

 An original point of view inside the SAR studies to develop new selective COX-2 inhibitors.
An original point of view inside the SAR studies to develop new selective COX-2 inhibitors.
Abstract
Most drugs used to treat pain and inflammation act through inhibition of the enzymes prostaglandin G/H synthase, commonly known as cyclooxygenase (COX). Among these, the simultaneous inhibition of cyclooxygenase 1 (COX-1) would explain the unwanted side effects in the gastrointestinal tract and many adverse cardiovascular effects, such as high blood pressure, myocardial infarction and thrombosis. These side effects led in time to the development of NSAIDs that behave as selective COX-2 inhibitors. This manuscript highlights the structure–activity relationships which characterize the chemical scaffolds endowed with selective COX-2 inhibition. Additionally, the role of COX-2 inhibitors in the pain phenomenon and cancer is discussed.
Introduction
Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) are one of the most highly prescribed drugs, commonly used for musculoskeletal pathologies such as rheumatoid arthritis and osteoarthritis. They act by inhibiting the biosynthesis of prostanoids,1 such as prostaglandins PGE2, PGF2α, PGD2, and PGI2 and thromboxane TXA2. Thus, they play an important role in many cellular responses and pathophysiological processes, such as modulation of the inflammatory reaction and its resolution, gastrointestinal cytoprotection and ulceration, angiogenesis and cancer, haemostasis and thrombosis, renal hemodynamics and progression of kidney disease, and atheroprotection and atherosclerosis.2 NSAIDs exert their action by inhibiting cyclooxygenase (COX), which converts arachidonic acid (AA) into prostanoids that act as physio-pathological effectors. The COX enzyme presents two isoforms: COX-1 and COX-2; even NSAIDs are classified into several classes, generally non-selective or selective for COX-2.3 COX is the enzyme that catalyses the synthesis of prostaglandins from the substrate AA, a fatty acid with 20 carbon atoms, present in the body. The products of this catalysis are involved in various physio-pathological conditions, and in particular the inhibition of this pathway provides good anti-inflammatory and anti-nociceptive activities.4
COX-1 and COX-2 share the same catalytic activities and generate the same products. Each isozyme performs different biological functions, due to several differences in the biology of COX isozymes, such as the regulation of gene expression, the stability of transcripts and proteins, and the requirement of different levels of hydroperoxides.5
COX-1 is a “housekeeper” molecule, involved in various physiological pathways. Specifically, it generates prostaglandins and lipid mediators, which regulate normal cell activities. Activating COX-1 results in the production of prostacyclin which, when released by the endothelium, is anti-thrombogenic, while, when released by the gastric mucosa, it is cytoprotective. This cytoprotection extends to both exogenous damaging substances on the gastric mucosa and endogenously produced gastric juice. Inhibition of gastric prostaglandin production is regarded as the cause of the most frequent and most dangerous side-effects of NSAIDs, gastric ulceration, bleeding and perforation. In platelets, COX-1 leads to TXA2 production, causing aggregation of the platelets to prevent inappropriate bleeding.6
Research shows that COX-2 is considered an inducible form of the enzyme, but in the past few years, its presence in specific tissues revealed how it is expressed also in physiological conditions. Numerous experiments have demonstrated that the two isoforms have a molecular weight of 71 kDa, and the amino acidic sequence of COX-2 presents a good homology (60%) with a non-inducible form. The amino acid structure revealed how Val523 is present in COX-2 instead of Ile523, making a lateral binding site not present in COX-1 available. COX-2 is found in small amounts in normal human lungs, rat kidneys and foetal membranes, but most of its activity seems to be the result of induction. While COX-1 can also change its expression, its activity may increase 2–3-fold, while that of COX-2 may increase more than 20-fold during the inflammatory reaction.7
Side effects associated with NSAID consumption spurred medicinal chemists to synthesize new molecules, selective COX-2 inhibitors, mainly to reduce gastrointestinal and renal side effects such as anorexia, nausea, vomiting, dyspepsia, and diarrhoea. In this context, COX-1 was inhibited in epithelial cells, where its inhibition depressed PGI2/PGE2 production, involved in gastroprotection.8 In 1999, to reduce side effects, the coxibs family was introduced, as selective COX-2 inhibitors, endowed with good anti-inflammatory and anti-nociceptive properties. The most famous one, celecoxib (Celebrex®), presents as a central core a 1,2-diarylheterocycle, with bulky side chains and a sulphonyl group. These compounds present another ring that binds the hydrophobic pocket in the canal, which links the COX-2 substrate, and is not present in COX-1.9 Only celecoxib and etoricoxib (Algix® or Arcoxia®, (Fig. 1)) are presently marketed; they are used for reducing the pain from osteoarthritis, rheumatoid arthritis and acute gouty arthritis; they are also used for treating ankylosing spondylitis and dysmenorrhea. Other coxibs such as firocoxib (Previcox®) are used for veterinary purposes, in particular for the treatment of dog and horse inflammatory conditions. However, COX-2 inhibitors present side effects such as arterial hypertension, myocardial infarction, stroke, and heart failure. At the gastrointestinal level, belching, dyspepsia, dysphagia, stomatitis, vomiting, flatulence, abdominal pain, diarrhea or constipation, gastritis, duodenal ulcers, gastrointestinal bleeding and perforation may occur.
Fig. 1. Celecoxib and etoricoxib, typical COX-2 inhibitors.

