction of RPA with several possible effectors. Meanwhile, the Fesik Laboratory has contributed the first submicromolar inhibitor of PPIs mediated by RPA, and, using an innovative technology, the first stapled helix peptide inhibitor of RPA. These molecules will allow researchers to continue to probe the outcomes and consequences of selective RPA inhibition in cells and animal models, which will lead to continued interest and research in the selective inhibition of RPA-mediated PPIs as a potential cancer therapy.
Acknowledgments
We would like to thank Dr. Gregory Oakley for graciously providing the predicted binding modes of fumaropimaric acid and HAMNO.
Biographies

James D. Patrone
James D. Patrone earned a PhD in Medicinal Chemistry from the University of Michigan in 2010. He then joined the group of Professor Stephen Fesik at Vanderbilt University, where he engaged in the design and synthesis of inhibitors of RPA-mediated protein–protein interactions. In 2015, James became an Assistant Professor of Chemistry at Rollins College in Orlando, Florida, USA. His group is interested in the application of fragment-based ligand discovery (FBLD) toward the discovery of chemical matter for challenging targets in drug discovery and synthesis of novel heterocyclic fragment molecules.

Alex G. Waterson
Alex G. Waterson obtained his PhD at Emory University under Dr. Albert Padwa and performed postdoctoral research with the late Dr. Albert Meyers. Alex joined GlaxoSmithKline in 2001, working on kinase-based oncology programs, including contributions to the discovery of Dabrafenib. In 2008, he moved to Vanderbilt to direct the Chemical Synthesis Core and co-lead a center within the NCI's Chemical Biology Consortium, and later joined with the Fesik lab. Alex is currently Research Associate Professor of Pharmacology and Chemistry, and continues to lead Fesik lab projects as well as Vanderbilt's Center for Cancer Drug Discovery in the NExT consortium.

Stephen W. Fesik
Stephen W. Fesik is the Orrin H. Ingram, II Chair in Cancer Research and a Professor of Biochemistry, Pharmacology, and Chemistry at Vanderbilt University. He is also a member of the Vanderbilt Ingram Cancer Center, the Vanderbilt Institute of Chemical Biology, and the Center for Structural Biology. The focus of his research is on cancer drug discovery using fragment-based methods and structure-based design. Prior to joining Vanderbilt in May 2009, Dr. Fesik was the Divisional Vice President of Cancer Research at Abbott Laboratories (now AbbVie).
Footnotes
†The authors declare no competing interests.
References
- Wold M. S., Kelly T. Proc. Natl. Acad. Sci. U. S. A. 1988;85:2523–2527. doi: 10.1073/pnas.85.8.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold M. S. Annu. Rev. Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- Bochkarev A., Bochkareva E., Frappier L., Edwards A. M. EMBO J. 1999;18:4498–4504. doi: 10.1093/emboj/18.16.4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iftode C., Borowiec J. A. Biochemistry. 2000;39:11970–11981. doi: 10.1021/bi0005761. [DOI] [PubMed] [Google Scholar]
- Kim C. S., Paulus B. F., Wold M. S. Biochemistry. 1994;33:14197–14206. doi: 10.1021/bi00251a031. [DOI] [PubMed] [Google Scholar]
- Brill S. J., Bastin-Shanower S. Mol. Cell. Biol. 1998;18:7225–7234. doi: 10.1128/mcb.18.12.7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipova D., Mullen J. R., Maniar H. S., Lu J., Gu C., Brill S. J. Genes Dev. 1996;10:2222–2233. doi: 10.1101/gad.10.17.2222. [DOI] [PubMed] [Google Scholar]
