

 Twenty-three monoketone derivatives of curcumin were synthesized to restore the effectiveness of fluconazole against fluconazole-resistant Candida spp.
Twenty-three monoketone derivatives of curcumin were synthesized to restore the effectiveness of fluconazole against fluconazole-resistant Candida spp.
Abstract
Twenty-three monoketone derivatives of curcumin were synthesized to investigate the synergy with fluconazole against fluconazole-resistant Candida spp. The minimal inhibitory concentration (MIC80) and the fractional inhibitory concentration index (FICI) of the antifungal synergist fluconazole were measured against fluconazole-resistant C. albicans, C. tropicalis and C. krusei in vitro. Most of these compounds showed good synergistic activities against C. tropicalis. Among them, compound 9 exhibited significant synergistic activities against Candida spp. SARs were also discussed. In particular, a cell growth test exhibited that a combination of 1 μg ml–1 fluconazole and 64 μg ml–1 or 128 μg ml–1 compound 9 showed the most potent fungicidal effect against C. tropicalis. The synergistic effect may be associated with the changes of the intracellular ATP content and cell membrane permeability. Our results provided a basis for future evaluation and development of these compounds as leads for therapeutics for fluconazole-resistant candidiasis.
1. Introduction
Fungal infections pose a continuous and serious threat to human health and life especially to immunocompromised patients. During the past decades, the incidence of systemic candidiasis has consistently increased. Candida species such as Candida albicans, Candida tropicalis and Candida krusei are known as the main pathogens which may cause superficial and systemic infections. Antifungal therapy with topical or systemic agents can be effective in the control and treatment of Candida infection. Fluconazole (FLC) is the most commonly used drug in the prophylaxis and therapy of Candida infection; however, widespread and repeated use of FLC resulted in resistance to or failure of FLC therapy. To combat the FLC-resistant Candida spp., much attention has been paid to the synergism of FLC with other agents. As synergists, drugs such as tetracyclic indoles,1 amiodarone,2etc. can significantly sensitize fungi to FLC.
Naturally derived products are a valuable source of drugs or lead compounds.3–5 Curcumin (CCM, 1,7-bis-(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione), a naturally occurring phenolic compound, is a dietary constituent of turmeric and is well known for its broad bioactivities. Earlier studies have revealed that CCM and its related compounds possess multiple pharmaceutical applications owing to their anti-cancer,6 antibacterial7 and antifungal8,9 properties. Studies indicated that CCM acts as an antifungal agent via various mechanisms, such as generation of oxidative stress, inhibition of hyphae development,10 and alteration of membrane-associated properties of ATPase activity, ergosterol biosynthesis, and proteinase secretion.8 Clinical trials have reported that ingestion of a significant dose of CCM (12 g per day) had no side effect.11 Considering the biological importance and low toxicity of CCM, many efforts have been devoted to CCM analogues. So far, there is no report about the antifungal structure–activity relationship (SAR) of CCM as well as its synergistic antifungal activity. Inspired by these, it was of our interest to investigate the antifungal activity of CCM and understand its preliminary SAR. Structurally, CCM is a symmetrical methoxyphenolic dienone with an active methylene center (Table 1). Evidence from both in vitro and in vivo studies showed that the β-diketone moiety is responsible for its instability and weak pharmacokinetics.11–17 During the past decades, structural modifications of CCM to enhance its bioactivities suggested that the stability and metabolic profile of CCM could be improved by deleting the β-diketone moiety.11–17 Therefore, monoketone derivatives of CCM were synthesized in our lab and evaluated for their antifungal activity alone and synergistic antifungal activity with fluconazole against fluconazole-resistant Candida spp., and the structure–activity relationship (SAR) was also investigated and discussed.
Table 1. Structure and interaction modes of the title compounds and their MIC80 and FICI values.
| No. | Structure | MIC80 (μg ml–1) |
||||||||
|
C. albicans 103 |
C. tropicalis 087 |
C. krusei 2159 |
||||||||
| With FLC a | FICI | Mode of interaction b | With FLC a | FICI | Mode of interaction b | With FLC a | FICI | Mode of interaction b | ||
| 1 |
|
>64 | >1.06 | I | 16 | 0.313 | S | >64 | >1.06 | I |
| 2 |
|
>64 | >1.06 | I | 32 | 0.563 | I | >64 | >1.06 | I |
| 3 |
|
>64 | >1.06 | I | 16 | 0.313 | S | >64 | >1.06 | I |
| 4 |
|
>64 | >1.06 | I | 32 | 0.563 | I | >64 | >1.06 | I |
| 5 |
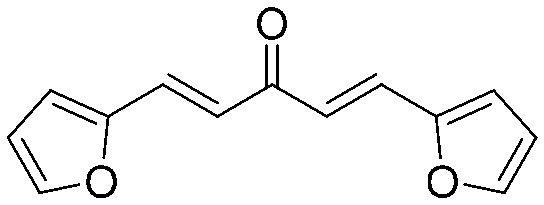
|
>64 | >1.06 | I | 16 | 0.313 | S | >64 | >1.06 | I |
| 6 |