Coxibs are frequently used to treat inflammation and pain, difficult social problems which need novel strategies and therapies. In particular, pain management is the research goal of various scientists. More and more are the targets implicated in pain management, such as Monoacylglycerol Lipase (MAGL),10 Cannabinoid Receptors 1/2 (CB 1/2),11 Transient Receptor Potential Vanilloid Channel 1 (TRPV1),12 and Fatty Acid Amide Hydrolase (FAAH).13 However, there is growing interest towards dual molecules such as FAAH/TRPV1 blockers and also FAAH/COX-2 inhibitors;14 specifically, it is shown how COX-2-mediated transformation of anandamide into pro-algesic prostamides has led to the hypothesis of the presence of deeper functional connections between the endocannabinoid and prostanoid systems.15 The interest in COX-2 inhibition has been emphasized by the fact that it is often involved in oxidative stress16 and also in cancer progression. In fact, studies have demonstrated how prediagnostic use of NSAIDs, such as aspirin, and selective COX-2 inhibitors was, however, associated with a reduced rate of breast cancer recurrence.17 Treatment with celecoxib significantly decreased the induced tumor size and metastasis of PyMT/Col1a1 tumors, by decreasing the expression levels of COX-2, PGE2, and Ki-67. COX-2 has had a direct role in modulating tumor progression and it may be an effective therapeutic target for women with dense breast tissue and early-stage breast cancer.18 In benign prostatic hyperplasia (BPH), 5α-reductase (5AR) inhibitors (i.e. dutasteride and finasteride) induced apoptosis and repression of the cell-adhesion protein E-cadherin, requiring both ERβ and TGFβ. Dutasteride also induced COX-2, which functions in a negative-feedback loop in TGFβ and ERβ signaling pathways as evidenced by the potentiation of apoptosis induced by dutasteride or finasteride upon pharmacological inhibition or shRNA-mediated ablation of COX-2.19 Also, COX-2 is involved in the regulation of tumorigenic Wnt signalling with 5-lipoxygenase.20
The best scaffold for selective COX-2 inhibitors
Side effects associated with well-known COX-2 inhibitors prompted chemists to develop new molecules, hoping to limit them. At the current state, COX-2 inhibitors can be categorized as diarylheterocycles or non-diarylheterocycles, and the largest proportion of selective COX-2 inhibitors comprises diarylheterocycles with a five-membered core.21
From a medicinal chemist's point of view, selective COX-2 inhibition was achieved by a series of structural devices. In particular, in the past few years, many molecules have been synthesized with heterocyclic rings and polar groups acting as selective inhibitors with low IC50 values.22 Heterocyclic compounds are very important in drug discovery because they interact with several biological pathways. In particular, coxibs have structural moieties that emerged as new interesting scaffolds in the research of novel selective COX-2 inhibitors.
With the aim of identifying new COX-2 inhibitors endowed with thromboxane prostanoid receptor antagonism, celecoxib has been exploited and modified to obtain new analogues with reduced side effects. N-(2-Chloro-6-fluorophenyl)-4-methyl-2-(1H-tetrazol-5-ylmethyl)benzenamine, in particular, demonstrated good and selective COX-2 inhibition (IC50 = 0.014 ± 23 μM). In a typical structure, it is possible to find three aryl rings, although in this molecule, the tetrazole is not the central ring, and no –SO2CH3/–SO2NH2 moiety can be found, which is present instead in N-[[2-[(2-chloro-6-fluorophenyl)amino]-5-methylphenyl]methyl]-1,1,1-trifluoromethanesulfonamide.23
Two famous coxibs, lumiracoxib and valdecoxib, were used as models in the in silico search and optimization of new selective COX-2 inhibitors. The search utilized in situ library scrutiny and produced many structures similar to lumiracoxib, whose common characteristics are two aromatic rings, at least two oxygen atoms, at least one carboxyl group, and at least one –OH or –NH(n) group. Other atoms present but not universal are halogen atoms, nitrogen atoms, methoxide, alkene, and amide groups. On the other hand, valdecoxib, which has a similar structure (sulphonamide-based), presents two aromatic rings, at least two oxygen atoms, one sulphonamide group, and at least one nitrogen atom. Other substituents present but not universal are the chlorine atom, oxadiazole substituent, and benzimidazole groups.24
Another chemically different big group, merged as a series of 1-N-substituted-3,5-diphenyl-2-pyrazoline derivatives, was designed and synthesized to inhibit COX-2 selectively. Effectively, all derivatives were found to be inactive as COX-1 inhibitors, while N-acetyl derivatives (1a–i) resulted to be more potent than the corresponding N-carbamoyl derivatives. Compound 1g (IC50 = 3.20 ± 0.24 μM) was the most potent derivative identified and, with regard to its structure, this confirmed the fact that a good COX-2 inhibitor must possess three spaced cycles and in this field, the most potent N-carbamoyl derivative, 1k (IC50 = 9.35 ± 0.76 μM), instead possesses an electron-withdrawing group. In the active site, on the other hand, compound 1g produced hydrogen bonds with Arg513 and Phe518; also, the phenoxy group was accommodated into an aromatic cage delimited by Phe518, Trp387, Tyr385 and Phe384.25
