

 Studies on FAAH inhibitory potency and metabolic stability of indolyl-substituted alkyl- and alkylpiperidine carbamates are described.
Studies on FAAH inhibitory potency and metabolic stability of indolyl-substituted alkyl- and alkylpiperidine carbamates are described.
Abstract
A series of phenyl 4-[(indol-1-yl)alkyl]piperidine carbamates was synthesized and tested for inhibition of the endocannabinoid degrading enzyme fatty acid amide hydrolase (FAAH) and for metabolic stability in rat liver S9 fractions and porcine blood plasma. Structure–activity relationship studies revealed that variation of the length of the alkyl spacer connecting the indole and the piperidine heterocycle, introduction of substituents into the indole ring, replacement of the piperidine by a piperazine scaffold as well as opening of the piperidine ring system affect activity significantly. The metabolic stability of this compound class proved to be significantly higher than that of corresponding phenyl N-(indol-1-ylalkyl)carbamates.
The endocannabinoid system is involved in numerous physiological and pathophysiological processes. Central elements of this system are the cannabinoid receptors CB1 and CB2, endogenous ligands such as anandamide and 2-arachidonoyl glycerol, enzymes that synthesize and degrade those ligands, as well as proteins for their transportation.1–3 An important player in the endocannabinoid metabolism is fatty acid amide hydrolase (FAAH).4 This enzyme rapidly cleaves anandamide into arachidonic acid and ethanolamine and, by this way, it terminates its beneficial effects, like analgesia and reduction of inflammation. Since inhibition of FAAH is supposed to prolong and enhance the action of anandamide, inhibitors of FAAH may represent new agents against pain and inflammation.5 In the past years many potent inhibitors of FAAH have been found6 like α-ketoheterocycles,7 heteroarylpropan-2-ones,8 urea compounds9 and carbamate derivatives.10 Despite some drawbacks during the evaluation of such agents in man,3,11,12 FAAH still can be considered as an interesting target for new therapeutic approaches.3,13,14
The carbamate inhibitors of FAAH can be divided in compounds, in which the carbamate nitrogen is part of a cyclic structure such as piperidine or piperazine (e.g. compounds 1 and 2),15–17 and in derivatives, in which this nitrogen is substituted with one alkyl or cycloalkyl residue (e.g. compounds 3 and 4) (Fig. 1).18–20
Fig. 1. Inhibitors of FAAH with carbamate structure.

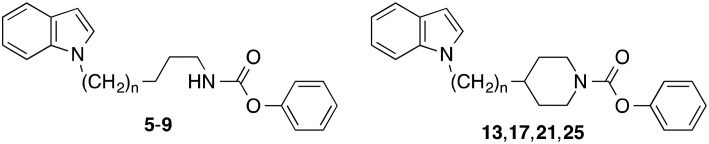
We have developed FAAH inhibitors of the latter class with N-alkyl substituents bearing heteroaryl-residues at the terminal alkylcarbon like the indolylalkylcarbamates 5–9 (Fig. 1).21 In the present investigation we wanted to evaluate the effect of an incorporation of the carbamate nitrogen and the adjoining alkyl carbons of these alkylcarbamates into a piperidine ring system on their FAAH inhibitory potency and in vitro metabolic stability. Furthermore, more detailed structure–activity relationship studies with this type of compounds should be performed.
The derivative of compound 5, in which the first three carbon atoms and the nitrogen of the butylcarbamate chain were integrated into a piperidine ring was synthesized as outlined in Scheme 1. Piperidin-4-ylmethanol was reacted with phenyl chloroformate to give the phenyl carbamate 11. The alcohol group of this compound was substituted with bromine in an Appel reaction using tetrabromomethane and triphenylphosphine. Treatment of obtained intermediate 12 with indole, deprotonated by NaH in DMF, afforded the target compound 13.
Scheme 1. Reagents and conditions: (a) phenyl chloroformate, N-ethyl-N,N-diisopropylamine, THF, room temperature, 12 h; (b) tetrabromomethane, triphenylphosphine, CH2Cl2, room temperature, 4 d; (c) indole, NaH, DMF, room temperature, 20 h.

