

Abstract
Ridinilazole, a novel targeted antibacterial being developed for the treatment of C. difficile infection (CDI) and prevention of recurrence, was shown in a recent Phase 2 study to be superior to vancomycin with regard to the primary efficacy measure, sustained clinical response (SCR), with the superiority being driven primarily by marked reductions in the rates of CDI recurrence within 30 days. Tolerability of ridinilazole was comparable to that of vancomycin. The current nested cohort study compared the effects of ridinilazole and vancomycin on fecal microbiota during and after treatment among participants in the Phase 2 study. Changes in the microbiota were assessed using qPCR and high-throughput sequencing on participants’ stools collected at multiple time-points (baseline [Day 1], Day 5, end-of-treatment [EOT; Day 10], Day 25, end-of-study [EOS; Day 40], and at CDI recurrence). qPCR analyses showed profound losses of Bacteroides, C. coccoides, C. leptum, and Prevotella groups at EOT with vancomycin treatment, while ridinilazole-treated participants had a modest decrease in C. leptum group levels at EOT, with levels recovering by Day 25. Vancomycin-treated participants had a significant increase in the Enterobacteriaceae group, with this increase persisting beyond EOT. At EOT, alpha diversity decreased with both antibiotics, though to a significantly lesser extent with ridinilazole (p <0.0001). Beta diversity analysis showed a significantly larger weighted Unifrac distance from baseline-to-EOT with vancomycin. Taxonomically, ridinilazole had a markedly narrower impact, with modest reductions in relative abundance in Firmicutes taxa. Microbiota composition returned to baseline sooner with ridinilazole than with vancomycin. Vancomycin treatment resulted in microbiome-wide changes, with significant reductions in relative abundances of Firmicutes, Bacteroidetes, Actinobacteria, and a profound increase in abundance of Proteobacteria. These findings demonstrate that ridinilazole is significantly less disruptive to microbiota than vancomycin, which may contribute to the reduced CDI recurrence observed in the Phase 2 study.
Introduction
Over 450,000 cases of CDI occur in the US annually, with over 80,000 first recurrences and approximately 29,000 deaths [1]. The most common precipitant is antibiotic use. Antibiotics cause loss of colonization resistance with the potential establishment of a long-lasting, species-poor microbiota susceptible to pathogen invasion [2–5]. Oral vancomycin and metronidazole treatment are associated with high CDI recurrence rates, likely due to deleterious effects on resident colonic flora [6, 7]. Recurrences are costly in terms of both clinical burden and healthcare resource utilization [8, 9]. In one study, readmission was required in approximately one-third of recurrence cases [8].
Both microbiota biomass and composition at the intestinal-bacterial interface likely influence the C. difficile colonization niche [7]. Although colonization resistance has been associated with specific taxa [10–15], it is likely that different, yet diverse, microbiota community structures can confer protection. Consistent characteristics of communities susceptible to CDI are low diversity levels and diminished metabolic function [16] with loss of relative abundance of members of the Bacteroidetes and Firmicutes phyla and increases in that of Proteobacteria [17–21]. Fecal microbiota transplantation (FMT) normalizes these features and breaks the CDI recurrence cycle [22–26].
In aggregate, these data support a role for CDI agents with minimal effects on indigenous microbiota to reduce risk of recurrence. Ridinilazole (SMT19969) is a narrow-spectrum, non-absorbable, potent C. difficile-targeting antimicrobial [27]. In a recent Phase 2 randomized, controlled, double-blinded clinical trial comparing its efficacy to vancomycin, ridinilazole was associated with marked reduction in rates of recurrent disease (14.3% vs. 34.8%) [28]. The goal of the present study was to compare effects over time and treatment group on host fecal microbiota biomass and taxonomic composition in participants from the Phase 2 study. Fecal biomasses of specific microbial families (Eubacteria and 5 different microbiota groups; Bacteroides, C. coccoides, C. leptum, Enterobacteriaceae, and Prevotella) were quantified by qPCR. Relative abundances of resident taxa, microbiota diversity, and taxonomic composition were assessed by high-throughput sequencing.
Materials and methods
Participant enrollment and sample collection
One hundred patients were enrolled and randomized 1:1 to receive 10 days of either vancomycin or ridinilazole [28]. Institutional review boards at each center (see S1 Table for details) provided ethics approval. The study complied with ethical principles expressed in the Declaration of Helsinki and followed all principles of good clinical practice. Written informed consent was obtained from all participants. Stool samples were obtained at study entry (baseline, D1), day 5 (D5), day 10 (D10, EOT), day 25 (D25) and day 40 post-entry (D40, end-of-study, EOS), and if recurrence was suspected. In order to provide a healthy control benchmark for this study, control stool samples were obtained from age-, gender- and location-similar volunteers enrolled in a separate study [29], and handled similarly as the samples from CDI subjects in this study. Of 100 patients enrolled in the clinical trial, 18 did not provide at least 3 stool samples for analysis (Fig 1). Due to the fact that stool microbiota is highly individual-specific, the appropriate comparison is of the patient to themselves over the time of the study. Thus,18 subjects from whom a limited number of stools were collected were eliminated from this study. Of the remaining 82 participants, there were 41 per treatment arm. As any antibiotic therapy can alter microbiota, thus introducing confounding effects, some of the Phase 2 study subjects and samples were excluded from this nested cohort study in order to confine the analysis to studying the effect of ridinilazole or vancomycin only. Some subjects received concomitant non-CDI antibiotics during the study, so any sample obtained after initiation of treatment with non-CDI antibiotics was excluded. Similarly, samples were not included in the analysis if participants were being treated for a suspected CDI recurrence. Some subjects received up to 24 hours of standard CDI treatment prior to enrollment, which was subsequently determined to have an effect on baseline microbiota (see results). These subjects were studied separately and removed from the primary analysis.
Fig 1. Consort diagram for participant inclusion in the nested cohort.

Abbreviations: RDZ, ridinilazole; VAN, vancomycin.
Stool DNA extraction
DNA was extracted using the QIAquick FAST DNA Stool kit (QIAGEN, Mississauga, Canada) with these modifications: ~250mg of frozen stool was suspended in 1.4mL of Inhibitex buffer (QIAGEN) supplemented with 50μg of lysozyme (Sigma) and 13.5U of lysostaphin (Sigma), then subjected to 5 minutes of bead-beating with 500mg of 0.1mm silica/zirconium beads (BioSpec Corp) on a Vortex Genie with a MoBio adapter. The supernatant was incubated at 70°C for 10 minutes with 100μg Proteinase K QIAGEN) and 400μg RNase A (Sigma), microfuged for 1 minute and transferred to a clean tube, after which the manufacturer’s protocol was resumed. DNA was eluted in 100μL of buffer. If the concentration as measured on a Nanodrop 2000 was <10μg/mL, the extraction was repeated. A 50μL portion of the extracted DNA was removed for qPCR analysis and the remainder was used for 16S rDNA amplicon generation.