Examples of 2-pyrazolines and pyrazoles were designed as celecoxib analogues; their scaffold consists of two adjacent aryl rings attached to a five-membered ring with a COX-2 pharmacophore (–SO2Me in 2a–f and 3a–f, –SO2NH2 in 2g–l and 3g–l). The best compounds were 2d and 3a (IC50 = 0.97 μM). About the structural modifications, the methyl group on the second aryl ring was replaced with a trifluoromethyl moiety; in addition, the trifluoromethyl moiety at C-3 of the central five-membered ring was replaced with a substituted aryl moiety since it was reported that the substituent at C-3 of the central ring has very few steric restrictions compared to COX-2 binding.26
At the same time, the pyrazole structure was evaluated for its ability to inhibit COX-2 by the synthesis of 1,3,5-triarylpyrazoline (compounds 4a–m) and 1,5-diarylpyrazole (compounds 5a–d) derivatives. For the triarylpyrazolines, the thienyl analogue is more potent than its furyl counterpart; for the diarylpyrazoles, similarly, the thienyl analogue was more potent than its furyl counterpart. Therefore, the presence of the sulphur atom not only increases the selectivity towards COX-2, but also increases the anti-inflammatory action. Furthermore, in all cases of substitution, the triarylpyrazolines resulted to be more potent than the corresponding diaryl derivatives, demonstrating the fact that three aryl rings could improve COX-2 inhibition.27
The dihydropyrazole group has been used as the five-membered core ring with a diarylheterocycle scaffold, and additionally a sulphonamide group was attached to the para position of one aryl ring. The resulting structure has little resemblance to the other inhibitors but contains the same types of substituents; in fact, the most potent compound, 6d (IC50 = 0.08 ± 0.03 μM), contains an aryl ring bearing two fluorine atoms.28
Even coumarin has found to be the best scaffold for new molecules acting as selective COX-2 inhibitors. In particular, 4-chlorocoumarin-substituted 1,5-diarylpyrazole benzenesulfonamide derivatives have been synthesized. The presence of electron-withdrawing groups on the para position is more preferable than that of electron-donating groups. The most potent compound was 7t (IC50 = 0.09 ± 0.01 μM) and the order of substituents to improve COX-2 inhibition was CF3 > NO2 > F > H, CH3 > OCH3, OCH2CH3, OH. A fundamental position was R3 in the para position (Fig. 2).29
Fig. 2. Examples of pyrazole derivatives.

It is well known that the indole nucleus is an important heterocyclic compound and a useful scaffold present in numerous natural and synthetic molecules. This scaffold still represents an attractive target for medicinal chemists, due to its polyhedric behaviour against most physio-pathological pathways.30 In particular, many researchers have evaluated its ability to inhibit COX-2,31 and 2-[4-(aminosulfonyl) phenyl]-3-(4-methoxyphenyl)-1H-indole was identified as an excellent COX-2 inhibitor (IC50 = 0.006 nM), endowed with imaging properties.32
Furthermore, indole has been studied and well used to ameliorate COX-2 inhibitor scaffolds and structure–activity relationships. Specifically, the famous two aryl rings were connected to a tricyclic core in the forms of -pyrrole and -dihydropyrrolo[3,2,1-hi]indole. In this context, the oxidized forms presented very good COX-2 inhibition; in fact, the best compound was found to be 9d (IC50 = 0.02 μM). This indicates not only steric but also electronic influences resulting from the extended π-system. The R-substituents are the typical moieties which provide COX-2 selectivity, i.e. H, Me, F, SO2CH3, SO2NH2, and OEtF (Fig. 3).33
Fig. 3. Indole-based molecules.

Taking into consideration that several 5-membered carbocycles and heterocycles are able to interact with the COX active site, 1,4- and 1,5-diaryl-substituted 1,2,3-triazoles supported previous studies on COX inhibition. In particular, they possess a SO2Me group as a COX-2 pharmacophore at the para position of one of the aryl rings. In all the compounds 10a–f, small electron-withdrawing groups (F, Cl) gave IC50 values in the submicromolar range (0.15–0.20 μM). Compounds 11a–f presented the common vicinal substitution pattern of potent and selective COX-2 inhibitors. As expected, the most potent were chlorine- and fluorine-substituted compounds. It is therefore implied that the order of COX-2 inhibitory potency was F > Cl > H > Me > OMe > NMe2 (Fig. 4).34
Fig. 4. Triazole derivatives.

In this chemically different scenario, it is fundamental to mention pyrrole, a small molecule, which is a versatile and useful tool in several biological pathways. There are many examples in which COX inhibition was pursued with a series of pyrrole-based esters,35 negatively affected by a low in vivo profile, due to their gastrointestinal hydrolysis. The new molecules present a typical tricyclic structure with pyrrole as the central core but decorated with two aryl rings. In this case, the innovation is an amide function versus ester, which gives the molecule a safer biological profile that induces inhibition of COX with modest IC50 values (the best molecule, 12e, had an IC50 value of 0.92 ± 0.05 μM).36
In this context, the basic structure has been improved, in order to increase the selectivity towards COX-2 and enhance the anti-inflammatory activity of the pyrrole-based molecules. In particular, it has been investigated whether or not the presence of fluorine atoms was able to increase or reduce COX-2 selectivity. The compounds obtained, 1,5-diarylpyrrole-3-alkoxyethyl ether derivatives, were used in inflammation models, showing good anti-inflammatory and anti-nociceptive activities, with an IC50 value of 0.007 μM (SI > 14.285) for the best compound 14b. The data has demonstrated how the presence of two fluorine atoms in the same molecule could positively affect COX-2 inhibition, with good results also in in vivo models of pain (Fig. 5).37
Fig. 5. Pyrrole-based molecules.