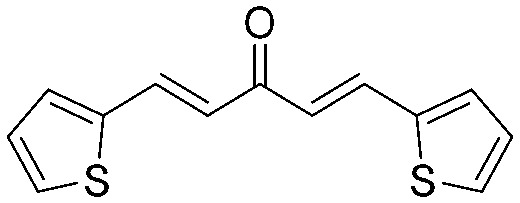
|
8 | 0.188 | S | >64 | >1.06 | I | >64 | >1.06 | I |
| 7 |
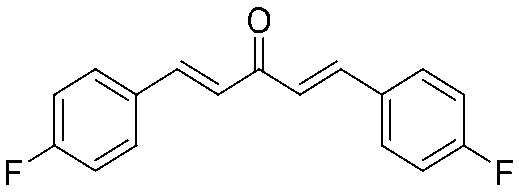
|
>64 | >1.06 | I | 8 | 0.188 | S | >64 | >1.06 | I |
| 8 |
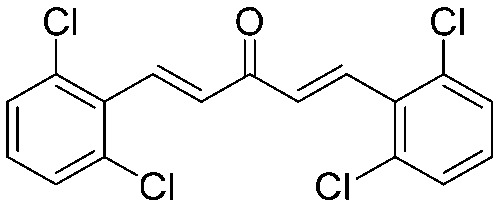
|
>64 | >1.06 | I | >64 | >1.06 | I | >64 | >1.06 | I |
| 9 |
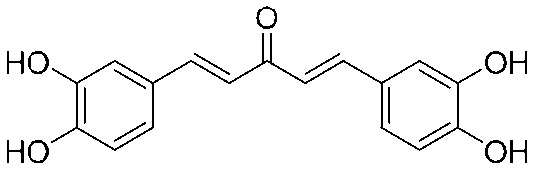
|
4 | 0.125 | S | 4 | 0.125 | S | 8 | 0.188 | S |
| 10 |
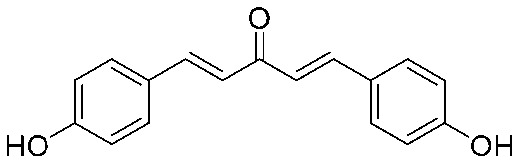
|
>64 | >1.06 | I | 8 | 0.188 | S | >64 | >1.06 | I |
| 11 |
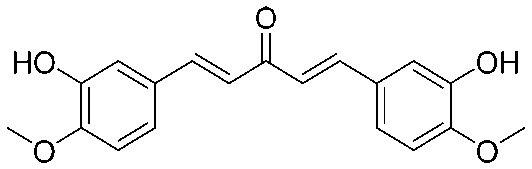
|
32 | 0.563 | I | 8 | 0.188 | S | >64 | >1.06 | I |
| 12 |
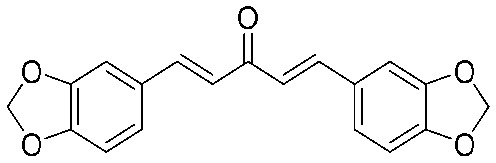
|
32 | 0.563 | I | 16 | 0.313 | S | 2 | 0.094 | S |
| 13 |
|
>64 | >1.06 | I | >64 | >1.06 | I | >64 | >1.06 | I |
| 14 |
|
>64 | >1.06 | I | 4 | 0.125 | S | >64 | >1.06 | I |
| 15 |
|
>64 | >1.06 | I | >64 | >1.06 | I | >64 | >1.06 | I |
| 16 |
|
>64 | >1.06 | I | 8 | 0.188 | S | >64 | >1.06 | I |
| 17 |
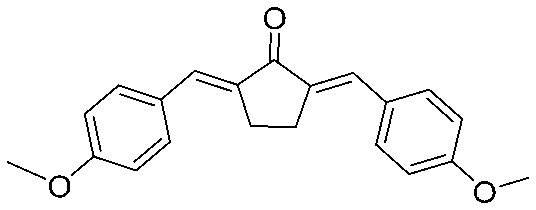
|
>64 | >1.06 | I | 64 | 1.06 | I | >64 | >1.06 | I |
| 18 |
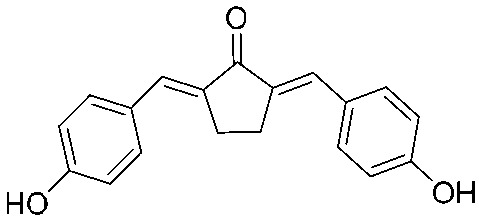
|
>64 | >1.06 | I | 8 | 0.188 | S | >64 | >1.06 | I |
| 19 |
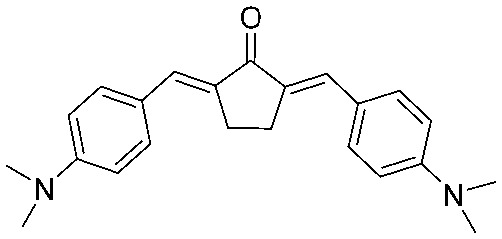
|
>64 | >1.06 | I | 64 | 1.06 | I | >64 | >1.06 | I |
| 20 |
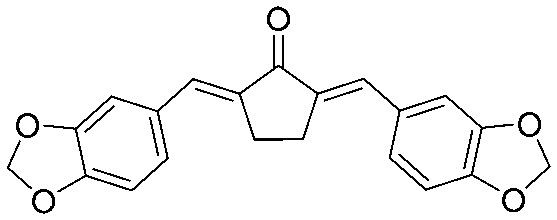
|
>64 | >1.06 | I | 64 | 1.06 | I | >64 | >1.06 | I |
| 21 |
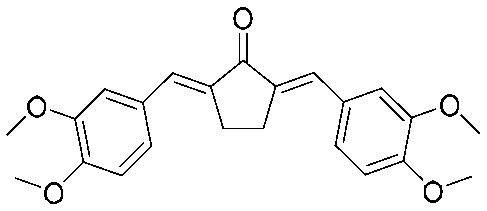
|
>64 | >1.06 | I | 16 | 0.313 | S | >64 | >1.06 | I |
| 22 |
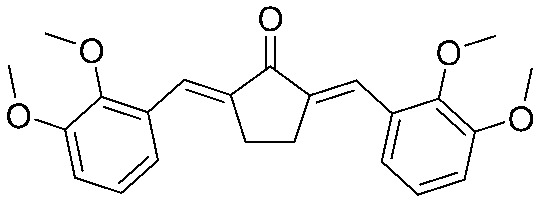
|
>64 | >1.06 | I | >64 | >1.06 | I | >64 | >1.06 | I |
| 23 |
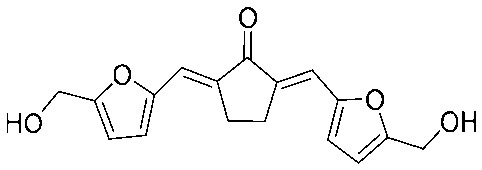
|
>64 | >1.06 | I | 4 | 0.125 | S | >64 | >1.06 | I |
| 24 |
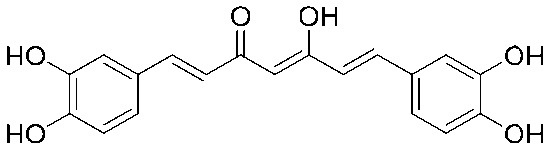
|
4 | 0.125 | S | 4 | 0.125 | S | 2 | 0.563 | I |
| CCM |
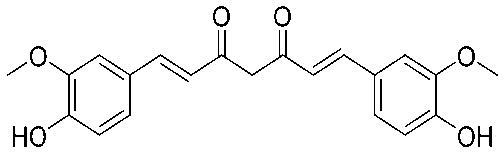
|
8 | 0.188 | S | 8 | 0.188 | S | 8 | 0.188 | S |
aMIC80 value of compound in column 2 in combination with 8.0 μg ml–1 fluconazole.
bI: indifferent; S: synergy.
2. Results and discussion
Most of the title compounds were synthesized by aldol condensation of substituted benzaldehydes or heteroaromatic aldehydes with acetone or cyclohexanone in ethyl alcohol catalyzed by 10–20% NaOH aqueous solution with good yields (Scheme 1). However, under these conditions the benzaldehydes substituted with hydroxyl groups hardly yielded the corresponding products, and even no reaction occurred at all. Thus this kind of products were obtained by aldol condensation catalyzed by SOCl2 in anhydrous ethyl alcohol. The synthetic method of the title compounds has the advantages of simple operation, mild conditions and easy work-up. The structures of the synthesized compounds were confirmed by MS and 1H NMR. The Z/E configuration of compounds 1–15 can be easily confirmed as E-configuration from the coupling constants (J = about 15.9 Hz) in 1H NMR spectra. In order to confirm the Z/E configuration of compounds 16–23, compound 20 was taken as an example to perform NOESY experiments. A clear NOESY correlation between H-1′, H-2′′, and H-6′′ demonstrated that compound 20 was in E-configuration. Thus it can be concluded that compounds 16–23 were in E-configuration.
Scheme 1. Synthetic route for the title compounds. Reagents and conditions: (a) NaOH, EtOH, rt, 12–24 h; (b) SOCl2, anhydrous EtOH, rt, 12–24 h.

The in vitro synergistic antifungal activities of the title compounds were tested using the microbroth dilution method according to the standards of the Clinical and Laboratory Standards Institute, USA.14 The MIC80 of FLC against the FLC-resistant C. albicans (clinical isolate 103), C. tropicalis (clinical isolate 087) and C. krusei (clinical isolate 2159) was determined to be 128.0 μg ml–1. The MIC80 values of the title compounds with or without FLC (8.0 μg ml–1) are described in Table 1. Furthermore, the fractional inhibitory concentration index (FICI) of each agent was calculated by adding the MIC80 (with FLC)/MIC80 (used alone) ratios. The interaction modes, synergistic or indifferent, were defined according to FICI values of ≤0.5 or >0.5, respectively.18 The structures of the title compounds and their synergistic antifungal activities are summarized in Table 1.
Firstly, compounds 1–15 were synthesized and their synergistic antifungal activities were tested. CCM had synergistic antifungal activity against all the screened Candida spp. with a FICI value of 0.188. Most of the title compounds exhibited selectivity against Candida spp.; usually they displayed a more potent synergy against C. tropicalis than C. albicans and C. krusei. For example, except for compounds 2, 4, 6, 8, 13 and 15, all the other compounds displayed synergistic activity against C. tropicalis, while only two compounds (6 and 9) showed synergy against C. albicans and only two compounds (9 and 12) showed synergy against C. krusei. These results demonstrated that the monoketone moiety was more beneficial for the synergy against C. tropicalis, but not for C. albicans and C. krusei.