Quantitative PCR (qPCR) analysis
After ethanol precipitation of the extracted DNA to eliminate inhibitors, the reconstituted DNA was checked for purity and concentration using a Synergy H1 Hybrid Reader with Take3 Micro-Volume plates (BioTek, Winooski, VT) and then diluted with PCR-grade water to 5ng/μL. The DNA levels of bacterial groups were assessed using well-established PCR primers/conditions (S2 Table) [30–34]. Using the Mx3000P qPCR System (Agilent Technologies, Santa Clara, CA), qPCR was performed on each sample in triplicate in a final volume of 20μL containing 25ng DNA template, primers at 0.3μM, and SYBR Green 2x Master Mix (QIAGEN), with a FAM-tagged probe at 0.25μM for Eubacteria. Threshold cycle values were converted to copies per ng of DNA using a standard curve. Standards were prepared by performing PCR using the above group-specific primers on appropriate bacterial strains or DNA from normal stool. The products were cloned using Invitrogen Zero Blunt TOPO PCR Cloning Kit (ThermoFisher Scientific, Waltham, MA), and inserts were verified by sequencing at the Tufts University Core Facility. A Basic Local Alignment Search Tool (BLAST) search was performed to identify the closest matching database sequence (S3 Table). A range of 10-fold serially diluted plasmid standard DNA (5X108 to 500 copies) was run on each qPCR plate in triplicate. Standard curve R2 values ranged from 0.990–0.999, with inter-assay variability and coefficient of variance below 6% and 0.07, respectively. Copies per gram of stool were then calculated, accounting for initial sample DNA concentrations and stool weights. The change in bacterial levels (Δlog10 copies/gram stool) from entry level to each available successive time-point was determined for each participant and median changes were then calculated.
High-throughput sequencing
Amplicons of the V4 region of bacterial 16S rDNA were generated from extracted DNA using primers described by Caporaso [35]. Triplicate PCR amplifications on 50ng of DNA were performed using a 5’ HotStarTaq Master Mix kit (Thermo Fisher) with the following conditions: 94°C for 3 minutes followed by 35 cycles of 94°C for 45 seconds, 50°C for 60 seconds, and 72°C for 90 seconds, with a final extension of 72°C for 10 min. Amplicon DNA concentrations were determined by Quant-iT assay (Invitrogen, Carlsbad, CA), pooled in equimolar concentrations, purified (Qiaquick PCR Purification Kit, QIAGEN), and then eluted from Agencourt Ampure XP beads (Beckman-Coulter, Beverly, MA). Amplicon pools of ~100 samples each were sequenced on an Illumina MiSeq at the Tufts University Core Facility by a standard 250bp paired-end Illumina protocol. Initial data was processed using QIIME Version 1.8.0, an open- source software on the Galaxy website (http://huttenhower.sph.harvard.edu/galaxy/) [35, 36]. Operational taxonomic units (OTUs) were defined by 99% identity; taxonomic assignment was by closed reference with GreenGenes 13_8. To analyze differences between treatment groups and across timepoints, the OTU table was normalized to the lowest number of sequences per sample, then consolidated by summing to species level and to each successively higher taxonomic level. The alpha diversity parameters Chao and Shannon (based on number of OTUs) and Phylogentic Diversity (PD) (based on phylogenetic relationships), were generated in QIIME; alpha diversity is widely regarded as an indication of microbiota “health”. For beta diversity, which describes the relationships between samples, weighted Unifrac distance matrices were generated in QIIME.
Statistical analyses
For both qPCR and high-throughput sequencing data, significance of differences between treatment groups at each time point was calculated using Mann-Whitney U test while within each treatment arm, significance of differences between time-points was assessed by the Wilcoxon Signed Rank test; both were performed with GraphPad Prism (GraphPad, San Diego, CA) on log-transformed data.
For high-throughput sequencing data, the LEfSe (Linear Effect Size) algorithm [37] was used to identify significant differences in microbiota composition between baseline and each study timepoint, followed by Wilcoxon Signed Rank tests on identified taxa. The Benjamini-Hochberg procedure was used to control the false discovery rate at 0.10 [38]. The MaAsLin algorithm on the Galaxy website was used to find associations of taxa with treatment. For beta diversity, principal co-ordinates analyses on weighted Unifrac distance matrices generated in QIIME were performed in the vegan package in the statistical program R. For qPCR data, to analyze whether trajectories across time differed between treatment groups, repeated measures models with time and group as categorical fixed factors [39] were used on log-transformed data, with diagnostics to assess the impact of potential influential points.
Results
Pre-enrollment standard CDI therapy resulted in different baseline microbiota
Twenty-two participants in the trial had received up to 24 hours of metronidazole and/or vancomycin immediately prior to randomization. The qPCR analysis of their baseline samples revealed that C. coccoides and C. leptum groups were 0.76 and 0.42 log10 copies/gram lower (p = 0.004 and 0.025, respectively) and Enterobacteriaceae was 1.28 log10 copies/gram higher (p = 0.018) than those who did not receive pre-enrollment treatment. Their baseline microbiota relative abundance profiles also differed dramatically (S1 Fig). Therefore, the pretreated participants were excluded from the primary qPCR and microbiota data analysis, leaving 22 participants in each treatment arm (Fig 1). There were no significant differences in age, sex, BMI, or use of PPIs, NSAIDS, opioids or probiotics between treatment groups.
Baseline microbiota were not different between treatment groups
At study entry, there was no difference by qPCR in log10 copies/gram stool in any bacterial group between ridinilazole- and vancomycin-treated participants (Table 1; see S4 Table for supporting data). Study participants’ baseline samples had significantly lower copies/gram stool of all groups tested (p <0.5–<0.001) except Enterobacteriaceae, when compared to a population of 14 healthy volunteers as a benchmark.