Among the heterocyclic compounds, the 2-imidazoline core has been outlined as the main group linked to two aryl substituents, one of which bears the sulfonic group while the other bears electron-withdrawing substituents such as bromine, chlorine, fluorine, and trifluoromethyl groups. A good compound was identified, 15ab, with an IC50 value of 0.3 μM.38
An interesting series of compounds was represented by 1,5-diaryl-substituted tetrazoles, synthesized by introducing the SO2NH2 COX-2 pharmacophore to position C-5 of the tetrazole moiety via the phenyl group, while a selection of different para substituents (H, CH3, OCH3, Cl, F, CF3 or N(CH3)2) was used to decorate the other aryl substituent. The in vitro bioassay studies showed that only azoles 16c, 16d, and 16g displayed inhibitory potency toward the COX-2 enzyme with IC50 values of 1.2, 4.8, and 5.7 μM, respectively, while no inhibition activity was detected toward the COX-1 enzyme. In this field, the primary scaffold has been improved to emphasize selective COX-2 inhibition; in some cases, a spacer has been introduced, such as the methylene group, between the tetrazole core and the pharmacophore represented by the phenyl ring with –SO2R in the para position. The other phenyl ring also presents in the para position the –OH substituent in all compounds (free or in an aliphatic chain). The best compound was found to be 17i, with a very low IC50 value versus COX-2 (IC50 = 3 μM, SI > 67) (Fig. 6).39
Fig. 6. Nitrogen derivatives.

The triazole nucleus has been further replaced by the oxadiazole nucleus. In particular, 4-(3,4-dimethylphenyl)-2(1H)-phthalazinone derivatives have been designed and synthesized as new anti-inflammatory agents, demonstrating good and selective COX-2 inhibition, especially in the case of compound 18b (IC50 = 0.59 μM), which presents in its structure a series of heterocyclic groups, particularly a 1,3,4-oxadiazole and a nitro group that have increased its selectivity rather than COX-1 inhibition.40
Also, a series of oxadiazole derivatives have been designed by linking them to two aromatic rings, decorated with electron-withdrawing groups or in the para position with –Cl/–NO2/–tBu, which enhanced COX-2 inhibition, while in the central core, the N-acetylation did not significantly affect the selectivity. Also, the presence of the –SO2CH3 group significantly increased the COX-2 selectivity compared to –SCH3. In this type of molecule, replacement of the aromatic ring with pyridine reduced the COX-2 activity. The best compound 21e presents IC50 value = 0.48 μM with SI = 132.83.41
Although pyridine in these oxadiazole derivatives reduced selective COX-2 inhibition, in the original scaffold, they have demonstrated good selectivity (decent IC50 values). In particular, a series of imidazopyrazolopyridines was designed endowed with good COX-2 inhibition due to the hydrogen-bond acceptor in the para position of the phenyl ring, connected with a diazo moiety (azo bridge); also, various substituents such as methoxide groups have been introduced which form hydrogen bonds with the guanidine moiety of Arg513 (ameliorating COX-2 selectivity) (Fig. 7).42
Fig. 7. Oxadiazole and imidazopyrazolopyridine scaffolds.

Furthermore, to investigate how other heterocycles should be used as new promising COX-2 inhibitors, the thiadiazole nucleus has also been used to synthesize a series of derivatives with good IC50 values. Four substances, belonging to two different classes, resulted to be very interesting. All of them showed low IC50 values (24b (0.11 μM, SI > 454.54), 24c (0.13 μM, SI > 384.61), 25c (0.14 μM, SI > 357.14), 25e (0.13 μM, SI > 384.62)) and good anti-inflammatory and analgesic activities (Fig. 8).43
Fig. 8. Thiadiazole derivatives.

Surprisingly, the quinoline scaffold as a novel COX-2 inhibitor has also been investigated as an anti-breast cancer agent (mediated by COX-2 inhibition). In particular, a new group of 4-(imidazolylmethyl)quinoline derivatives possessing a methylsulfonyl moiety at the para position of the C-2 phenyl ring demonstrated good inhibition with IC50 values in the potent range of 0.063–0.090 μM (Fig. 9).44
Fig. 9. Quinoline derivatives.

The intense search for new anti-inflammatory agents prompted medicinal chemists to investigate how all heterocyclic compounds may interfere with COXs. In this field, cyclic imides such as phthalimides have received great attention due to their COX-1/2 inhibition.45 Based on SC-558 and celecoxib, a new scaffold for small molecular COX inhibitors has been evaluated, bearing 3,4,5-trimethoxybenzyl, 4-methoxybenzyl, or 4-fluorobenzyl fragments, in association with various substituents, such as H, Me, NO2, Cl and t-butyl, at the cyclic imide core. The lead has been tapped and it is shown that diminishment of activity may be explained on the bases of non-aromatic features of the imide scaffold, while fluoro substituents on the N-benzyl moiety showed COX-2 inhibition loss compared to methoxide substituents. These compounds inhibited COX-2 with low IC50 values of 0.18–8.5 μM.46
By combining the maleimide ring and benzenesulfonamide moiety (COX-2 pharmacophore), other novel compounds have been synthesized. The unsubstituted 3,4-dichloromaleimido benzenesulfonamide (compound 29) showed poor COX-2 inhibition, while substitution of the phenyl ring attached at the fourth position of the maleimide ring resulted in a greater inhibitory power and selectivity toward COX-2; the presence of electronegative groups at the meta position improved inhibition compared to that at the para position, and cyclic amines affected COX-2 inhibition. From the modelling point of view, it was observed that one of the O-atoms and –NH2 of the SO2NH2 moiety exhibited hydrogen interactions with Gln192 and Ala516, and the phenyl ring showed hydrophobic interactions with Leu359, Leu352, Gln350, His356, Tyr355, Met522, Tyr504, Ala527 and Phe523 in the active site (Fig. 10).47
Fig. 10. Cyclic imides.