The synergistic antifungal effects of these active compounds against C. tropicalis were equal or even superior to that of CCM (FICI = 0.188). Compound 9 which was substituted with a 3′,4′-dihydroxyl group and compound 14 which was substituted with a 3′,4′-dimethoxy group are the most potent synergists (FICI = 0.125). Compounds with monohydroxyl (10) and 3′-hydroxyl-4′-methoxyl (11) substitutions also exhibited potent synergistic activities (FICI = 0.188). However, compounds with halogen atoms (1, 2, 3, 8 and 15) and dimethylamine (4) showed weak or no activity, while compounds with heteroaryl groups (5, 6) showed almost no inhibitory activity. These results demonstrated that the hydroxyl substitution was beneficial for the synergistic antifungal effects against C. tropicalis.
Although only two compounds, 6 and 9, showed synergy against C. albicans and two compounds, 9 and 12, showed synergy against C. krusei, the synergistic antifungal effects of these active compounds were equal or superior to that of CCM. The FICI values of compounds 6 and 9 against C. albicans were 0.188 and 0.125, respectively; and those of compounds 9 and 12 against C. krusei were 0.188 and 0.094, respectively. A preliminary conclusion could be made that the catechol moiety was favorable for the synergistic antifungal activity against C. albicans, C. tropicalis and C. krusei; and the catechol methylene ether moiety was favorable for the synergistic antifungal activity against C. krusei. A control compound 24 (3′,4′-dihydroxyl curcumin) was synthesized to further evaluate the monoketone scaffold. The FICI values of compounds 24 and 9 against C. albicans and C. tropicalis were both 0.125 except that compound 24 when used alone against C. krusei showed potent antifungal activity with MIC80 = 4 μg ml–1. These results further confirmed that the catechol moiety was important for synergy, and the β-diketone moiety could be changed into monoketone.
In order to further study the SARs, some monoketone analogues (16–23) of CCM which were incorporated with a five-membered ring were synthesized and their synergistic antifungal activity was evaluated. We hoped this modification could help to enhance the activity by restricting the conformation of the diarylpentanoid system to increase the rigidity of the molecules, but to our disappointment, this modification resulted in the loss of activity for all compounds 16–23 against C. albicans and C. krusei. As for C. tropicalis, only four compounds (16, 18, 21 and 23) showed synergistic antifungal activity. Among them, compound 18 substituted with free hydroxyl showed potent synergy (MIC = 8 μg ml–1, FICI = 0.188) and compound 23 substituted with 5′-hydroxymethyl furan was the most active (MIC = 4 μg ml–1, FICI = 0.125), even compared with CCM. These results demonstrated that the introduction of a five-membered ring would not help to improve the activity in general, and the free hydroxyl or hydroxymethyl furan moiety is beneficial for the synergistic activity against C. tropicalis.
To visualize and further confirm the synergistic effect of the most active compound 9 with FLC, antifungal susceptibility assays were conducted using a turbidity observation method and a cell growth test (growth curves). The results are summarized in Fig. 1 and 2. The suspension of cells treated with 9 alone at the concentration of 64 μg ml–1 showed a markedly reduced turbidity (Fig. 1), while the suspension was thick when cells were treated with FLC at the maximum concentration of 1 μg ml–1, but in combination with FLC, 9 exhibited an antifungal effect on FLC-resistant C. tropicalis 087 at the concentration of 4 μg ml–1. This result suggested that 9 showed a strong antifungal effect at the concentration of 64 μg ml–1 alone or 4 μg ml–1 in combination with FLC against FLC-resistant C. tropicalis 087. In the growth curves (Fig. 2), the synergistic fungicidal effect of 9 with fluconazole was further confirmed. As shown in Fig. 2A, 9 at the concentration of 64 μg ml–1 exhibited the same inhibition effect as that of FLC at 0.5 μg ml–1. When the concentration of 9 reached 128 μg ml–1, cells were killed or cell apoptosis was promoted. The synergistic effect of 9 on growth could be obviously seen from the other curves. When treated with a combination of 9 and FLC at low concentrations, C. tropicalis 087 was significantly grown at lower rates. In particular, the combination of 1 μg ml–1 FLC and 9 (64 μg ml–1 or 128 μg ml–1) showed the most potent antifungal effect (Fig. 2B). Together, the results suggested that 9 showed a strong synergistic fungicidal effect against FLC-resistant C. tropicalis 087.
Fig. 1. Antifungal susceptibility assays of FLC alone or in combination with compound 9. FLC-resistant C. tropicalis 087 was treated with or without different concentrations of compound 9 (0, 4, 8, 16, 32, 64, 128 μg ml–1) and FLC (0, 0.25, 0.5, 1 μg ml–1) alone or combinations of FLC and compound 9 in a shaking incubator. The turbidity of the suspension was observed and pictures were taken after 16 h of incubation.

Fig. 2. Growth curves of cells with different treatments. (A) Time–growth curve for C. tropicalis 087 treated with compound 9 (64, 128 μg ml–1) or FLC (0.25, 0.5 μg ml–1) alone in RPMI 1640 medium at 30 °C for 48 h. The OD was measured at 630 nm. (B) Time–growth curve for C. tropicalis 087 treated with FLC (1 μg ml–1) or in combination with compound 9 (64, 128 μg ml–1). (C) Time–growth curve for C. tropicalis 087 treated with FLC (0.5 μg ml–1) or in combination with compound 9 (64, 128 μg ml–1). (D) Time–growth curve for C. tropicalis 087 treated with FLC (0.25 μg ml–1) or in combination with compound 9 (64, 128 μg ml–1). FLC, fluconazole; compd. 9, compound 9. Data are shown as the means ± SD from three independent experiments.

In order to explore the synergistic antifungal mechanism of 9, we measured the intracellular ATP levels and membrane permeability as reported before.19,20 As shown in Fig. 3A, CCM obviously inhibited intracellular ATP production alone or in combination with FLC. However, a significant difference was not detected between CCM alone and in combination with FLC, whereas the combination of compound 9 and FLC could obviously decrease the intracellular ATP content significantly compared with compound 9 or FLC used alone (P < 0.01, Fig. 3B). The results indicated that the reduction of intracellular ATP was not the synergistic mechanism but an individual antifungal mechanism for CCM. Nevertheless, the difference between the intracellular ATP contents when compound 9 was used alone and in combination with FLC suggested that the synergy of compound 9 may be associated with the inhibition of ATP production.
Fig. 3. Intracellular ATP content in C. tropicalis 087 treated with different treatments. (A) FLC (4 μg ml–1), CCM (32, 64, 128 μg ml–1) or a combination for 8 hours. (B) C. tropicalis 087 was treated with fluconazole (4 μg ml–1), compound 9 (32, 64, 128 μg ml–1) or a combination for 8 hours. Fluorescence intensities were determined on a TD 20/20 luminometer (Turner Biosystem, Sunnyvale, CA). Each experiment was independently repeated three times. * indicates P < 0.05 compared with drug-free control; ** indicates P < 0.01 compared with drug-free control.

Since the changes of cell membrane permeability were targets for many antifungal agents, a cell membrane permeability assay was carried out to analyze the membrane damage caused by compound 9 and fluconazole. PI (propidium iodide) is a kind of nucleic acid staining dye and can enter only into cells which have permeable membranes. When the cell membrane permeability changes, the dye would bind to nucleic acid and show red fluorescence. As shown in Fig. 4A1, we noticed that rare PI fluorescence was observed in untreated cells, but increased PI fluorescence was observed when cells were incubated with 32 μg ml–1 compound 9 alone (Fig. 4A2). In addition, fluconazole, at 2 μg ml–1, could have an effect on the permeability of the cell membranes (Fig. 4A3). Significantly, much more PI fluorescence was observed in the combination-treated (32 μg ml–1 compound 9 plus 2 μg ml–1 fluconazole) cells (Fig. 4A4). The changes and difference could also be seen in Fig. 4B. In summary, our results suggested that the synergistic effect may also be associated with the changes of cell membrane permeability.
Fig. 4. Membrane permeability changes induced by compound 9 and fluconazole in C. tropicalis 087. (A) PI staining for detection of disruption of membrane permeability in C. tropicalis 087. (1) Cells were treated with no agents, (2) cells were treated with compound 9 (32 μg ml–1), (3) cells were treated with fluconazole (2 μg ml–1) and (4) cells were treated with the combination of compound 9 (32 μg ml–1) and fluconazole (2 μg ml–1). (B) PI staining for detection of increase in membrane permeability in C. tropicalis 087 treated with compounds. Black, cells were treated with no agents; yellow, cells were treated with compound 9 (32 μg ml–1) alone; blue, cells were treated with fluconazole (2 μg ml–1) alone; red, cells were treated with the combination of fluconazole and compound 9.