Table 1. Comparison of bacterial group median quantities at each timepoint within treatment arms.
| Ridinilazole | Vancomycin | Controls | |||||||
|---|---|---|---|---|---|---|---|---|---|
| # of subjects→ | n = 21 | n = 18 | n = 14 | ||||||
| Baseline | Day 5 | Δlog10 | p-value | Baseline | Day 5 | Δlog10 | p-value | ||
| Bacteroides | 6.15E+09 | 1.19E+10 | 0.28 | ns | 6.28E+09 | 1.02E+06 | −3.47 | <0.0001 | 1.07E+10 |
| Eubacteria | 5.48E+09 | 7.35E+09 | 0.08 | ns | 5.60E+09 | 1.59E+09 | −0.43 | 0.016 | 1.77E+10 |
| Enterobacteriaceae | 6.98E+07 | 1.18E+08 | 0.02 | ns | 4.79E+07 | 3.61E+08 | 0.60 | <0.001 | 5.95E+06 |
| C. coccoides | 9.14E+08 | 4.11E+08 | −0.47 | ns | 1.40E+09 | 1.86E+06 | −3.00 | <0.0001 | 5.75E+09 |
| C. leptum | 5.83E+08 | 1.85E+08 | −0.28 | ns | 2.67E+08 | 2.33E+05 | −3.02 | <0.0001 | 2.33E+09 |
| Prevotella | 2.95E+09 | 3.39E+09 | 0.06 | ns | 1.10E+09 | 5.52E+06 | −2.30 | <0.0001 | 1.02E+10 |
| # of subjects→ | n = 21 | n = 19 | |||||||
| Baseline | Day 10 | Δlog10 | p-value | Baseline | Day 10 | Δlog10 | p-value | ||
| Bacteroides | 6.15E+09 | 1.12E+10 | 0.09 | ns | 8.52E+09 | 1.14E+06 | −3.42 | <0.0001 | |
| Eubacteria | 5.48E+09 | 6.98E+10 | 0.01 | ns | 5.74E+09 | 1.81E+06 | −0.53 | 0.006 | |
| Enterobacteriaceae | 6.98E+07 | 1.69E+08 | −0.01 | ns | 3.38E+07 | 5.38E+08 | 0.51 | <0.0001 | |
| C. coccoides | 9.14E+08 | 5.57E+08 | −0.29 | ns | 1.48E+09 | 1.89E+06 | −2.83 | <0.0001 | |
| C. leptum | 5.83E+08 | 9.21E+07 | −0.71 | 0.003 | 3.05E+08 | 2.41E+05 | −2.94 | <0.0001 | |
| Prevotella | 2.95E+09 | 5.01E+09 | 0.12 | ns | 1.16E+09 | 6.99E+06 | −1.97 | 0.003 | |
| # of subjects→ | n = 18 | n = 15 | |||||||
| Baseline | Day 25 | Δlog10 | p-value | Baseline | Day 25 | Δlog10 | p-value | ||
| Bacteroides | 5.02E+09 | 5.82E+09 | −0.01 | ns | 6.28E+09 | 9.98E+08 | −0.05 | ns | |
| Eubacteria | 5.45E+09 | 7.18E+09 | 0.09 | ns | 5.63E+09 | 5.86E+09 | 0.05 | ns | |
| Enterobacteriaceae | 6.03E+07 | 5.18E+07 | 0.08 | ns | 1.15E+07 | 4.35E+08 | 1.03 | 0.017 | |
| C. coccoides | 9.04E+08 | 1.88E+09 | 0.16 | ns | 1.62E+09 | 1.07E+09 | 0.05 | ns | |
| C. leptum | 6.42E+08 | 5.61E+08 | −0.04 | ns | 3.43E+09 | 3.80E+08 | −0.36 | ns | |
| Prevotella | 1.31E+09 | 2.45E+09 | 0.10 | ns | 1.57E+09 | 5.72E+08 | 0.03 | ns | |
| # of subjects→ | n = 17 | n = 10 | |||||||
| Baseline | EOS | Δlog10 | p-value | Baseline | EOS | Δlog10 | p-value | ||
| Bacteroides | 6.15E+09 | 1.02E+10 | 0.15 | ns | 5.12E+09 | 9.77E+09 | 0.26 | ns | |
| Eubacteria | 5.48E+09 | 9.10E+09 | 0.01 | ns | 5.31E+09 | 6.70E+09 | 0.01 | ns | |
| Enterobacteriaceae | 5.08E+07 | 2.19E+07 | 0.26 | ns | 3.68E+07 | 1.31E+08 | 1.10 | ns | |
| C. coccoides | 7.28E+08 | 1.93E+09 | 0.36 | ns | 1.62E+09 | 2.72E+09 | 0.27 | ns | |
| C. leptum | 6.42E+08 | 5.22E+08 | −0.11 | ns | 3.43E+08 | 1.77E+08 | −0.78 | ns | |
| Prevotella | 1.63E+09 | 4.49E+09 | 0.14 | ns | 1.53E+09 | 3.45E+09 | 0.21 | ns | |
Medians calculated using subjects with samples at both timepoints, and for 14 controls.
Fold change expressed as log10 copies/gram.
Significance determined by Wilcoxon matched pairs signed rank test.
Controls = healthy volunteers as described in methods.
Ridinilazole has minimal impact on microbiota group levels compared to vancomycin
Vancomycin-treated participants had a small but statistically significant loss in total bacterial biomass as measured by qPCR, with median decreases of 0.43 and 0.53 log10 copies/gram of stool for Eubacteria at D5 and D10, respectively (Table 1; see S4 Table for supporting data). This was not observed in ridinilazole-treated participants. Profound effects on Bacteroides group were seen in vancomycin-treated participants, with median decreases of > 3 log10 copies/gram stool at both D5 and D10 compared to baseline. Similar, statistically significant decreases were found in the C. coccoides and C. leptum groups and in Prevotella (3.00, 3.02, and 2.30 log10 copies/g at D5, and 2.83, 2.94, and 1.97 log10 copies/g at D10, respectively). In marked contrast, ridinilazole-treated participants had no significant differences in microbiota groups assessed at any time point, except for a modest decrease in C. leptum levels at D10 (−0.71 log10 copies/g; p = 0.05), which recovered completely by D25.
Conversely, vancomycin-treated participants had significant increases in Enterobacteriaceae which persisted beyond end of treatment, with a maximum increase of more than 1 log10 at D25 when compared to baseline. In ridinilazole-treated participants, no significant changes in Enterobacteriaceae were observed throughout the study.
To test whether the trajectories across time for any microbiota group differed between treatments, p-values for the interaction of time and treatment group were calculated using repeated measures model on log-transformed data (Fig 2; see S4 Table for supporting data) from all participants at every time point. P-values were highly statistically significant for Bacteroides, C. leptum, Enterobacteriaceae, and Prevotella groups (all <0.0001), but not for the C. coccoides group (p = 0.06) or Eubacteria (p = 0.07). Exclusion of potential influential points did not change results.