The majority of selective COX-2 inhibitors belong to diarylheterocycles that contain vicinal diaryl substituents attached to a central ring system, mainly a mono- or bicyclic ring. A particular class is represented by 3′-(4-substituted phenyl)-4′-(4-(methylsulfonyl)phenyl)spiroisoxazoline derivatives containing naphthalene and chromanonespiro-bridges. These compounds have been synthesized as the result of bioisosteric replacement of the central tricyclic bridge with a spiroisoxazoline motif. The structure–activity relationship in this set of molecules provides good selective COX-2 inhibition, which was affected by the type of substituent at the para position of the C-30 phenyl ring. Accordingly, compounds having smaller groups (38a, 38b and 38g) were more potent and selective COX-2 inhibitors compared to other analogues, which had larger ones (Fig. 11).48
Fig. 11. Spiroisoxazoline scaffold.

Following the trail of drug repurposing, the presence of nido-dicarbaborate in the structure of indomethacin has also been considered in the substitution of the phenyl ring, which could improve the potent and selective COX-2 inhibition compared to other Non-Steroidal Anti-Inflammatory Drugs (NSAIDs).49
The use of natural products as scaffolds for biological targets is a procedure well exploited by medicinal chemists. In addition, to inhibit COX-2 selectively, natural compounds such as oleuropein were used. This natural compound was the subject of several chemical modifications aimed at creating semisynthetic derivatives50 and α-amino acids, which were docked, as new tripeptides, in the COX-2 active site to ensure inhibition.51 Hydrocinnamic acid has been used to design a series of various ether and ester derivatives acting as COX-2 inhibitors, with a good result for caffeic acid diethyl ester (CA-DE), which forms 3 hydrogen bonds with the active site of COX-2 (4-OH···OH-Tyr355, 4-OH···NH-Arg120 and C O···OH-Tyr385).52 In addition, other small peptides have been designed to improve the potent anti-inflammatory action via COX-2 inhibition; specifically, H2N–Gly–Gly–Phe–Leu–OMe with an IC50 value of 0.06 μM demonstrates good selective COX-2 inhibition and good anti-inflammatory activity.53
Investigating the natural compounds, flavonoids also have a chemical structure which can mimic the inhibitory activity against COX-2,54 and in this context, by maintaining fluorine and methoxide groups as substituents, a novel scaffold based on chromen-4-one has been docked into the COX-2 active site demonstrating good inhibitory activity. In particular, in the compound 2-(3,4-dimethoxyphenyl)-3-(4-fluorophenyl)-6-methoxy-4H-chromen-4-one, the central chromone ring is well positioned in a hydrophobic pocket formed by Met113, Val116, Val349, Tyr355, Leu359, and Ala527 residues; it could also form cation–π interactions with the guanidium group of Arg120 (Fig. 12 and Table 1).55
Fig. 12. Chromenone scaffold.
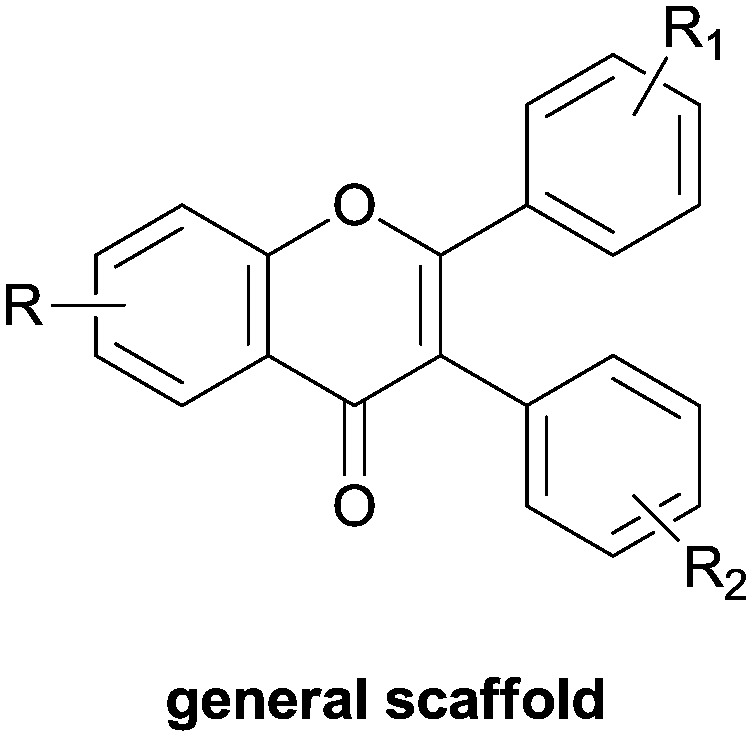
Table 1. Most potent compounds acting as selective COX-2 inhibitors.