Further cytotoxicity activities of the active compounds 6, 7, 9, 10, 12, 14, 16, 18, 23 and CCM were investigated in human umbilical vein endothelial cells (HUVECs) by the MTT assay. The results are reported in Table 2. Curcumin exhibited a significantly stronger cytotoxic effect at the concentration of 32 μg ml–1 with a viability of 22.0%. This is in agreement with previous research.21 However, compounds 6, 7, 9, 10, 12, 14, 16, 18, and 23 showed a moderate cytotoxic effect at 32 μg ml–1 with viabilities of over 50%. Meanwhile, compounds 6, 9, 10, 12, and 16 exhibited lower cytotoxic effects than curcumin at the concentration of 16 μg ml–1. Among these synergistic compounds, compound 9 showed the lowest cytotoxicity against HUVECs. Taken together, our results suggested that these monoketone derivatives of CCM exhibited lower cytotoxicity than CCM.
Table 2. Cytotoxic activity of curcumin and its analogues on HUVECs.
| Compd. | Viability
a
[%] |
|
| 32 μg ml–1 | 16 μg ml–1 | |
| CCM | 22 ± 1.8 | 80 ± 1.7 |
| 6 | 49 ± 2.5 | 84 ± 1.1 |
| 7 | 43 ± 1.0 | 64 ± 1.1 |
| 9 | 75 ± 1.6 | 91 ± 2.5 |
| 10 | 75 ± 2.7 | 87 ± 4.2 |
| 12 | 71 ± 2.9 | 88 ± 4.0 |
| 14 | 55 ± 2.6 | 67 ± 3.0 |
| 16 | 69 ± 2.6 | 86 ± 4.0 |
| 18 | 60 ± 0.3 | 69 ± 1.8 |
| 23 | 57 ± 4.0 | 68 ± 0.3 |
aData are shown as the means ± SD from three independent experiments.
3. Conclusion
Our study may be useful to find more advanced FLC synergists against FLC-resistant Candida spp. Our results showed that monoketone derivatives of CCM displayed selectivity against C. albicans, C. tropicalis and C. krusei, and most of the compounds displayed good synergistic antifungal effects on C. tropicalis and no effect on C. albicans 103 and C. krusei 2159. The SAR study indicates that substituted hydroxyl or methoxyl are important groups in monoketone derivatives. Compound 9 had the most potently synergistic effect on C. tropicalis 087 and C. albicans 103 (FICI = 0.188) in combination with FLC in vitro. These results were further confirmed by the turbidity observation method and growth curves. The synergistic antifungal effect may be associated with the changes of the intracellular ATP content and the changes of membrane permeability. The synergistic antifungal mechanism of curcumin that had been reported could be due to the modulation of ABC multidrug transporters22 and the overexpression of the MFS pump CaMdr1p,23 but to the best of our knowledge, this study reports for the first time the ability of compound 9, a curcumin derivative, to change the intracellular ATP content and membrane permeability. Therefore, we propose that compound 9 be considered a potential candidate in the development of a novel synergistic antimicrobial agent on account of its significant antimicrobial activity.
4. Experimental
4.1. General
Reagents were purchased from common commercial suppliers and were used without further purification. Analytical TLC was carried out on silica gel F254 precoated (0.2 mm thickness) plastic TLC sheets. The TLC plates were spotted with samples using a fine glass capillary tube and developed in a chromatographic tank saturated with solvent vapor at room temperature. Melting points were determined in open capillary tubes and were uncorrected. 1H NMR and 2D NMR spectra were recorded on a Bruker Ac 300 or 600 MHz spectrometer in DMSO-d6 or CDCl3 solution. Chemical shifts were recorded in δ ppm relative to internal TMS. Mass spectra (MS and HRMS) were obtained by electron spray ionization (ESI) in positive or negative mode using an LCQ DECA XP LC-MS and an Agilent UHPLC 1290 system and a Q-TOF 6538 MS-MS spectrometer.
4.2. Experimental procedures
4.2.1. General procedure for the synthesis of compounds 1–8 and 12–23
To a stirred solution of the appropriate ketone (1 mmol, 1 equiv.) and the corresponding aromatic aldehyde (2 mmol, 2 equiv.) in ethanol (10 ml) at room temperature was added 10% NaOH (aqueous) solution (4 mmol, 4 equiv.) slowly and stirring continued at room temperature. Upon completion (monitored by TLC), the mixture was poured into an excess amount of ice water and then filtered, dried and recrystallized from ethanol or DMF to give the title compounds.
4.2.2. General procedure for the synthesis of compounds 9–11
To a stirred solution of the appropriate ketone (1 mmol, 1 equiv.) and the corresponding aromatic aldehyde containing hydroxyl groups (2 mmol, 2 equiv.) in anhydrous ethanol (10 ml) at room temperature was added SOCl2 (2 mmol, 2 equiv.) slowly and stirring continued at room temperature. Upon completion (monitored by TLC), the mixture was poured into an excess amount of ice water and then filtered, dried and recrystallized from ethanol or DMF to give the title compounds.
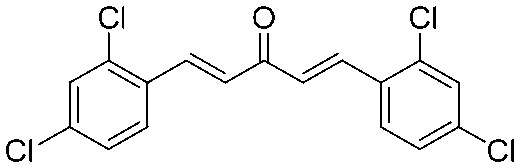
4.2.3. (1E,4E)-1,5-Bis(2,4-dichlorophenyl)penta-1,4-dien-3-one (1)
Yield 77%, mp: 207–210 °C. 1H NMR (300 MHz, CDCl3) δ 8.07 (2H, d, J = 18.0 Hz, H-β), 7.67 (2H, d, J = 6.0 Hz, Ar–H), 7.50 (2H, d, J = 3.0 Hz, Ar–H), 7.35 (2H, dd, J1 = 6.0 Hz, J2 = 6.0 Hz, Ar–H), 7.05 (2H, d, J = 18.0 Hz, H-α). 13C NMR (75 MHz, CDCl3) δ 188.2, 138.3 (×2), 136.7 (×2), 136.0 (×2), 131.5 (×2), 130.2 (×2), 128.4 (×2), 127.7 (×4). HRMS (ESI): [M + H]+ = 370.9564 (calcd. for C17H11Cl4O 370.9559).
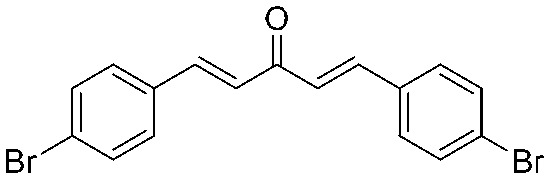
4.2.4. (1E,4E)-1,5-Bis(4-bromophenyl)penta-1,4-dien-3-one (2)
Yield 82%, mp: 146–148 °C. 1H NMR (300 MHz, CDCl3) δ 7.69 (2H, d, J = 15.9 Hz, H-β), 7.57 (4H, d, J = 8.4 Hz, Ar–H), 7.49 (4H, d, J = 8.4 Hz, Ar–H), 7.06 (2H, d, J = 15.9 Hz, H-α). 13C NMR (75 MHz, CDCl3) δ 188.3, 142.2 (×2), 133.6 (×2), 132.3 (×4), 129.8 (×4), 125.8 (×2), 124.8 (×2). HRMS (ESI): [M + H]+ = 390.9333 (calcd. for C17H13Br2O 390.9333).