Fig 2. Microbiota levels belonging to different taxonomic groups measured by qPCR in samples from study participants and 14 healthy controls.

Red circles represent participants treated with ridinilazole and blue circles represent participants treated with vancomycin. Bolded bars represent the median of all samples at that time point.
Ridinilazole has less impact on alpha and beta diversity than vancomycin
All high-throughput sequences generated in this study were submitted to the NCBI SRA site and accepted as study SRP110532. Taxonomy was assigned to a total of 10,757 OTUs. The OTU table was rarefied to 20,900 sequences per sample, resulting in a lower limit of detection (LLD) of a percent relative abundance of 0.00478%. Prior to analysis of differences in microbiota composition between samples, taxa which were present in <10% of samples were trimmed from the OTU table; the resulting table used in LEfSe analyses comprised 224 taxa levels.
As microbiota are disrupted at the time of CDI diagnosis, alpha diversity measurements are shown from healthy controls for comparison. As expected, baseline alpha diversity indices for study participants compared to healthy controls (Table 2) were significantly lower (p <0.0001) in all measured parameters. While both study antibiotics resulted in significant decreases in both OTU-based alpha diversity parameters by EOT, mean values were significantly lower following vancomycin than ridinilazole (range p <0.01 to p <0.0001). When the alpha diversity index phylogenetic diversity was considered, the difference between antibiotics was dramatic. Ridinilazole treatment resulted in no significant change in PD between baseline and EOT, whereas a major loss in diversity was observed with vancomycin (p >0.0001). Excluding those participants who experienced recurrence, effects on alpha diversity had resolved by day 25 following either treatment (data not shown).
Table 2. Comparison of alpha diversity measures between baseline and end-of-treatment (EOT) within each treatment arm (horizontal), and between treatment arms (vertical).
| Alpha Diversity Measure | Baseline | EOT | |||
|---|---|---|---|---|---|
| Chao | Mean | Stdev* | Mean | Stdev | p-value (Baseline:EOT)** |
| Control | 1039 | 217.0 | |||
| Ridinilazole | 485.6 | 188.6 | 387.7 | 108.7 | <0.05 |
| Vancomycin | 490.8 | 164.3 | 295.5 | 99.0 | <0.001 |
| p-value (Rid:Vanco)*** | NS | <0.0001 | |||
| Shannon | Mean | Stdev | Mean | Stdev | |
| Control | 6.18 | 0.67 | |||
| Ridinilazole | 4.43 | 1.40 | 3.98 | 1.08 | <0.05 |
| Vancomycin | 3.96 | 1.14 | 3.11 | 0.92 | <0.05 |
| p-value (Rid:Vanco) | NS | <0.01 | |||
| PD | Mean | Stdev | Mean | Stdev | |
| Control | 32.50 | 6.83 | |||
| Ridinilazole | 14.26 | 4.87 | 12.09 | 3.58 | NS |
| Vancomycin | 14.66 | 3.79 | 7.70 | 2.15 | <0.0001 |
| p-value (Rid:Vanco)*** | NS | <0.0001 | |||
*Stdev = Standard deviation.
**Comparison of baseline to EOT for each drug treatment.
***Comparison of ridinilazole to vancomycin.
p-values (Rid:Vanco and Baseline:EOT) derived by Mann-Whitney test.
PD = Phylogenetic diversity.
Beta diversity analyses confirmed that study participants’ baseline microbiota were significantly different from controls, though not from each other (Fig 3). Following antibiotic treatment, the effect of ridinilazole on microbial community structure was minimal, while that of vancomycin was profound. The cluster of vancomycin-treated participants at EOT is significantly different from those at baseline, is further from controls, and demonstrates the deleterious impact on community structure by vancomycin; in contrast, the ridinilazole cluster was not significantly shifted during treatment.
Fig 3. Beta diversity between participants receiving ridinilazole or vancomycin and normal controls.

Principal co-ordinate analysis was performed between samples at baseline and EOT from participants receiving ridinilazole or vancomycin in the vegan package in R on weighted Unifrac distances generated in QIIME. For another comparator, healthy controls are also shown. Ellipses represent 95% confidence interval of each cluster. Abbreviations: RDZ, ridinilazole; VAN, vancomycin; PC1, first Principal co-ordinate; PC2, second principal co-ordinate.
Taxonomic composition is modestly affected by ridinilazole compared to vancomycin
As in the qPCR analysis, the effect of study antibiotic on microbial composition was assessed by comparing samples at baseline to samples from the same participant at each available successive time point. Vancomycin had a wide-ranging effect (Fig 4), with 103 taxonomic designations identified by LEfSe as discriminating between baseline and day 10 at a linear discriminant analysis (LDA) value of 2. Although the change in abundance of the phylum Firmicutes was not significant, significant decreases were detected in four Firmicutes families by the Wilcoxon ranked sum test after controlling the false discovery rate using the Benjamini-Hochberg procedure. These families were: Peptostreptococcaceae (the family of which C. difficile is a member), Ruminococcaceae, Erysipelothrichaceae and Lachnospiraceae (Table 3). In the Lachnospiraceae and Ruminococcaceae families, abundances dropped 3 and 2 logs respectively, to below the lower limit of detection (LLD). Genera of interest within Lachnospiraceae that fell below the LLD were the butyrate-producing Blautia, Coprococcus, Dorea, Roseburia, and the species Ruminococcus gnavus, all associated with a “healthy” gut. In Ruminococcaceae, the genera Oscillospira and Ruminococcus also fell below the LLD. Concurrently, abundances of the genera Veillonella (family Veillonellaceae) and Lactobacillus (class Bacilli) increased by over a log.
Fig 4. Effects of vancomycin and ridinilazole on relative abundance of taxa at baseline vs end-of-therapy.
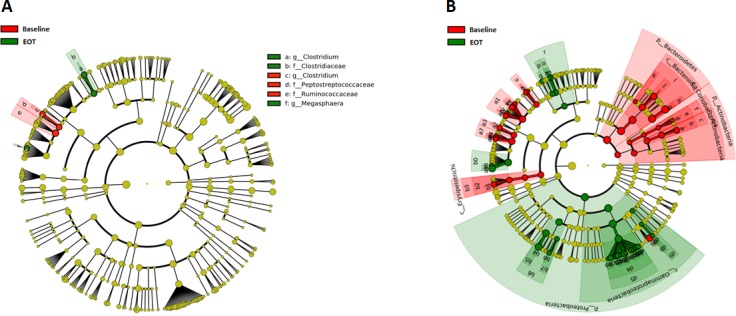
Cladograms were generated by LEfSe showing taxa with significantly higher relative abundance at baseline in red, and those with significantly higher relative abundance at end-of-therapy (EOT) in green. The phylogenetic tree is represented by concentric rings with phyla located at the innermost ring and subsequent taxonomic levels descend outwards to the species level. Panel A: Ridinilazole; Panel B: Vancomycin.