| Compound | Original number of compound in the referenced paper | Celecoxib COX-2 IC50 (μM) | COX-1 IC50 (μM) | COX-2 IC50 (μM) | Selectivity index (SI) | Analysis | Ref. |
| 2d | 14d | 0.87 | 5.6 | 0.97 | 5.77 | a | 26 |
| 3a | 15a | 0.87 | 5.9 | 0.97 | 6.08 | a | 26 |
| 4j | 10j | 0.87 | 2.14 | 0.56 | 3.82 | a | 27 |
| 6d | 4d | 0.07 ± 0.01 | 36.11 ± 0.56 | 0.08 ± 0.03 | ∼451 | b | 28 |
| 7t | 7t | 0.31 ± 0.12 | 48.20 ± 1.30 | 0.09 ± 0.01 | >500 | b | 29 |
| 9d | 8d | 0.06 | >100 | 0.02 | >5000 | c | 33 |
| 11f | 11f | 0.07 | 0.91 | 0.03 | 30.3 | d | 34 |
| 12e | 5 | — | No inhibition at 10 μM | 0.92 ± 0.05 | — | e | 36 |
| 16c | 3c | 0.02 | No inhibition in the range 10–9–10–3 M | 1.2 | — | c | 39 |
| 17i | 4i | 0.02 | >200 | 3 | >67 | a | 39 |
| 23e | 6e | 0.1 | 63.76 | 0.48 | 132.83 | e | 41 |
| 24b | 7b | 0.18 | >50 | 0.11 | >454.54 | f | 43 |
| 24c | 7c | 0.18 | >50 | 0.13 | >384.61 | f | 43 |
| 25c | 13c | 0.18 | >50 | 0.14 | >357.14 | f | 43 |
| 25e | 13e | 0.18 | >50 | 0.13 | >384.62 | f | 43 |
| 26d | 9d | — | 34.5 | 0.063 | 547.6 | f | 44 |
| 37b | 7b | — | 10.46 | 0.09 | 116.2 | f | 48 |
Conclusions
In this manuscript, the authors' aim was to highlight the main chemical features that are able to show a selective COX-2 inhibitory activity. The selectivity is still the major goal that drives the design of new molecules. In this context, if it is possible to reach a higher COX-2 selectivity, there are many more possibilities to prepare drugs endowed with detectable action, without or minimal side effects. Like a perfect recipe, by dosing the correct ingredients, the pharmacophores' features are described to be a useful guide to retrieve the most selective COX-2 inhibitor scaffolds and chemical features. Even the repurposing theory that employs some of the side effects shown by the different drugs used indicates that a window may be opened for the world of research on selective COX-2 inhibitors. Nowadays, the selective COX-2 inhibition is an innovative strategy for pain management and cancer treatment, therefore it is crucial and necessary to develop these types of drugs.
Footnotes
†The authors declare no competing interests.
References
- Capone M. L., Tacconelli S., Di Francesco L., Sacchetti A., Sciulli M. G., Patrignani P. Prostaglandins Other Lipid Mediators. 2013;82:85. doi: 10.1016/j.prostaglandins.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Smyth E. M., Grosser T., Wang M., Yu Y., Fitzgerald G. A. J. Lipid Res. 2009;50:S423. doi: 10.1194/jlr.R800094-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzer C. A., Marnett L. J. J. Lipid Res. 2009;50:S29. doi: 10.1194/jlr.R800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrignani P., Patrono C. Biochim. Biophys. Acta, Gen. Subj. 2015;1851:422. doi: 10.1016/j.bbalip.2014.09.016. [DOI] [PubMed] [Google Scholar]
- Botting R. M. J. Therm. Biol. 2006;31:208. [Google Scholar]
- Garavito R. M., Mulichak A. M. Annu. Rev. Biophys. Biomol. Struct. 2003;32:183. doi: 10.1146/annurev.biophys.32.110601.141906. [DOI] [PubMed] [Google Scholar]
- Consalvi S., Biava M., Poce G. Expert Opin. Ther. Pat. 2015;25:1357. doi: 10.1517/13543776.2015.1090973. [DOI] [PubMed] [Google Scholar]
- Ricciotti E., Fitzgerald G. A. Arterioscler., Thromb., Vasc. Biol. 2011;31:986. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S., Klotz U. Eur. J. Clin. Pharmacol. 2008;64:233. doi: 10.1007/s00228-007-0400-7. [DOI] [PubMed] [Google Scholar]
- Brindisi M., Maramai S., Gemma S., Brogi S., Grillo A., Di Cesare Mannelli L., Gabellieri E., Lamponi S., Saponara S., Gorelli B., Tedesco D., Bonfiglio T., Landry C., Jung K., Armirotti A., Luongo L., Ligresti A., Piscitelli F., Bertucci C., Dehouck M., Campiani G., Maione S., Ghelardini C., Pittaluga A., Piomelli D., Di Marzo V., Butini S. J. Med. Chem. 2016;59:2612. doi: 10.1021/acs.jmedchem.5b01812. [DOI] [PubMed] [Google Scholar]
- Brizzi A., Aiello F., Marini P., Cascio M. G., Corelli F., Brizzi V., De Petrocellis L., Ligresti A., Luongo L., Lamponi S., Maione S., Pertwee R. G., Di Marzo V. Bioorg. Med. Chem. 2014;22:4770. doi: 10.1016/j.bmc.2014.07.006. [DOI] [PubMed] [Google Scholar]
- Aiello F., Badolato M., Pessina F., Sticozzi C., Maestrini V., Aldinucci C., Luongo L., Guida F., Ligresti A., Artese A., Allarà M., Costa G., Frosini M., Schiano Moriello A., De Petrocellis L., Valacchi G., Alcaro S., Maione S., Di Marzo V., Corelli F., Brizzi A. ACS Chem. Neurosci. 2016;7:737. doi: 10.1021/acschemneuro.5b00333. [DOI] [PubMed] [Google Scholar]