- Daughdrill G. W., Ackerman J., Isern N. G., Botuyan M. V., Arrowsmith C., Wold M. S., Lowry D. F. Nucleic Acids Res. 2001;29:3270–3276. doi: 10.1093/nar/29.15.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali S. I., Shin J. S., Bae S. H., Kim B., Choi B. S. Int. J. Biochem. Cell Biol. 2010;42:1210–1215. doi: 10.1016/j.biocel.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Ciccia A., Bredemeyer A. L., Sowa M. E., Terret M. E., Jallepalli P. V., Harper J. W., Elledge S. J. Genes Dev. 2009;23:2415–2425. doi: 10.1101/gad.1832309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansbach C. E., Betous R., Lovejoy C. A., Glick G. G., Cortez D. Genes Dev. 2009;23:2405–2414. doi: 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mer G., Bochkarev A., Gupta R., Bochkareva E., Frappier L., Ingles C. J., Edwards A. M., Chazin W. J. Cell. 2000;103:449–456. doi: 10.1016/s0092-8674(00)00136-7. [DOI] [PubMed] [Google Scholar]
- Block W. D., Yu Y., Lees-Miller S. P. Nucleic Acids Res. 2004;32:997–1005. doi: 10.1093/nar/gkh265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley G. G., Patrick S. M., Yao J., Carty M. P., Turchi J. J., Dixon K. Biochemistry. 2003;42:3255–3264. doi: 10.1021/bi026377u. [DOI] [PubMed] [Google Scholar]
- Barr S. M., Leung C. G., Chang E. E., Cimprich K. A. Curr. Biol. 2003;13:1047–1051. doi: 10.1016/s0960-9822(03)00376-2. [DOI] [PubMed] [Google Scholar]
- Wang H., Guan J., Wang H., Perrault A. R., Wang Y., Iliakis G. Cancer Res. 2001;61:8554–8563. [PubMed] [Google Scholar]
- Oakley G. G., Loberg L. I., Yao J., Risinger M. A., Yunker R. L., Zernik-Kobak M., Khanna K. K., Lavin M. F., Carty M. P., Dixon K. Mol. Biol. Cell. 2001;12:1199–1213. doi: 10.1091/mbc.12.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao R. G., Cao C. X., Zhang H., Kohn K. W., Wold M. S., Pommier Y. EMBO J. 1999;18:1397–1406. doi: 10.1093/emboj/18.5.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang F., Newport J. W. J. Cell Sci. 1993;106(Pt 3):983–994. doi: 10.1242/jcs.106.3.983. [DOI] [PubMed] [Google Scholar]
- Din S., Brill S. J., Fairman M. P., Stillman B. Genes Dev. 1990;4:968–977. doi: 10.1101/gad.4.6.968. [DOI] [PubMed] [Google Scholar]
- Bochkareva E., Korolev S., Lees-Miller S. P., Bochkarev A. EMBO J. 2002;21:1855–1863. doi: 10.1093/emboj/21.7.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh R. M., Orren D. K., Nehlin J. O., Ravn P. H., Kenny M. K., Machwe A., Bohr V. A. J. Biol. Chem. 1999;274:18341–18350. doi: 10.1074/jbc.274.26.18341. [DOI] [PubMed] [Google Scholar]
- Bochkareva E., Kaustov L., Ayed A., Yi G. S., Lu Y., Pineda-Lucena A., Liao J. C. C., Okorokov A. L., Milner J., Arrowsmith C. H., Bochkarev A. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15412–15417. doi: 10.1073/pnas.0504614102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Vaithiyalingam S., Glick G. G., Mordes D. A., Chazin W. J., Cortez D. Mol. Cell. Biol. 2008;28:7345–7353. doi: 10.1128/MCB.01079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namiki Y., Zou L. Proc. Natl. Acad. Sci. U. S. A. 2006;103:580–585. doi: 10.1073/pnas.0510223103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning E., Klimovich V., Nager A. R. Nucleic Acids Res. 2006;34:4126–4137. doi: 10.1093/nar/gkl550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daughdrill G. W., Buchko G. W., Botuyan M. V., Arrowsmith C., Wold M. S., Kennedy M. A., Lowry D. F. Nucleic Acids Res. 2003;31:4176–4183. doi: 10.1093/nar/gkg451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs D. M., Lipton A. S., Isern N. G., Daughdrill G. W., Lowry D. F., Gomes X., Wold M. S. J. Biomol. NMR. 1999;14:321–331. doi: 10.1023/a:1008373009786. [DOI] [PubMed] [Google Scholar]
- Braun K. A., Lao Y., He Z. G., Ingles C. J., Wold M. S. Biochemistry. 1997;36:8443–8454. doi: 10.1021/bi970473r. [DOI] [PubMed] [Google Scholar]
- Ball H. L., Myers J. S., Cortez D. Mol. Biol. Cell. 2005;16:2372–2381. doi: 10.1091/mbc.E04-11-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanzer J. G., Liu S. Q., Oakley G. G. Bioorg. Med. Chem. 2011;19:2589–2595. doi: 10.1016/j.bmc.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuck S. C., Turchi J. J. Cancer Res. 2010;70:3189–3198. doi: 10.1158/0008-5472.CAN-09-3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A. K., Dormi S. S., Turchi A. M., Woods D. S., Turchi J. J. Biochem. Pharmacol. 2015;93:25–33. doi: 10.1016/j.bcp.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher T. M., Bodenmiller D., Fitch R. W., Jalal S. I., Turchi J. J. Mol. Cancer Ther. 2011;10:1796–1806. doi: 10.1158/1535-7163.MCT-11-0303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss W. A., Taylor S. S., Shokat K. M. Nat. Chem. Biol. 2007;3:739–744. doi: 10.1038/nchembio1207-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao Y., Lee C. G., Wold M. S. Biochemistry. 1999;38:3974–3984. doi: 10.1021/bi982371m. [DOI] [PubMed] [Google Scholar]