Table 3. Taxa to the genus level with ≥2-fold change in relative abundance from baseline to end of treatment (EOT) with vancomycin or ridinilazole.
| Vancomycin | Ridinilazole | |||||||
|---|---|---|---|---|---|---|---|---|
| Taxa | Baseline | EOT | Fold change | q value | Baseline | EOT | Fold change | q value |
| Actinobacteria | 1.49E-04 | 9.57E-05 | 4.9 | 0.021 | ||||
| c_Actinobacteria g_Bifidobacterium | 2.14E-04 | <LLD | 4.5 | 0.029 | ||||
| c_Coriobacteria f_Coriobacteriaceae | 1.35E-04 | <LLD | 2.8 | 0.025 | ||||
| Bacteroidetes | 7.96E-01 | 4.49E-04 | 1773.5 | 0.006 | ||||
| f_Bacteroidaceae g_Bacteroides | 7.36E-01 | 1.17E-04 | 6274.8 | 0.008 | ||||
| f_Porphyromonadaceae | 5.42E-03 | <LLD | 113.5 | 0.034 | ||||
| Firmicutes | ||||||||
| c_Bacilli f_Lactobacillaceae | 4.79E-05 | 2.43E-03 | 50.8 | 0.019 | ||||
| c_Clostridia f_Clostridiaceae g_Clostridium | 2.39E-04 | <LLD | 5.0 | 0.027 | 9.57E-04 | 9.57E-05 | 10.0 | 0.002 |
| c_Clostridia f_Lachnospiraceae | 6.76E-02 | <LLD | 1415.8 | 0.003 | ||||
| g_Blautia | 7.59E-03 | <LLD | 158.9 | 0.004 | ||||
| g_Coprococcus | 6.55E-04 | <LLD | 13.7 | 0.007 | ||||
| g_Dorea | 4.98E-04 | <LLD | 10.4 | 0.016 | ||||
| g_Roseburia | 1.91E-04 | <LLD | 4.0 | 0.022 | ||||
| g_Ruminococcus | 3.23E-03 | <LLD | 67.6 | 0.014 | ||||
| c_Clostridia f_Peptostreptococcaceae | 8.55E-04 | <LLD | 17.9 | 0.005 | 1.53E-03 | <LLD | 35.8 | <0.001 |
| Clostridium difficile | 1.91E-04 | <LLD | 4.0 | 0.026 | 7.18E-04 | <LLD | 15.0 | 0.001 |
| c_Clostridia f_Ruminococcaceae | 2.72E-02 | <LLD | 570.2 | 0.002 | 4.13E-02 | 2.82E-03 | 14.6 | 0.002 |
| g_Faecalibacterium | 1.46E-03 | <LLD | 30.5 | 0.013 | ||||
| g_Oscillospira | 2.14E-03 | <LLD | 44.8 | 0.015 | ||||
| g_Ruminococcus | 1.91E-04 | <LLD | 4.0 | 0.021 | ||||
| c_Clostridia f_Veillonellaceae g_Veillonella | 2.51E-03 | 3.05E-02 | 12.2 | 0.025 | ||||
| c_Erysipelotrichi f_Erysipelotrichiaceae | 2.22E-03 | <LLD | 46.6 | 0.005 | ||||
| g_Eubacterium | 8.13E-04 | <LLD | 17.0 | 0.022 | ||||
| Proteobacteria | 2.86E-02 | 6.55E-01 | 22.9 | 0.002 | ||||
| f_Enterobacteriaceae | 2.16E-03 | 4.73E-01 | 219.4 | 0.001 | ||||
| g_Citrobacter | 4.79E-05 | 1.23E-02 | 257.6 | 0.012 | ||||
| g_Enterobacter | <LLD | 7.60E-04 | 15.9 | 0.008 | ||||
| g_Erwinia | <LLD | 4.06E-04 | 8.5 | 0.010 | ||||
| g_Escherichia | 2.34E-04 | 1.30E-02 | 55.6 | 0.009 | ||||
| g_Klebsiella | 4.79E-05 | 0.05445 | 1137.6 | 0.010 | ||||
| g_Salmonella | <LLD | 3.58E-04 | 7.5 | 0.011 | ||||
| g_Serratia | <LLD | 5.74E-04 | 12.0 | 0.016 | ||||
| g_Trabulsiella | <LLD | 1.35E-04 | 2.8 | 0.027 | ||||
| f_Pasteurellaceae g_Haemophilus | 2.14E-04 | <LLD | 4.5 | 0.033 | ||||
| Decrease | Increase | |||||||
| <5 fold | <5 fold | |||||||
| >5 to <10 fold | >5 to <10 fold | |||||||
| >10 fold | >10 fold | |||||||
| >100 fold | >100 fold | |||||||
| >1000 fold | >1000 fold | |||||||
P-values were determined by the Wilcoxon rank sum test, following which q values were derived using the Benjamini-Hochberg procedure to hold the false discovery rate to 0.10
c = class, o = order, f = family, g = genus, LLD = lower limit of detection
In other phyla, the genus Bifidobacterium (phylum Actinobacteria) decreased to below the LLD, a reduction of over 4-fold. Mirroring our qPCR data, the most dramatic losses were in the Bacteroidetes, which was the most abundant phylum at baseline and dropped by over 3 logs, from 79% to 0.03% relative abundance. Simultaneously, a dramatic increase in Proteobacteria was observed, primarily in the family Enterobacteriaceae, where the relative abundance at EOT was 219 times higher than baseline. The median abundance of several taxa increased from undetectable to levels >10x LLD, including the pathogens Citrobacter, Enterobacter, Serratia and Klebsiella, with abundance of the latter expanding by over 3 logs. Univariate analysis in MaAsLin assessing associations of taxa with either treatment group at baseline and EOT was confirmatory of LEfSe results (S5 Table and S6 Table). The highest negative correlations with vancomycin were those of Bacteroides, Ruminococcaceae, and Lachnospiraceae and the highest positive correlation, that of Enterobacteriaceae.