- Nasirinezhad F., Jergova S., Pearson J. P., Sagen J. Neuropharmacology. 2015;95:100. doi: 10.1016/j.neuropharm.2014.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello F., Carullo G., Badolato M., Brizzi A., Malek N., Starowicz K. ChemMedChem. Front. Pharmacol. 2016;2016;117:1686. 257. doi: 10.1002/cmdc.201600111. [DOI] [PubMed] [Google Scholar]
- Scarpelli R., Sasso O., Piomelli D. ChemMedChem. 2016;11:1242. doi: 10.1002/cmdc.201500395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laube M., Kniess T., Pietzsch J. Antioxidants. 2016;5:14. doi: 10.3390/antiox5020014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin-Fenton D. P., Heide-Jørgensen U., Ahern T. P., Lash T. L., Christiansen P., Ejlersten B., Sørensen H. T. Epidemiology. 2016;27(4):586. doi: 10.1097/EDE.0000000000000480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esbona K., Inman D., Saha S., Jeffrey J., Schedin P., Wilke L., Keely P. Breast Cancer Res. 2016;18:35. doi: 10.1186/s13058-016-0695-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T. T., Grubisha M. J., Frahm K. A., Wendell S. G., Liu J., Ricke W. A., Auchus R. J. and DeFranco D. B., JBC Papers, http://www.jbc.org/cgi/doi/10.1074/jbc.M115.711515. [DOI] [PMC free article] [PubMed]
- Roos J., Grösch S., Werz O., Schröder P., Ziegler S., Fulda S., Paulus P., Urbschat A., Kühn B., Maucher I., Fettel J., Vorup-Jensen T., Piesche M., Matrone C., Steinhilber D., Parnham M. J., Maier T. J. Pharmacol. Ther. 2016;157:43. doi: 10.1016/j.pharmthera.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Gomes R. A., Genesi G. L., Maltarollo V. G., Trossini G. H. G. J. Biomol. Struct. Dyn. 2016 doi: 10.1080/07391102.2016.1185379. [DOI] [PubMed] [Google Scholar]
- Zarghi A., Arfaei S. Iran. J. Pharm. Res. 2011;10:655. [PMC free article] [PubMed] [Google Scholar]
- Bertinaria M., Shaikh M. A., Buccellati C., Cena C., Rolando B., Lazzarato I., Fruttero R., Gasco A., Hoxha M., Capra V., Sala A., Rovati G. E., Hoxha M., Buccellati C., Capra V., Garella D., Cena C., Rolando B., Fruttero R., Carnevali S., Sala A., Rovati G. E., Bertinaria M. ChemMedChem. Pharmacol. Res. 2012;2016;7103(9):1647. 132. doi: 10.1002/cmdc.201200272. [DOI] [PubMed] [Google Scholar]
- Bartzatt R. Anti-Inflammatory Anti-Allergy Agents Med. Chem. 2014;13:17. doi: 10.2174/18715230113129990019. [DOI] [PubMed] [Google Scholar]
- Fioravanti R., Bolasco A., Manna F., Rossi F., Orallo F., Ortuso F., Alcaro S., Cirilli R. Eur. J. Med. Chem. 2010;45:6135. doi: 10.1016/j.ejmech.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Abdellatif K. R. A., Elshemy H. A. H., Azoz A. A. Bioorg. Chem. 2015;63:13. doi: 10.1016/j.bioorg.2015.09.002. [DOI] [PubMed] [Google Scholar]
- Abdellatif K. R. A., Abdelall E. K. A., Fadaly W. A. A., Kamel G. M. Bioorg. Med. Chem. Lett. 2016;26:406. doi: 10.1016/j.bmcl.2015.11.105. [DOI] [PubMed] [Google Scholar]
- Qiu H., Wang P., Li Z., Ma J., Wang X., Yang Y., Zhu H. Pharmacol. Res. 2016;104:86. doi: 10.1016/j.phrs.2015.12.025. [DOI] [PubMed] [Google Scholar]
- Lu X., Wang Z., Ren S., Shen F., Man R., Zhu H. Bioorg. Med. Chem. Lett. 2016;26:3491. doi: 10.1016/j.bmcl.2016.06.037. [DOI] [PubMed] [Google Scholar]
- Aiello F., Valacchi G. Curr. Top. Med. Chem. 2014;14(22):2576. doi: 10.2174/1568026614666141203142926. [DOI] [PubMed] [Google Scholar]
- Laube M., Tondera C., Sharma S. K., Bechmann N., Pietzsch F.-J., Pigorsch A., Köckerling M., Wuest F., Pietzsch J., Kniess T., Bhardwaj A., Kaur J., Wuest F., Knaus E. E. RSC Adv. ChemMedChem. 2014;2014;49:38726. 109. [Google Scholar]
- Tondera C., Laube M., Wimmer C., Kniess T., Mosch B., Großmann K., Pietzsch J., Tondera C., Ullm S., Laube M., Meister S., Neuber C., Mosch B., Kniess T., Pietzsch J. Biochem. Biophys. Res. Commun. Biochem. Biophys. Res. Commun. 2013;2015;430458:301. 40. doi: 10.1016/j.bbrc.2012.10.133. [DOI] [PubMed] [Google Scholar]
- Laube M., Gassner C., Sharma S. K., Günther R., Pigorsch A., König J., Köckerling M., Wuest F., Pietzsch J., Kniess T. J. Org. Chem. 2015;80:5611. doi: 10.1021/acs.joc.5b00537. [DOI] [PubMed] [Google Scholar]
- Wuest F., Tang X., Kniess T., Pietzsch J., Suresh M. Bioorg. Med. Chem. 2009;17:1146. doi: 10.1016/j.bmc.2008.12.032. [DOI] [PubMed] [Google Scholar]