The homologous derivatives of 13, in which the methylene group linking the indole with the piperidine ring was replaced by ethylene and propylene bridges, respectively, were prepared starting from the piperidinyl alcohols 14 and 18 (Scheme 2). Treatment with phenyl chloroformate afforded the carbamates 15 and 19. The primary alcohol groups of these compounds were transformed into mesyl esters utilizing methanesulfonyl chloride and triethylamine. Finally, the mesylates 16 and 20 were reacted with the sodium salt of indole to yield the desired indolylalkyl-substituted piperidine carbamates 17 and 21.
Scheme 2. Reagents and conditions: (a) 15: phenyl chloroformate, triethylamine, CH2Cl2, room temperature, overnight; 19: phenyl chloroformate, N-ethyl-N,N-diisopropylamine, THF, room temperature, 12 h; (b) methanesulfonyl chloride, triethylamine, THF, room temperature, 2 h; (c) indole, NaH, KI, DMF, 60 °C, 2 h.

For the synthesis of the homologous derivative with butyl spacer between indole and piperidine, the BOC-protected piperidinylbutanol 22 (ref. 22 and 23) was reacted with methanesulfonyl chloride and triethylamine (Scheme 3). Substitution of the mesylate group of obtained intermediate 23 by indole followed by cleavage of the BOC-group with conc. HCl in methanol led to the indolylbutyl-substituted piperidine 24. Carbamoylation of the piperidine nitrogen of 24 with phenyl chloroformate afforded the target carbamate 25.
Scheme 3. Reagents and conditions: (a) methanesulfonyl chloride, triethylamine, CH2Cl2, room temperature, 5 h; (b) indole, NaH, DMF, 60 °C, 5 h followed by conc. HCl, methanol, room temperature, 2 h; (c) phenyl chloroformate, N-ethyl-N,N-diisopropylamine, CH2Cl2, room temperature, 5 h.

The derivatives of 17 with varying substituents at the phenyl ring of the indole scaffold (26–34) and the azaindole compounds 39–42 were synthesized by reaction of 16 with the appropriate indole employing conditions similar to those described for the preparation of 17.
The piperazine carbamate 38 (Scheme 4) was prepared from 35 using a reaction sequence analogous to that outlined for the synthesis of 25 (Scheme 3).
Scheme 4. Reagents and conditions: (a) methanesulfonyl chloride, triethylamine, CH2Cl2, room temperature, 3 h; (b) 6-fluoroindole, NaH, DMSO, 60 °C, 6 h; (c) conc. HCl, methanol, room temperature, 24 h followed by phenyl chloroformate, N-ethyl-N,N-diisopropylamine, THF, room temperature, 5 h.

The synthesis of the N-methylated pentylcarbamte 46 started with the alkylation of 6-fluoroindole (43) with N-(6-bromohexyl)phthalimide under basic conditions (Scheme 5). Cleavage of the phthalimide protecting group of obtained intermediate 44 with hydrazine provided the amine 45, which was methylated with paraformaldehyde/NaBH4 (ref. 24) and finally reacted with phenyl chloroformate to yield the carbamate 46.
Scheme 5. Reagents and conditions: (a) NaH, 5-bromopentylphthalimide, DMSO, 60 °C, 6 h and 90 °C, 3 h; (b) hydrazine hydrate (24%), ethanol, reflux, 3 h; (c) paraformaldehyde, NaOCH3, methanol, room temperature, 5 h, NaBH4, room temperature, 30 min followed by phenyl chloroformate, N-ethyl-N,N-diisopropylamine, THF, room temperature, 3 h.