Some differences persisted at D25, two weeks after end of treatment, with the Enterobacteriaceae E. coli and Klebsiella being 19- and 43-fold higher than baseline, although these differences were not significant when corrected for false discovery. These changes had resolved by EOS. The levels of C. difficile on days 25 and 40 were not significantly different from baseline.
In marked contrast, the antibacterial effect of ridinilazole was confined to the phylum Firmicutes (Fig 4, Table 3). By EOT, the median abundance of C. difficile was reduced to below the LLD and remained undetectable thereafter. Modest changes were detected in the Clostridiaceae and Ruminococcaceae families, with 10-and 14-fold decreases in relative abundance from baseline, respectively. By day 25, and at EOS the only changes from baseline were the sustained loss of detectable C. difficile and a 13-fold increase in the genus Ruminococcus in the family Ruminococcaceae (data not shown).
Subjects with recurrence had both lower alpha diversity scores and taxonomic composition differences compared to those without recurrence
Due to the limited number (only 2) of ridinilazole-treated participants with recurrence in the primary analysis, analysis of differences between the treatment arms in the microbiota of participants with recurrence could not be performed. To explore factors associated with recurrence (or recurrence protection) we considered the status of the microbiota of the entire clinical study cohort (82 patients) at EOT. Alpha diversity of participants who had a subsequent recurrence was significantly lower at EOT in all three parameters measured than that of those who did not experience recurrence (Table 4). Lachnospiraceae and Ruminococcaceae were higher in those who did not experience recurrence, while several genera of Enterobacteriaceae were higher in those who did (S2 Fig).
Table 4. Differences at end of treatment in alpha diversity and median relative abundance of taxa between subjects who had or did not have a confirmed recurrence of C. difficile during the study.
| Alpha diversity | Recurrence | No recurrence | P-value | |
| n | 11 | 69 | ||
| Chao | Mean | 280 | 365 | <0.05 |
| Stdev* | 81.6 | 133 | ||
| Shannon | Mean | 2.89 | 3.67 | <0.05 |
| Stdev | 0.89 | 1.11 | ||
| PD | Mean | 7.33 | 10.56 | <0.01 |
| Stdev | 1.04 | 3.94 | ||
| Median Relative Abundance | Recurrence | No recurrence | P-value | |
| Taxa higher in subjects without recurrence | ||||
| Firmicutes | 3.18E-02 | 1.11E-01 | <0.05 | |
| o_Clostridiales | 1.57E-02 | 8.85E-02 | <0.05 | |
| f_Lachnospiraceae | <LLD | 2.68E-03 | <0.05 | |
| g_Blautia | <LLD | 1.91E-04 | <0.05 | |
| f_Ruminococcaceae | <LLD | 6.70E-04 | <0.05 | |
| Taxa higher in subjects with recurrence | ||||
| Proteobacteria | ||||
| c_Gammaproteobacteria | 5.70E-01 | 1.60E-01 | <0.05 | |
| f_Enterobacteriaceae | 5.70E-01 | 1.05E-01 | <0.05 | |
| g_Erwinia | 5.74E-04 | 9.57E-05 | <0.05 | |
| g_Klebsiella | 7.57E-02 | 5.74E-03 | <0.05 | |
| Klebsiella oxytoca | 3.11E-03 | 9.57E-05 | <0.05 | |
| g_Salmonella | 5.26E-04 | <LLD | <0.01 | |
| Salmonella enterica | 5.26E-04 | <LLD | <0.01 |
*Stdev = standard deviation.
PD = phylogenetic diversity.
c = class, o = order, f = family, g = genus, s = species.
Discussion
Our observations regarding vancomycin’s effects on the intestinal microbiota are in accord with those of previous studies [11, 40]. We found that vancomycin treatment resulted in a biomass decrease of Bacteroides and Prevotella groups, and that of both Firmicutes subgroups we examined, as well as a concomitant increase in the biomass of the Enterobacteriaceae group. High-throughput sequencing showed numerous changes following vancomycin, including decreased alpha diversity parameters, significant changes in beta diversity and decreased relative abundance of Bifidobacterium, two Bacteroidetes families and four Firmicutes families, as well as increases in Enterobacteriaceae. These alterations in microbial community structure have been associated with susceptibility to CDI (16–20). Utilizing fluorescent in situ hybridization/flow cytometry, Tannock et al observed a similar decrease in commensal clostridial species following vancomycin treatment, with concomitant outgrowth of Enterobacteriaceae [11]. Louie et al conducted a qPCR-based study of the effects of fidaxomicin and vancomycin treatment of CDI on quantities of major microbiota [31]. Interestingly, they found persistent Bacteroides decreases several weeks after completion of vancomycin treatment compared to fidaxomicin-treated samples. Although our qPCR analysis did not detect a delayed recovery, our high-throughput sequencing was suggestive of this, also revealing the significant persistence of Enterobacteriaceae.
This novel finding of this study is the limited disruption of intestinal microbiota following ridinilazole treatment, both in terms of biomass (how many are there) and composition (which taxonomic groups are represented). Both are likely important in preventing C. difficile colonization, disease, and recurrence, by preserving sufficient density of the correct type(s) of species to create an environment not conducive to C. difficile expansion. Loss of alpha diversity was significantly lower with ridinilazole than with vancomycin, indicating a smaller impact on microbiota health and preservation of colonization resistance. This was confirmed by the absence of an expansion of the phylum Proteobacteria. The only microbiota biomass loss was with the C. leptum group at D10, with this loss being modest compared to that seen with vancomycin. While high-throughput sequencing revealed significant decreases in some Clostridiales taxa other than C. difficile, this was not unexpected, as ridinilazole shows some effect on total colony counts of Clostridia species [41]. The changes in Clostridiaceae and Ruminococcaceae were lower than those resulting from vancomycin and resolved with cessation of antibiotic. The Lachnospiraceae, known to enhance colonic epithelial cell health and immune function, in part by generating the short-chain fatty acid butyrate, were unaffected by ridinilazole. In contrast to findings in vancomycin-treated participants, the median relative abundance of C. difficile in ridinilazole-treated participants fell below LLD by D5, remaining there until EOS.
In sum, ridinilazole is a promising, effective CDI antimicrobial with minimal effects on bystander indigenous colonic flora. Its microbiota-preserving narrow-spectrum activity is likely responsible for the observed decreased recurrence rate compared to vancomycin treatment.
Supporting information
Cladogram generated by LefSe showing taxa in baseline samples with a significantly higher percent relative abundance when comparing participants who did (Yes = green) or did not (No = red) receive anti-CDI treatment within 24 hours of study enrollment. The phylogenetic tree is represented by concentric rings, with phyla at the innermost ring and lower taxonomic levels in the rings tiered successively outwards.