- Battilocchio C., Poce G., Alfonso S., Porretta G. C., Consalvi S., Sautebin L., Pace S., Rossi A., Ghelardini C., Di Cesare Mannelli L., Schenone S., Giordano A., Di Francesco L., Patrignani P., Biava M. Bioorg. Med. Chem. 2013;21:3695. doi: 10.1016/j.bmc.2013.04.031. [DOI] [PubMed] [Google Scholar]
- Consalvi S., Alfonso A., Di Capua A., Poce G., Pirolli A., Sabatino M., Ragno R., Anzini M., Sartini S., La Motta C., Di Cesare Mannelli L., Ghelardini C., Biava M. Bioorg. Med. Chem. 2015;23:810. doi: 10.1016/j.bmc.2014.12.041. [DOI] [PubMed] [Google Scholar]
- Di Capua A., Sticozzi C., Brogi S., Brindisi M., Cappelli A., Sautebin L., Rossi A., Pace S., Ghelardini C., Di Cesare Mannelli L., Valacchi G., Giorgi G., Giordani A., Poce G., Biava M., Anzini M. Eur. J. Med. Chem. 2016;109:99. doi: 10.1016/j.ejmech.2015.12.044. [DOI] [PubMed] [Google Scholar]
- Sarnpitak P., Mujumdar P., Morisseau C., Hwang S. H., Hammock B., Iurchenko V., Zozulya S., Gavalas A., Geronikaki A., Ivanenkov Y., Krasavin M. Eur. J. Med. Chem. 2014;84:160. doi: 10.1016/j.ejmech.2014.07.023. [DOI] [PubMed] [Google Scholar]
- Jawabrah Al-Hourani B., Sharma S. K., Kaur J., Wuest F., Jawabrah Al-Hourani B., Al-Awaida W., Matalka K. Z., El-Barghouthi M. I., Alsoubani F., Wuest F. Med. Chem. Res. Bioorg. Med. Chem. Lett. 2015;2016;2426:78. 4757. [Google Scholar]
- Hasabelnaby S., Mohi El-Deen E. M., Goudah A. Anti-Inflammatory Anti-Allergy Agents Med. Chem. 2015;14(3):148. doi: 10.2174/1871523014666151103113544. [DOI] [PubMed] [Google Scholar]
- Grover J., Bhatt N., Kumar V., Patel N. K., Gondaliya B. J., Sobhia M. E., Bhutani K. K., Jachak S. M., Consalvi S., Poce G., Ragno R., Sabatino M., La Motta C., Sartini S., Calderone V., Martelli A., Ghelardini C., Di Cesare Mannelli L., Biava M. RSC Adv. ChemMedChem. 2015;2016;511:45535. 1804. [Google Scholar]
- Badrey M. G., Abdel-Aziz H. M., Gomha S. M., Abdalla M. M., Mayhoub A. S. Molecules. 2015;20:15287. doi: 10.3390/molecules200815287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragab F. A., Heiba H. I., El-Gazzar M. G., Abou-Seri S. M., El-Sabbagh W. A., El-Hazek R. M. MedChemComm. 2016;7:2309. [Google Scholar]
- Ghodsi R., Azizi E., Zarghi A. Iran. J. Pharm. Res. 2016;15(1):169. [PMC free article] [PubMed] [Google Scholar]
- Al-Suwaidan J. A., Alanazi A. M., El-Azab A. S., Al-Obaid A. M., ElTahir K. E., Maarouf A. R., Abu El-Enin M. A., Abdel-Aziz A. A.-M. Bioorg. Med. Chem. Lett. 2013;23:2601. doi: 10.1016/j.bmcl.2013.02.107. [DOI] [PubMed] [Google Scholar]
- Alanazi A. M., El-Azab A. S., Al-Suwaidan I. A., ElTahir K. E. H., Asiri Y. A., Abdel-Aziz N. I., Abdel-Aziz A. A.-M. Eur. J. Med. Chem. 2015;92:115. doi: 10.1016/j.ejmech.2014.12.039. [DOI] [PubMed] [Google Scholar]
- Firke S. D., Bari S. B. Bioorg. Med. Chem. 2015;23:5273. doi: 10.1016/j.bmc.2015.07.070. [DOI] [PubMed] [Google Scholar]
- Adolhasani H., Dastmalchi S., Hamzeh-Mivehroud M., Daraei B., Zarghi A. Med. Chem. Res. 2016;28:858. [Google Scholar]
- Neumann W., Xu S., Sárosi M. B., Scholz M. S., Crews B. C., Ghebreselasie K., Banerjee S., Marnett L. J., Hey-Hawkins E. ChemMedChem. 2016;11:175. doi: 10.1002/cmdc.201500199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procopio A., Alcaro S., Nardi M., Oliverio M., Ortuso F., Sacchetta P., Pieragostino D., Sindona G. J. Agric. Food Chem. 2009;57:11161. doi: 10.1021/jf9033305. [DOI] [PubMed] [Google Scholar]
- Vernieri E., Gomez-Monterrey I., Milite C., Grieco P., Musella S., Bertamino A., Scognamiglio I., Alcaro S., Artese A., Ortuso F., Novellino E., Sala M., Campiglia P. J. Amino Acids. 2013;2013:606282. doi: 10.1155/2013/606282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva T., Borges F., Edraki N., Alizadeh M., Miri R., Saso L., Firuzi O. RSC Adv. 2015;5:58902. [Google Scholar]
- Singh P., Kaur S., Kaur J., Singh G., Bhatti R. J. Med. Chem. 2016;59:3920. doi: 10.1021/acs.jmedchem.6b00134. [DOI] [PubMed] [Google Scholar]
- Dao T. T., Chi Y. S., Kim J., Kim H. P., Kim S., Park H. Bioorg. Med. Chem. Lett. 2004;14:1165. doi: 10.1016/j.bmcl.2003.12.087. [DOI] [PubMed] [Google Scholar]
- Rullah K., Aluwi M. F. F. M., Yamin B. M., Baharuddin M. S., Ismail N. H., Teruna H. Y., Bukhari S. N. A., Jantan I., Jalil J., Husain K., Wai L. K. J. Mol. Struct. 2015;1081:51. [Google Scholar]