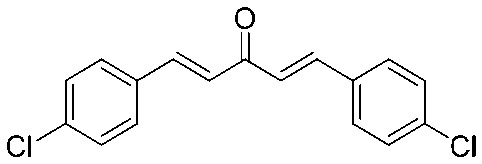
4.2.5. (1E,4E)-1,5-Bis(4-chlorophenyl)penta-1,4-dien-3-one (3)
Yield 79%, mp: 192–194 °C. 1H NMR (300 MHz, CDCl3) δ 7.70 (2H, d, J = 15.9 Hz, H-β), 7.56 (4H, d, J = 8.4 Hz, Ar–H), 7.41 (4H, d, J = 8.4 Hz, Ar–H), 7.05 (2H, d, J = 15.9 Hz, H-α). 13C NMR (75 MHz, CDCl3) δ 188.4, 142.1 (×2), 136.5 (×2), 133.2 (×2), 129.6 (×4), 129.3 (×4), 125.7 (×2). HRMS (ESI): [M + H]+ = 303.0343 (calcd. C17H13Cl2O 303.0343).
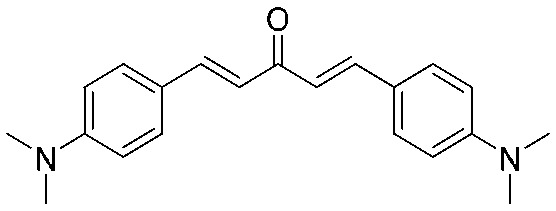
4.2.6. (1E,4E)-1,5-Bis(4-(dimethylamino)phenyl)penta-1,4-dien-3-one (4)
Yield 83%, mp: 188–190 °C. 1H NMR (300 MHz, CDCl3) δ 7.71 (2H, d, J = 15.9 Hz, H-β), 7.54 (4H, d, J = 8.7 Hz, Ar–H), 6.91 (2H, d, J = 15.9 Hz, H-α), 6.71 (4H, d, J = 8.7 Hz, Ar–H), 3.06 (12H, s, NCH3). 13C NMR (75 MHz, CDCl3) δ 188.9, 151.8 (×2), 143.0 (×2), 130.1 (×4), 122.9 (×2), 121.3 (×2), 111.9 (×4), 40.2 (×4). HRMS (ESI): [M + H]+ = 321.1965 (calcd. for C21H25N2O 321.1967).
4.2.7. (1E,4E)-1,5-Di(furan-2-yl)penta-1,4-dien-3-one (5)
Yield 65%, mp: 58–60 °C. 1H NMR (300 MHz, CDCl3) δ 6.51 (2H, m, Ar–H), 6.71 (2H, d, J = 3.6 Hz, Ar–H), 6.93 (2H, d, J = 15.6 Hz, H-α), 7.50 (2H, overlapped, Ar–H), 7.50 (2H, d, J = 15.9 Hz, H-β). 13C NMR (75 MHz, CDCl3) δ 188.2, 151.5 (×2), 145.0 (×2), 129.3 (×2), 123.2 (×2), 116.0 (×2), 112.7 (×2). HRMS (ESI): [M + H]+ = 215.0711 (calcd. for C13H11O5 215.0708).
4.2.8. (1E,4E)-1,5-Di(thiophen-2-yl)penta-1,4-dien-3-one (6)
Yield 70%, mp: 112–114 °C. 1H NMR (300 MHz, CDCl3) δ 7.86 (2H, d, J = 15.6 Hz, H-β), 7.43 (2H, d, J = 5.1 Hz, Ar–H), 7.35 (2H, d, J = 3.6 Hz, Ar–H), 7.10 (2H, t, J1 = 4.8 Hz, J2 = 8.7 Hz, Ar–H), 6.84 (2H, d, J = 15.6 Hz, H-α). 13C NMR (75 MHz, CDCl3) δ 187.7, 140.3 (×2), 135.6 (×2), 131.9 (×2), 128.9 (×2), 128.4 (×2), 124.4 (×2). HRMS (ESI): [M + H]+ = 247.0249 (calcd. for C13H11OS2 247.0251).
4.2.9. (1E,4E)-1,5-Bis(4-fluorophenyl)penta-1,4-dien-3-one (7)
Yield 63%, mp: 150–152 °C. 1H NMR (300 MHz, CDCl3) δ 7.72 (2H, d, J = 15.9 Hz, H-β), 7.63 (4H, dd, J1 = 5.4, J2 = 8.7 Hz, Ar–H), 7.13 (4H, t, J1 = 8.7 Hz, J2 = 17.4 Hz, Ar–H), 7.01 (2H, d, J = 15.9 Hz, H-α). 13C NMR (75 MHz, CDCl3) δ 188.5, 165.7–162.4 (×2, d, 1JCF = 250.2 Hz), 142.1 (×2), 130.9 (×2, d, JCF = 3.2 Hz), 130.4 (×4, d, JCF = 8.7 Hz), 125.1, 125.0, 116.2 (×4, d, JCF = 21.8 Hz). HRMS (ESI): [M + H]+ = 271.0936 (calcd. for C17H13F2O 271.0934).
4.2.10. (1E,4E)-1,5-Bis(2,6-dichlorophenyl)penta-1,4-dien-3-one (8)
Yield 78%, mp: 144–146 °C. 1H NMR (300 MHz, CDCl3) δ 7.84 (2H, d, J = 16.5 Hz, H-β), 7.40 (4H, d, J = 7.8 Hz, Ar–H), 7.23 (2H, d, J = 16.5 Hz, H-α), 7.23 (2H, d, J1 = 12.3 Hz, J2 = 16.2, Ar–H). 13C NMR (75 MHz, CDCl3) δ 188.9, 137.4 (×2), 135.2 (×2), 133.1 (×2), 132.3 (×4), 130.0 (×2), 128.9 (×4). HRMS (ESI): [M + H]+ = 370.9559 (calcd. for C17H11Cl4O 370.9564).
4.2.11. (1E,4E)-1,5-Bis(3,4-dihydroxyphenyl)penta-1,4-dien-3-one (9)
Yield 60%, mp: 151–153 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.63 (2H, s), 9.15 (2H, s), 7.57 (2H, d, J = 15.9 Hz, H-β), 7.11 (4H, t, J1 = 15.9 Hz, J2 = 24.0 Hz, Ar–H), 7.00 (2H, d, J = 15.9 Hz, H-α), 6.80 (2H, d, J = 8.1 Hz, Ar–H). 13C NMR (75 MHz, MeOH) δ 190.2, 148.5 (×2), 145.4 (×2), 144.2 (×2), 126.9 (×2), 122.2 (×2), 122.0 (×2), 115.2 (×2), 114.2 (×2). HRMS (ESI): [M + H]+ = 299.0919 (calcd. for C17H17O5 299.0920).
4.2.12. (1E,4E)-1,5-Bis(4-hydroxyphenyl)penta-1,4-dien-3-one (10)
Yield 70%, mp: 234–237 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.03 (2H, s, –OH), 7.66 (2H, d, J = 16.2 Hz, H-β), 7.63 (4H, d, J = 8.4 Hz, Ar–H), 7.10 (2H, d, J = 16.2 Hz, H-α), 6.84 (4H, d, J = 8.4 Hz, Ar–H). 13C NMR (75 MHz, MeOH) δ 190.3, 160.2 (×2), 143.8 (×2), 130.3 (×4), 126.3 (×2), 122.0 (×2), 115.5 (×4). HRMS (ESI): [M – H]– = 265.0867 (calcd. for C17 H13O3 265.0865).
4.2.13. (1E,4E)-1,5-Bis(3-hydroxy-4-methoxyphenyl)penta-1,4-dien-3-one (11)
Yield 78%, mp: 195–197 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.64 (2H, s, –OH), 7.66 (2H, d, J = 15.9 Hz, H-β), 7.38 (2H, d, J = 3.0 Hz, Ar–H), 7.21 (2H, d, J = 8.1 Hz, Ar–H), 7.15 (2H, d, J = 15.9 Hz, H-α), 6.84 (2H, d, J = 8.1 Hz, Ar–H), 3.86 (6H, s, OCH3). 13C NMR (75 MHz, DMSO-d6) δ 188.5, 149.9 (×2), 148.4 (×2), 143.2 (×2), 126.8 (×2), 123.8 (×2), 123.5 (×2), 116.1 (×2), 111.8 (×2), 56.2 (×2). HRMS (ESI): [M – H]– = 325.1075 (calcd. for C19H17O5 325.1076).