The target compounds were evaluated for FAAH inhibition utilizing microsomes from rat brain as enzyme source25 and the fluorogenic probe N-(2-hydroxyethyl)-4-pyren-1-ylbutanamide26 as substrate. To the incubation buffer the detergent Triton X-100 was added for solubilization of the substrate and for stabilization of the membrane bound enzyme.27–29 Inhibitory potency of the test substances was assessed by comparing the amount of 4-pyren-1-ylbutanoic acid released from the substrate in their absence and presence after an incubation time of 60 min by reversed phase HPLC with fluorescence detection applying 6-pyren-1-ylhexanoic acid as internal standard.25 The metabolic stability in rat liver S9 fractions and in porcine blood plasma was evaluated with reversed phase HPLC and MS-detection.30
Investigations on the change of inhibitory potency of the phenyl N-indolylalkylcarbamates 5–9 in dependence of the length of the alkyl chain exhibited two maxima of activity.21 With IC50 values of about 0.1 μM each, the compounds with pentyl and heptyl residues (6 and 8) possessed the highest inhibitory activities in this series of compounds (Table 1). Elongation or shortening of the alkyl spacer of 6 and 8, respectively, by one carbon reduced FAAH inhibition by approximately three- to tenfold as reflected by the IC50 values of the carbamates 5, 7 and 9. Similar structure–activity relationships were found for the analogous piperidine carbamates 13, 17, 21 and 25. Stepwise extension of the length of the alkyl linker between the indole and the piperidine ring from one to four carbons was also accompanied with a fluctuation in activity. The highest potencies were observed for the derivatives with ethyl and butyl chains 17 and 25. Notably, in these two (indolylalkyl)piperidine carbamates the number of carbons connecting the indole and the carbamate nitrogens is identical with that in the most active indolylalkylcarbamates 6 and 8. Furthermore, it can be seen that incorporation of the carbamate nitrogen and the adjoining α-, β- and γ-alkyl carbons of the butyl, hexyl and heptyl chain of 5, 7 and 8 respectively, into a piperidine ring led to a two- to tenfold decrease of activity, while in case of the derivate with a pentyl spacer (6) this structural modification did not have a significant impact on inhibitory potency as shown by the similar IC50 values of 6 and 17.
Table 1. Inhibitory potency against FAAH and stability in biological environments.
| ||||
| Compound | n | Inhibition of FAAH IC50 a (μM) | Stability in liver S9 fractions b (%) | Stability in blood plasma c (%) |
| 5 | 1 | 0.96 | 53 ± 5 | 82 ± 9 |
| 6 | 2 | 0.094 | 52 ± 2 | 91 ± 7 |
| 7 | 3 | 0.25 | 50 ± 2 | 68 ± 8 |
| 8 | 4 | 0.090 | 59 ± 3 | 39 ± 7 |
| 9 | 5 | 0.24 | 49 ± 1 | 64 ± 4 |
| 13 | 1 | 9.8 | 63 ± 5 | 93 ± 4 |
| 17 | 2 | 0.11 | 87 ± 4 | 94 ± 4 |
| 21 | 3 | 0.57 | 80 ± 1 | 93 ± 3 |
| 25 | 4 | 0.18 | 78 ± 6 | 87 ± 8 |
| 3 (URB597) | 0.060 | 82 ± 4 | 77 ± 6 | |
aValues are the means of at least two independent determinations, errors are within ±20%; the deviations of the IC50 value of URB597 from published data8,25,26,30,34 are due to variations in the assay procedure (for details see ESI of ref. 20).
bPercent of parent remaining after incubation with rat liver S9 fractions for 30 min in presence of the co-factor NADPH; values are means ± standard deviations of independent determinations (n = 3); the lower stability value published for URB597 recently21 was an artefact.
cPercent of parent remaining after incubation with porcine blood plasma for 30 min; values are means ± standard deviations of independent determinations (n = 3).
Recently, we have found that the stability of the alkylcarbamates 5–9 against metabolic phase I reactions in rat liver S9 fractions was quite similar. About 50–60% of these compounds remained unmetabolized after incubation in liver homogenate in presence of NADPH. The corresponding piperidine carbamates 13, 17, 21 and 25 exhibited a higher stability under the same conditions. Here, 60–90% of the parent compounds could be recovered at the end of the metabolic reactions (Table 1). In porcine plasma the alkycarbamates 5–9 showed varying stabilities. While the concentration of the butyl and pentylcarbamates 5 and 6 decreased only slightly, the heptylcarbamate 8 was metabolized by about 60%. This significant instability of 8 was due to hydrolysis by plasma paraoxonase and bis(p-nitrophenyl)phosphate-sensitive carboxylesterases as proven with inhibitors of these enzymes.30 In contrast, all new piperidine carbamates displayed a pronounced stability in porcine blood plasma.
The piperidine carbamate with the highest FAAH inhibitory activity was the derivative with ethyl spacer between piperidine and indole residue. Since this compound (17) possessed a high metabolic stability in liver homogenate as well as in blood plasma, it was chosen for further inhibitor development. First, different residues were introduced at indole 5-position. While chloro, methyl, methoxy and cyano substituents reduced inhibition of FAAH by four- to tenfold, a fluoro atom did not affect activity significantly (Table 2). Next, a fluoro atom was introduced in the other three free positions of the phenyl part of the indole. In position 4, this atom also did not change FAAH inhibitory potency. In contrast, fluoro atoms in position 6 (32) and 7 (33) enhanced activity about twofold. As shown by the IC50 values of 34 and 38, exchange of the indole ring of 17 by a carbazole heterocycle, and replacement of the piperidine ring of 32 by a piperazine system resulted in an about fourfold drop of potency. The four azaindole-substituted derivatives 39–42 (Table 3) were also less active against FAAH than the corresponding indole 17 (Table 2), with the compounds bearing nitrogen atoms in indole 6 and 7 position (41 and 42) possessing a higher activity than the 5- and 6-substituted analogues 39 and 40.
Table 2. Inhibitory potency against FAAH.