(TIF)
Cladogram generated by LefSe showing taxa at end-of-therapy samples with a significantly higher percent relative abundance when comparing participants who did (green) or who did not (red) recur. The phylogenetic tree is represented by concentric rings, with phyla at the innermost ring and lower taxonomic levels in the rings tiered successively outwards.
(TIF)
(DOCX)
(DOCX)
(DOCX)
The mean and median values are calculated only for those subjects who did not receive anti-CDI antibiotic prior to enrollment.
(PDF)
(DOCX)
(DOCX)
Acknowledgments
We are grateful to Ms. Lori Lyn Price, of the Tufts Clinical and Translational Science Institute for assistance with statistical analyses. We thank Dr. Honorine Ward for critically reading the manuscript. Assistance in the preparation of this manuscript was provided by Dr. Prasad Kulkarni and Ms. Alexandra Rayser of the Healthcare Alliance Group, Voorhees, New Jersey, USA and was funded by Summit Therapeutics Plc.
Data Availability
Relevant data are available at NCBI's SRA database under the accession number SRP110532, as well as in the paper and its Supporting Information files.
Funding Statement
CMT, AVK, and DRS have received research grant funding from Summit Therapeutics Plc (http://www.summitplc.com/). CMT has served as a consultant for Summit Therapeutics Plc. RJV is an employee of Summit Therapeutics Plc., which has received a Translation Award from the Wellcome Trust (Grant Number 099444; https://wellcome.ac.uk/). The sponsor (Summit Therapeutics Plc) had a role in conceptualization of the study, analysis of the data, and preparation of the manuscript.
References
- 1.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(9):825–34. 10.1056/NEJMoa1408913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vollaard EJ, Clasener HA. Colonization resistance. Antimicrob Agents Chemother. 1994;38(3):409–14. Epub 1994/03/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol. 2013;13(11):790–801. 10.1038/nri3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489(7415):220–30. Epub 2012/09/14. 10.1038/nature11550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4554–61. Epub 2010/09/18. 10.1073/pnas.1000087107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis BB, Buffie CG, Carter RA, Leiner I, Toussaint NC, Miller LC, et al. Loss of Microbiota-Mediated Colonization Resistance to Clostridium difficile Infection With Oral Vancomycin Compared With Metronidazole. J Infect Dis. 2015;212(10):1656–65. Epub 2015/04/30. 10.1093/infdis/jiv256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vincent C, Manges AR. Antimicrobial Use, Human Gut Microbiota and Clostridium difficile Colonization and Infection. Antibiotics (Basel, Switzerland). 2015;4(3):230–53. Epub 2015/01/01. 10.3390/antibiotics4030230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheitoyan-Pesant C, Abou Chakra CN, Pepin J, Marcil-Heguy A, Nault V, Valiquette L. Clinical and Healthcare Burden of Multiple Recurrences of Clostridium difficile Infection. Clin Infect Dis. 2016;62(5):574–80. Epub 2015/11/20. 10.1093/cid/civ958 [DOI] [PubMed] [Google Scholar]
- 9.Nanwa N, Kendzerska T, Krahn M, Kwong JC, Daneman N, Witteman W, et al. The economic impact of Clostridium difficile infection: a systematic review. Am J Gastroenterol. 2015;110(4):511–9. 10.1038/ajg.2015.48 [DOI] [PubMed] [Google Scholar]
- 10.Skraban J, Dzeroski S, Zenko B, Mongus D, Gangl S, Rupnik M. Gut microbiota patterns associated with colonization of different Clostridium difficile ribotypes. PLoS One. 2013;8(2):e58005 Epub 2013/03/08. 10.1371/journal.pone.0058005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tannock GW, Munro K, Taylor C, Lawley B, Young W, Byrne B, et al. A new macrocyclic antibiotic, fidaxomicin (OPT-80), causes less alteration to the bowel microbiota of Clostridium difficile-infected patients than does vancomycin. Microbiology (Reading, England). 2010;156(Pt 11):3354–9. Epub 2010/08/21. [DOI] [PubMed] [Google Scholar]
- 12.Vincent C, Stephens DA, Loo VG, Edens TJ, Behr MA, Dewar K, et al. Reductions in intestinal Clostridiales precede the development of nosocomial Clostridium difficile infection. Microbiome. 2013;1(1):18 Epub 2014/01/24. 10.1186/2049-2618-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517(7533):205–8. 10.1038/nature13828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antharam VC, Li EC, Ishmael A, Sharma A, Mai V, Rand KH, et al. Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J Clin Microbiol. 2013;51(9):2884–92. 10.1128/JCM.00845-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Louie TJ, Emery J, Krulicki W, Byrne B, Mah M. OPT-80 eliminates Clostridium difficile and is sparing of bacteroides species during treatment of C. difficile infection. Antimicrob Agents Chemother. 2009;53(1):261–3. Epub 2008/10/29. 10.1128/AAC.01443-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Theriot CM, Koenigsknecht MJ, Carlson PE Jr., Hatton GE, Nelson AM, Li B, et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun. 2014;5:3114 10.1038/ncomms4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L, Dong D, Jiang C, Li Z, Wang X, Peng Y. Insight into alteration of gut microbiota in Clostridium difficile infection and asymptomatic C. difficile colonization. Anaerobe. 2015;34:1–7. Epub 2015/03/31. 10.1016/j.anaerobe.2015.03.008 [DOI] [PubMed] [Google Scholar]
- 18.Gu S, Chen Y, Zhang X, Lu H, Lv T, Shen P, et al. Identification of key taxa that favor intestinal colonization of Clostridium difficile in an adult Chinese population. Microbes and infection. 2016;18(1):30–8. Epub 2015/09/19. 10.1016/j.micinf.2015.09.008 [DOI] [PubMed] [Google Scholar]