4.2.14. (1E,4E)-1,5-Bis(benzo[d][1,3]dioxol-5-yl)penta-1,4-dien-3-one (12)
Yield 79%, mp: 183–185 °C. 1H NMR (300 MHz, CDCl3) δ 7.66 (2H, d, J = 15.6 Hz, H-β), 7.14 (2H, s), 7.11 (2H, d, J = 8.1 Hz), 6.90 (2H, d, J = 15.6 Hz, H-α), 6.85 (2H, d, J = 8.1 Hz), 6.04 (4H, s, –OCH2O–). 13C NMR (75 MHz, CDCl3) δ 188.6, 149.8 (×2), 148.4 (×2), 142.9 (×2), 129.3 (×2), 125.1 (×2), 123.8 (×2), 108.7 (×2), 106.6 (×2), 101.6 (×2). HRMS (ESI): [M + H]+ = 323.0921 (calcd. for C19H15O5 323.0920).
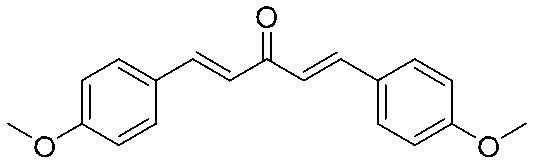
4.2.15. (1E,4E)-1,5-Bis(4-methoxyphenyl)penta-1,4-dien-3-one (13)
Yield 82%, mp: 129–131 °C. 1H NMR (300 MHz, CDCl3) δ 7.72 (2H, d, J = 16.2 Hz, H-β), 7.59 (4H, d, J = 8.7 Hz, Ar–H), 7.00 (2H, d, J = 16.2 Hz, H-α), 6.94 (4H, d, J1 = 5.7 Hz, J2 = 8.7 Hz, Ar–H), 3.87 (6H, s, OCH3). 13C NMR (75 MHz, CDCl3) 188.9, 161.5 (×2), 142.7 (×2), 130.1 (×4), 127.6 (×2), 123.5 (×2), 114.4 (×4), 55.4 (×2). HRMS (ESI): [M + H]+ = 295.1323 (calcd. for C19H19O3 295.1334).
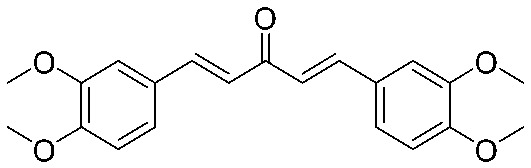
4.2.16. (1E,4E)-1,5-Bis(3,4-dimethoxyphenyl)penta-1,4-dien-3-one (14)
Yield 83%, mp: 82–84 °C. 1H NMR (300 MHz, CDCl3) δ 7.71 (2H, d, J = 15.9 Hz, H-β), 7.22 (2H, dd, J1 = 1.5 Hz, J2 = 8.1 Hz, Ar–H), 7.16 (s, 2H), 6.97 (2H, d, J = 15.9 Hz, H-α), 6.91 (2H, d, J = 8.4 Hz), 3.96 (6H, s, OCH3), 3.95 (6H, s, OCH3). 13C NMR (75 MHz, CDCl3) δ 188.7, 151.3 (×2), 149.2 (×2), 143.1 (×2), 127.8 (×2), 123.6 (×2), 123.1 (×2), 111.1 (×2), 109.8 (×2), 56.0 (×2), 55.9 (×2). HRMS (ESI): [M + H]+ = 355.1543 (calcd. for C21H23O5 355.1546).
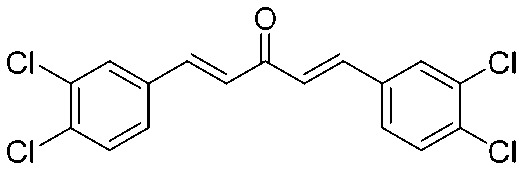
4.2.17. (1E,4E)-1,5-Bis(3,4-dichlorophenyl)penta-1,4-dien-3-one (15)
Yield 70%, mp: 191–193 °C. 1H NMR (300 MHz, CDCl3) δ 7.70 (2H, s), 7.62 (2H, d, J = 15.6 Hz, H-β). 7.48 (4H, m, Ar–H), 7.02 (2H, d, J = 15.6 Hz, H-α). 13C NMR (75 MHz, CDCl3) δ 187.7, 141.0 (×2), 134.6 (×2), 133.4 (×2), 131.0 (×2), 129.8 (×2), 129.2 (×2), 127.5 (×2), 126.5 (×2). HRMS (ESI): [M + H]+ = 370.9563 (calcd. for C17H11Cl4O 370.9564).
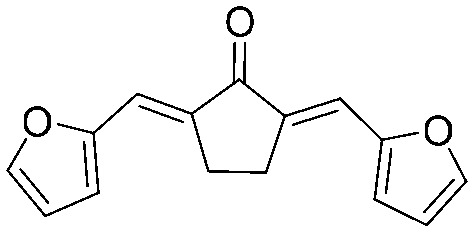
4.2.18. (2E,5E)-2,5-Bis(furan-2-ylmethylene)cyclopentan-1-one (16)
Yield 65%, mp: 166–168 °C. 1H NMR (300 MHz, CDCl3) δ 7.62 (2H, d, J = 1.8 Hz, Ar–H), 7.38 (2H, s, Ar–H), 6.72 (2H, d, J = 3.3 Hz, Ar–H), 6.57 (2H, m, Ar–H), 3.10 (4H, s, –CH2CH2–). 13C NMR (75 MHz, CDCl3) δ 195.4, 152.7 (×2), 145.1 (×2), 135.9 (×2), 119.8 (×2), 116.0 (×2), 112.7 (×2), 25.8 (×2). HRMS (ESI): [M + H]+ = 241.0859 (calcd. for C15H13O3 241.0865).
4.2.19. 2,5-Bis((E)-4-methoxybenzylidene)cyclopentan-1-one (17)
Yield 77%, mp: 212–214 °C. 1H NMR (300 MHz, CDCl3) δ 7.60 (6H, m), 6.99 (4H, d, J = 8.7 Hz, Ar–H), 3.89 (6H, s, OCH3), 3.10 (4H, s, –CH2CH2–). 13C NMR (75 MHz, CDCl3) δ 196.3, 160.5 (×2), 135.3 (×2), 133.3 (×2), 132.5 (×4), 128.8 (×2), 114.3 (×4), 55.4 (×2), 26.5 (×2). HRMS (ESI): [M + H]+ = 321.1485 (calcd. for C21H21O3 321.1491).
4.2.20. 2,5-Bis((E)-4-hydroxybenzylidene)cyclopentan-1-one (18)
Yield 67%, mp: >300 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.09 (2H, s), 7.56 (4H, d, J = 8.4 Hz), 7.36 (2H, s), 6.90 (4H, d, J = 8.1 Hz), 3.03 (4H, s, –CH2CH2–). 13C NMR (75 MHz, MeOD) δ 199.3, 163.6 (×2), 139.2 (×2), 137.4 (×4), 137.1 (×2), 131.3 (×2), 120.6 (×4), 30.6 (×2). HRMS (ESI): [M + H]+ = 293.1172 (calcd. for C19 H17O3 293.1178).
4.2.21. 2,5-Bis((E)-4-(dimethylamino)benzylidene)cyclopentan-1-one (19)
Yield 82%, mp: 118–120 °C. 1H NMR (300 MHz, CDCl3) δ 7.57 (6H, m), 6.77 (4H, d, J = 8.4 Hz), 3.10 (4H, s, –CH2CH2–), 3.07 (12H, s, OCH3). 13C NMR (75 MHz, CDCl3) δ 196.1, 150.7 (×2), 133.5 (×2), 132.6 (×2), 132.6 (×4), 124.3 (×2), 111.9 (×4), 40.1 (×2), 26.7 (×4). HRMS (ESI): [M + H]+ = 347.2123 (calcd. for C23H27N2O 347.2123).