- Iftode C., Borowiec J. A. Nucleic Acids Res. 1998;26:5636–5643. doi: 10.1093/nar/26.24.5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O., Olson A. J. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanzer J. G., Carnes K. A., Soto P., Liu S., Parkhurst L. J., Oakley G. G. Nucleic Acids Res. 2013;41:2047–2059. doi: 10.1093/nar/gks1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanzer J. G., Liu S., Wang L., Mosel A., Peng A., Oakley G. G. Cancer Res. 2014;74:5165–5172. doi: 10.1158/0008-5472.CAN-14-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Opiyo S. O., Manthey K., Glanzer J. G., Ashley A. K., Amerin C., Troksa K., Shrivastav M., Nickoloff J. A., Oakley G. G. Nucleic Acids Res. 2012;40:10780–10794. doi: 10.1093/nar/gks849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo L. I., Murga M., Zur R., Soria R., Rodriguez A., Martinez S., Oyarzabal J., Pastor J., Bischoff J. R., Fernandez-Capetillo O. Nat. Struct. Mol. Biol. 2011;18:721–727. doi: 10.1038/nsmb.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward I. M., Chen J. J. Biol. Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- Patrone J. D., Pelz N. F., Bates B. S., Souza-Fagundes E. M., Vangamudi B., Camper D. V., Kuznetsov A. G., Browning C. F., Feldkamp M. D., Frank A. O., Gilston B. A., Olejniczak E. T., Rossanese O. W., Waterson A. G., Chazin W. J., Fesik S. W. ChemMedChem. 2016;11:893–899. doi: 10.1002/cmdc.201500479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza-Fagundes E. M., Frank A. O., Feldkamp M. D., Dorset D. C., Chazin W. J., Rossanese O. W., Olejniczak E. T., Fesik S. W. Anal. Biochem. 2012;421:742–749. doi: 10.1016/j.ab.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrone J. D., Kennedy J. P., Frank A. O., Feldkamp M. D., Vangamudi B., Pelz N. F., Rossanese O. W., Waterson A. G., Chazin W. J., Fesik S. W. ACS Med. Chem. Lett. 2013;4:36–40. doi: 10.1021/ml400032y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank A. O., Feldkamp M. D., Kennedy J. P., Waterson A. G., Pelz N. F., Patrone J. D., Vangamudi B., Camper D. V., Rossanese O. W., Chazin W. J., Fesik S. W. J. Med. Chem. 2013;56:9242–9250. doi: 10.1021/jm401333u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldkamp M. D., Frank A. O., Kennedy J. P., Patrone J. D., Vangamudi B., Waterson A. G., Fesik S. W., Chazin W. J. Biochemistry. 2013;52:6515–6524. doi: 10.1021/bi400542z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterson A. G., Kennedy J. P., Patrone J. D., Pelz N. F., Feldkamp M. D., Frank A. O., Vangamudi B., Souza-Fagundes E. M., Rossanese O. W., Chazin W. J., Fesik S. W. ACS Med. Chem. Lett. 2015;6:140–145. doi: 10.1021/ml5003629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank A. O., Vangamudi B., Feldkamp M. D., Souza-Fagundes E. M., Luzwick J. W., Cortez D., Olejniczak E. T., Waterson A. G., Rossanese O. W., Chazin W. J., Fesik S. W. J. Med. Chem. 2014;57:2455–2461. doi: 10.1021/jm401730y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank A. O., Feldkamp M. D., Kennedy J. P., Waterson A. G., Pelz N. F., Patrone J. D., Vangamudi B., Camper D. V., Rossanese O. W., Chazin W. J., Fesik S. W. J. Med. Chem. 2013;56:9242–9250. doi: 10.1021/jm401333u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky L. D., Bird G. H. J. Med. Chem. 2014;57:6275–6288. doi: 10.1021/jm4011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdine G. L., Hilinski G. J. Methods Enzymol. 2012;503:3–33. doi: 10.1016/B978-0-12-396962-0.00001-X. [DOI] [PubMed] [Google Scholar]