- 19.Allegretti JR, Kearney S, Li N, Bogart E, Bullock K, Gerber GK, et al. Recurrent Clostridium difficile infection associates with distinct bile acid and microbiome profiles. Alimentary pharmacology & therapeutics. 2016;43(11):1142–53. Epub 2016/04/19. 10.1111/apt.13616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seekatz AM, Rao AM, Santhosh K, Young VB. Dynamics of the fecal microbiome in patients with recurrent and nonrecurrent Clostridium difficile infection. Genome Med. 2016;8(1):47 10.1186/s13073-016-0298-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee YJ, Arguello EP, Jenq RR, Littmann E, Kim GJ, Miller LC, et al. Protective Factors in the Intestinal Microbiome Against Clostridium difficile infection in Recipients of Allogeneic Hematopoietic Stem Cell Transplantation. J Infect Dis. 2017. 10.1093/infdis/jix0111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seekatz AM, Aas J, Gessert CE, Rubin TA, Saman DM, Bakken JS, et al. Recovery of the gut microbiome following fecal microbiota transplantation. mBio. 2014;5(3):e00893–14. Epub 2014/06/19. 10.1128/mBio.00893-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song Y, Garg S, Girotra M, Maddox C, von Rosenvinge EC, Dutta A, et al. Microbiota dynamics in patients treated with fecal microbiota transplantation for recurrent Clostridium difficile infection. PLoS One. 2013;8(11):e81330 Epub 2013/12/05. 10.1371/journal.pone.0081330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamilton MJ, Weingarden AR, Unno T, Khoruts A, Sadowsky MJ. High-throughput DNA sequence analysis reveals stable engraftment of gut microbiota following transplantation of previously frozen fecal bacteria. Gut microbes. 2013;4(2):125–35. Epub 2013/01/22. 10.4161/gmic.23571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weingarden AR, Chen C, Bobr A, Yao D, Lu Y, Nelson VM, et al. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. American journal of physiology Gastrointestinal and liver physiology. 2014;306(4):G310–9. Epub 2013/11/29. 10.1152/ajpgi.00282.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Nood E, Dijkgraaf MG, Keller JJ. Duodenal infusion of feces for recurrent Clostridium difficile. N Engl J Med. 2013;368(22):2145 Epub 2013/05/31. 10.1056/NEJMc1303919 [DOI] [PubMed] [Google Scholar]
- 27.Vickers RJ, Tillotson G, Goldstein EJ, Citron DM, Garey KW, Wilcox MH. Ridinilazole: a novel therapy for Clostridium difficile infection. Int J Antimicrob Agents. 2016. 10.1016/j.ijantimicag.2016.04.026 [DOI] [PubMed] [Google Scholar]
- 28.Vickers RJ, Tillotson GS, Nathan R, Hazan S, Pullman J, Lucasti C, et al. Efficacy and safety of ridinilazole compared with vancomycin for the treatment of Clostridium difficile infection: a phase 2, randomised, double-blind, active-controlled, non-inferiority study. Lancet Infect Dis. 2017;17(7):735–44. Epub 2017/05/04. 10.1016/S1473-3099(17)30235-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vanegas SM, Meydani M, Barnett JB, Goldin B, Kane A, Rasmussen H, et al. Substituting whole grains for refined grains in a 6-wk randomized trial has a modest effect on gut microbiota and immune and inflammatory markers of healthy adults. The American journal of clinical nutrition. 2017;105(3):635–50. Epub 2017/02/10. 10.3945/ajcn.116.146928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bernhard AE, Field KG. A PCR assay To discriminate human and ruminant feces on the basis of host differences in Bacteroides-Prevotella genes encoding 16S rRNA. Applied and environmental microbiology. 2000;66(10):4571–4. Epub 2000/09/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Louie TJ, Cannon K, Byrne B, Emery J, Ward L, Eyben M, et al. Fidaxomicin preserves the intestinal microbiome during and after treatment of Clostridium difficile infection (CDI) and reduces both toxin reexpression and recurrence of CDI. Clin Infect Dis. 2012;55 Suppl 2:S132–42. Epub 2012/07/07. 10.1093/cid/cis338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsuki T, Watanabe K, Fujimoto J, Miyamoto Y, Takada T, Matsumoto K, et al. Development of 16S rRNA-gene-targeted group-specific primers for the detection and identification of predominant bacteria in human feces. Applied and environmental microbiology. 2002;68(11):5445–51. Epub 2002/10/31. 10.1128/AEM.68.11.5445-5451.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bartosch S, Fite A, Macfarlane GT, McMurdo ME. Characterization of bacterial communities in feces from healthy elderly volunteers and hospitalized elderly patients by using real-time PCR and effects of antibiotic treatment on the fecal microbiota. Applied and environmental microbiology. 2004;70(6):3575–81. Epub 2004/06/09. 10.1128/AEM.70.6.3575-3581.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang W, Lederman MM, Hunt P, Sieg SF, Haley K, Rodriguez B, et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J Infect Dis. 2009;199(8):1177–85. Epub 2009/03/07. 10.1086/597476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. The ISME journal. 2012;6(8):1621–4. Epub 2012/03/10. 10.1038/ismej.2012.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods. 2010;7(5):335–6. Epub 2010/04/13. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome biology. 2011;12(6):R60 Epub 2011/06/28. 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological) 1995;57(1):289–300. [Google Scholar]
- 39.Diggle PJ, Liang K-L, Zeger SL. Analysis of Longitudinal Data Oxford, UK: Oxford University Press; 1996. [Google Scholar]
- 40.Isaac S, Scher JU, Djukovic A, Jimenez N, Littman DR, Abramson SB, et al. Short- and long-term effects of oral vancomycin on the human intestinal microbiota. J Antimicrob Chemother. 2017;72(1):128–36. Epub 2016/10/07. 10.1093/jac/dkw383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baines SD, Crowther GS, Freeman J, Todhunter S, Vickers R, Wilcox MH. SMT19969 as a treatment for Clostridium difficile infection: an assessment of antimicrobial activity using conventional susceptibility testing and an in vitro gut model. J Antimicrob Chemother. 2015;70(1):182–9. 10.1093/jac/dku324 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cladogram generated by LefSe showing taxa in baseline samples with a significantly higher percent relative abundance when comparing participants who did (Yes = green) or did not (No = red) receive anti-CDI treatment within 24 hours of study enrollment. The phylogenetic tree is represented by concentric rings, with phyla at the innermost ring and lower taxonomic levels in the rings tiered successively outwards.
(TIF)
Cladogram generated by LefSe showing taxa at end-of-therapy samples with a significantly higher percent relative abundance when comparing participants who did (green) or who did not (red) recur. The phylogenetic tree is represented by concentric rings, with phyla at the innermost ring and lower taxonomic levels in the rings tiered successively outwards.
(TIF)
(DOCX)
(DOCX)
(DOCX)
The mean and median values are calculated only for those subjects who did not receive anti-CDI antibiotic prior to enrollment.
(PDF)
(DOCX)
(DOCX)
Data Availability Statement
Relevant data are available at NCBI's SRA database under the accession number SRP110532, as well as in the paper and its Supporting Information files.