4.2.22. (2E,5E)-2,5-Bis(benzo[d][1,3]dioxol-5-ylmethylene)cyclopentan-1-one (20)
Yield 78%, mp: 247–249 °C. 1H NMR (600 MHz, CDCl3) δ 7.50 (2H, s, Ar–H), 7.12 (2H, dd, J1 = 8.4 Hz, J2 = 1.8 Hz, Ar–H), 7.10 (2H, d, J = 1.8 Hz, Ar-H), 6.87 (2H, d, J = 8.4 Hz, Ar-H), 6.02 (4H, s, –OCH2O–), 3.06 (4H, s, –CH2CH2–). 13C NMR (75 MHz, CDCl3) δ 196.1, 148.7 (×2), 135.5 (×2), 133.6 (×2), 130.3 (×2), 126.7 (×2), 109.7 (×2), 108.8 (×2), 101.5 (×2), 29.7 (×2), 26.5 (×2). HRMS (ESI): [M + H]+ = 349.1080 (calcd. for C21H17O5 349.1076).
4.2.23. (2E,5E)-2,5-Bis(3,4-dimethoxybenzylidene)cyclopentan-1-one (21)
Yield 80%, mp: 155–157 °C. 1H NMR (300 MHz, CDCl3) δ 7.58 (2H, s), 7.28 (2H, m), 7.17 (2H, s), 6.97 (2H, d, J = 8.4 Hz), 3.97 (12H, s), 3.15 (4H, s, –CH2CH2–). 13C NMR (75 MHz, CDCl3) δ 196.1, 150.2 (×2), 148.9 (×2), 135.4 (×2), 133.7 (×2), 129.0 (×2), 124.6 (×2), 113.4 (×2), 111.1 (×2), 56.0 (×2), 55.9 (×2), 26.5 (×2). HRMS (ESI): [M + H]+ = 381.1698 (calcd. for C23H25O5 381.1702).
4.2.24. (2E,5E)-2,5-Bis(2,3-dimethoxybenzylidene)cyclopentanone (22)
Yield 77%, mp: 140–142 °C. H NMR (600 MHz, CDCl3): 7.92 (1H × 2, s, –CH ), 7.15 (1H × 2, dd, J1 = 7.8 Hz, J2 = 1.2 Hz, Ar-H), 7.08 (1H × 2, m, Ar-H), 6.94 (1H × 2, dd, J1 = 7.8 Hz, J2 = 1.2 Hz, Ar-H), 3.88 (6H × 2, s, –OCH3), 3.02 (4H, s, –CH2CH2–). 13C NMR (75 MHz, CDCl3) δ 196.3, 153.0 (×2), 149.2 (×2), 138.6 (×2), 130.2 (×2), 128.2 (×2), 123.8 (×2), 121.6 (×2), 113.4 (×2), 61.5 (×2), 55.9 (×2), 26.8 (×2). HRMS (ESI): [M + H]+ = 381.1700 (calcd. for C23H25O5 381.1702).
4.2.25. (2E,5E)-2,5-Bis((5-(hydroxymethyl)furan-2-yl)methylene)cyclopentan-1-one (23)
Yield 65%, mp: 151–153 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.18 (2H, s), 6.94 (2H, d, J = 3.3 Hz), 6.56 (2H, d, J = 3.3 Hz), 5.44 (2H, m), 4.50 (4H, d, J = 4.2 Hz), 3.02 (4H, s). 13C NMR (75 MHz, DMSO) δ 194.4, 159.3 (×2), 151.6 (×2), 135.9 (×2), 119.3 (×2), 118.1 (×2), 110.7 (×2), 56.4 (×2), 25.9 (×2). HRMS (ESI): [M + H]+ = 301.1072 (calcd. for C17H17O5 301.1076).
4.2.26. (1E,4Z,6E)-1,7-Bis(3,4-dihydroxyphenyl)-5-hydroxyhepta-1,4,6-trien-3-one (24)
A suspension of curcumin (368 mg, 1 mmol) in 20 mL of anhydrous dichloromethane was cooled to –20 °C under a nitrogen atmosphere and 6.5 mL of 1 M boron tribromide solution in dichloromethane (6.5 equiv.) was slowly added to this stirred suspension. After 1 h, the reaction was allowed to proceed at room temperature for 24 h. The reaction mixture was carefully poured into a saturated sodium bicarbonate solution with stirring. The water and dichloromethane layers were separated and the water layer was extracted three times with ethyl acetate. The combined dichloromethane layer and ethyl acetate layer were dried over anhydrous sodium sulfate for 12 h after filtration, and then removed using a rotary evaporator and the resulting solid was purified by silica gel column chromatography (CH2Cl2/MeOH; 40 : 1–5 : 1%) using a Sephadex LH-20 column.
Yield 55%, mp: >200 °C. 1H NMR (300 MHz, DMSO-d6) δ 6.08 (1H, s), 6.58 (2H, d, J = 15.6 Hz, H-α), 6.79 (2H, d, J = 8.1 Hz), 7.04 (4H, m), 7.46 (2H, d, J = 15.6 Hz, H-β), 9.20 (2H, s), 9.65 (2H, s).13C NMR (75 MHz, DMSO-d6) δ 183.6, 148.8 (×2), 146.1 (×2), 141.2 (×2), 126.7 (×2), 122.1 (×2), 121.1 (×2), 116.3 (×2), 115.1 (×2), 101.4 (×2). HRMS (ESI): [M + H]+ = 341.1023 (calcd. for C19H17O6 341.1025).
4.3. Biological activity assays
4.3.1. Antifungal activity (broth microdilution method)
Clinical isolates of fluconazole resistant C. albicans 103, C. tropicalis 087 and C. krusei 2159 (MIC80 = 128.0 μg ml–1) were used in this study, and C. albicans ATCC 90028 was used as a quality control. The antifungal properties of the title compounds were evaluated by the broth microdilution method according to the NCCLS reference document M27-A2.14 Curcumin was used as a positive control. The MIC80 of FLC against the FLC-resistant C. albicans (clinical isolate 103) was determined to be 128.0 μg ml–1. The MIC80 values of the title compounds when used alone and when combined with FLC (8.0 μg ml–1) are described in Table 1. Furthermore, the fractional inhibitory concentration index (FICI) of each agent was calculated by adding the MIC80 (with FLC)/MIC80 (used alone) ratios. The interaction modes—synergistic or indifferent—were defined according to FICI values of ≤0.5 or >0.5, respectively.
4.3.2. Antifungal susceptibility assays (turbidity observation method)
Antifungal susceptibility assays were done according to Zhao et al.24 with little modification. The initial concentration of the fluconazole-resistant C. tropicalis 087 suspension was 104 CFU ml–1 in RPMI 1640 medium in glass tubes. Different concentrations of compound 9 (0, 4, 8, 16, 32, 64, 128 μg ml–1) and fluconazole (0, 0.25, 0.5, 1, 2 μg ml–1) alone or in combination were added to the tubes. The test tubes were incubated at 30 °C, 200 rpm, for 16 h; the turbidity was monitored and pictures were taken.
4.3.3. Cell growth and cell death measurement (growth curves)
Growth curves were obtained as previously described24 with little modification. Cells were incubated in RPMI 1640 medium with increasing concentrations of fluconazole (0.25, 0.5, 1 μg ml–1) alone or in combination with compound 9 (64, 128 μg ml–1). The growth condition was determined at OD630 for an interval of 8 hours. Curves were generated using Prism 5.0 (Graph-Pad Software, Inc.)
4.3.4. Measurement of intracellular ATP levels
Measurement of intracellular ATP levels was performed as reported before.19C. tropicalis 087 was treated with fluconazole (4 μg ml–1), curcumin (32, 64, 128 μg ml–1) and compound 9 (32, 64, 128 μg ml–1) or a combination for 8 hours and then adjusted to 1 × 107 CFU ml–1 in PBS (phosphate buffered saline). Intracellular ROS of C. tropicalis 087 were observed with a confocal scanning laser microscope (excitation, 485 nm, emission, 538 nm; Leica TCS sp2). For the measurement of ATP level, a total volume of 100 μl BacTiter-Glo reagent (Promega Corparation, Madison, WI) was mixed completely with the same volume of cell suspension and incubated for 10 minutes at 30 °C. Fluorescence intensities were determined on a TD 20/20 luminometer (Turner Biosystem, Sunnyvale, CA) and ATP contents were calculated based on the ATP increment standard curve (1 μM to 10 pM). Each experiment was independently repeated three times.
4.3.5. Cell membrane permeability assay
The membrane permeability assay was performed according to a previous work.20C. tropicalis 087 (1 × 106) was suspended in RPMI 1640 medium and treated with fluconazole (2 μg ml–1) and compound 9 (32 μg ml–1) for 12 hours at 30 °C. After the treatment, cells were incubated in water with 1.49 μM PI at 30 °C for 50 minutes. C. tropicalis 087 was harvested by centrifugation and resuspended in PBS (phosphate buffered saline). Flow cytometric analysis was performed using a FACS Calibur flow cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA).
4.3.6. Cell culture and MTT cytotoxicity assay
Human umbilical vein endothelial cells (HUVECs) were cultured in DMEM (Gibco, USA) with 10% FCS (Gibco, CA, USA) and 100 μg ml–1 streptomycin in 5% CO2 at 37 °C. The cytotoxicity assay of curcumin and its analogues was based on the reduction of 3-(4,5-dimethyl-thiazol-2yl)-2,5-diphenyl tetrazolium bromide (MTT, Sigma, USA) as previously described.25 To determine the cytotoxicity, cells were exposed to different concentrations of compounds (16 and 32 μg ml–1) for 24 h. After the treatment, cells were incubated with MTT solution (5 mg ml–1) for 3 h. The optical density of different samples was measured with a microplate reader (Thermo, MA, USA) at 570 nm. The cytotoxic activities were compared with those of the drug-free control and expressed as the relative viability percentages.
Acknowledgments
This work was supported by a grant from the Key Program of the National Natural Science Foundation of China (81330083) and Undergraduates Innovation Practice Base of Second Military Medical University (YJ-2015-08, YJ-2015-07).
Footnotes
†The authors declare no competing interests.
References
- Youngsaye W., Dockendorff C., Vincent B., Hartland C. L., Bittker J. A., Dandapani S., Palmer M., Whitesell L., Lindquist S., Schreiber S. L., Munoz B. Bioorg. Med. Chem. Lett. 2012;22:3362–3365. doi: 10.1016/j.bmcl.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamarra S., Rocha E. M., Zhang Y. Q., Park S., Rao R., Perlin D. S. Antimicrob. Agents Chemother. 2010;54:1753–1761. doi: 10.1128/AAC.01728-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre-Godal Q., Dorsaz S., Queiroz E. F., Conan C., Marcourt L., Wardojo B. P. E., Voinesco F., Buchwalder A., Gindro K., Sanglard D., Wolfender J.-L. Phytochemistry. 2014;105:68–78. doi: 10.1016/j.phytochem.2014.06.004. [DOI] [PubMed] [Google Scholar]
- Cragg G. M., Newman D. J. Biochim. Biophys. Acta, Gen. Subj. 2013;1830:3670–3695. doi: 10.1016/j.bbagen.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey A. L. Drug Discovery Today. 2008;13:894–901. doi: 10.1016/j.drudis.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Nagahama K., Utsumi T., Kumano T., Maekawa S., Oyama N., Kawakami J. Sci. Rep. 2016;6:30962. doi: 10.1038/srep30962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De R., Kundu P., Swarnakar S., Ramamurthy T., Chowdhury A., Nair G. B., Mukhopadhyay A. K. Antimicrob. Agents Chemother. 2009;53:1592–1597. doi: 10.1128/AAC.01242-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelofar K., Shreaz S., Rimple B., Muralidhar S., Nikhat M., Khan L. A. Can. J. Microbiol. 2011;57:204–210. doi: 10.1139/W10-117. [DOI] [PubMed] [Google Scholar]
- Martins C. V., da Silva D. L., Neres A. T., Magalhaes T. F., Watanabe G. A., Modolo L. V., Sabino A. A., de Fatima A., de Resende M. A. J. Antimicrob. Chemother. 2009;63:337–339. doi: 10.1093/jac/dkn488. [DOI] [PubMed] [Google Scholar]
- Sharma M., Manoharlal R., Puri N., Prasad R. Biosci. Rep. 2010;30:391–404. doi: 10.1042/BSR20090151. [DOI] [PubMed] [Google Scholar]
- Anand P., Kunnumakkara A. B., Newman R. A., Aggarwal B. B. Mol. Pharmaceutics. 2007;4:807–818. doi: 10.1021/mp700113r. [DOI] [PubMed] [Google Scholar]
- Pan M. H., Huang T. M., Lin J. K. Drug Metab. Dispos. 1999;27:486–494. [PubMed] [Google Scholar]
- Sardjiman S. S., Reksohadiprodjo M. S., Hakim L., van der Goot H., Timmerman H. Eur. J. Med. Chem. 1997;32:625–630. [Google Scholar]
- Rex J., Alexander B. D., Andes D., Arthington-Skaggs B., Brown S. D., Chaturvedi V., Ghannoum M. A., Espinel-Ingrof A., Knapp C. C., Ostrosky-Zeichner L., Pfaller M. A., Sheehan D. J. and Walsh T. J., Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, Approved Standard, CLSI document M27-A3, Clinical and Laboratory Standards Institute, Wayne, PA, 3rd edn, 2008, pp. 6–25. [Google Scholar]
- Anand P., Thomas S. G., Kunnumakkara A. B., Sundaram C., Harikumar K. B., Sung B., Tharakan S. T., Misra K., Priyadarsini I. K., Rajasekharan K. N., Aggarwal B. B. Biochem. Pharmacol. 2008;76:1590–1611. doi: 10.1016/j.bcp.2008.08.008. [DOI] [PubMed] [Google Scholar]
- Straganz G. D., Nidetzky B. J. Am. Chem. Soc. 2005;127:12306–12314. doi: 10.1021/ja042313q. [DOI] [PubMed] [Google Scholar]
- Liang G., Shao L., Wang Y., Zhao C., Chu Y., Xiao J., Zhao Y., Li X., Yang S. Bioorg. Med. Chem. 2009;17:2623–2631. doi: 10.1016/j.bmc.2008.10.044. [DOI] [PubMed] [Google Scholar]
- Odds F. C. J. Antimicrob. Chemother. 2003;52:1. doi: 10.1093/jac/dkg301. [DOI] [PubMed] [Google Scholar]
- Xu Y., Wang Y., Yan L., Liang R.-M., Dai B.-D., Tang R.-J., Gao P.-H., Jiang Y.-Y. J. Proteome Res. 2009;8:5296–5304. doi: 10.1021/pr9005074. [DOI] [PubMed] [Google Scholar]
- Maurya I. K., Pathak S., Sharma M., Sanwal H., Chaudhary P., Tupe S., Deshpande M., Chauhan V. S., Prasad R. Peptides. 2011;32:1732–1740. doi: 10.1016/j.peptides.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Bai Y., Wang W., Sun G., Zhang M., Dong J. Cell Proliferation. 2016;49:751–762. doi: 10.1111/cpr.12289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M., Manoharlal R., Shukla S., Puri N., Prasad T., Ambudkar S. V., Prasad R. Antimicrob. Agents Chemother. 2009;53:3256–3265. doi: 10.1128/AAC.01497-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gomes A. S., Curvelo J. A., Soares R. M., Ferreira-Pereira A. Med. Mycol. 2012;50:26–32. doi: 10.3109/13693786.2011.578156. [DOI] [PubMed] [Google Scholar]
- Zhao L., Jiang J., Zhu Z., Liao Z., Yao X., Yang Y., Cao Y., Jiang Y. Acta Biochim. Biophys. Sin. 2016;48:182–193. doi: 10.1093/abbs/gmv125. [DOI] [PubMed] [Google Scholar]
- Margina D., Gradinaru D., Manda G., Neagoe I., Ilie M. Food Chem. Toxicol. 2013;61:86–93. doi: 10.1016/j.fct.2013.02.046. [DOI] [PubMed] [Google Scholar]