| ||
| Compound | R | Inhibition of FAAH IC50 a (μM) |
| 17 | H | 0.11 |
| 26 | 5-F | 0.13 |
| 27 | 5-Cl | 0.41 |
| 28 | 5-CH3 | 0.70 |
| 29 | 5-OCH3 | 0.36 |
| 30 | 5-CN | 0.97 |
| 31 | 4-F | 0.11 |
| 32 | 6-F | 0.046 |
| 33 | 7-F | 0.052 |
| 34 | 0.41 | |
| 38 | 0.18 | |
aValues are the means of at least two independent determinations, errors are within ±20%.
Table 3. Inhibitory potency of azaindole-substituted 4-ethylpiperidine carbamates against FAAH.
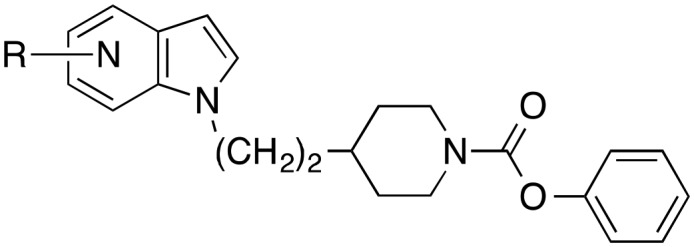
| ||
| Compound | Position of the nitrogen in the indole ring | Inhibition of FAAH IC50 a (μM) |
| 39 | 4 | 1.7 |
| 40 | 5 | 0.79 |
| 41 | 6 | 0.31 |
| 42 | 7 | 0.20 |
aValues are the means of at least two independent determinations, errors are within ±20%.
Finally, we were interested in the question, whether the intact piperidine ring of 32 is necessary for its pronounced activity. Therefore, we synthesized compound 46, in which carbon 3 of the piperidine ring is missing. This structural variation led to an about 35-fold decrease of activity (Table 4). Thus, the rigid carbamate structure is necessary for good enzyme inhibition. Viewed from the side of the pentyl carbamate 6 it can be said that methylation of the nitrogen of this aliphatic carbamate is not well tolerated by the enzyme.
Table 4. Inhibitory potency against FAAH and stability in biological environments.

| |||
| Compound | Inhibition of FAAH IC50 a (μM) | Stability in liver S9 fractions b (%) | Stability in blood plasma c (%) |
| 32 | 0.046 | 82 ± 1 | 93 ± 7 |
| 46 | 1.6 | 65 ± 5 | 93 ± 3 |
aValues are the means of at least two independent determinations, errors are within ±20%.
bPercent of parent remaining after incubation with rat liver S9 fractions for 30 min in presence of the co-factor NADPH; values are means ± standard deviations of independent determinations (n = 3).
cPercent of parent remaining after incubation with porcine blood plasma for 30 min; values are means ± standard deviations of independent determinations (n = 3).
FAAH belongs to the large group of serine hydrolase enzymes, which comprises of about 200 members.31 Phenyl carbamate inhibitors of FAAH have been shown to inactivate the enzyme by covalent carbamoylation of the enzyme's catalytic nucleophilic serine.17,32 Since other members of the serine hydrolase family can also react with carbamates in the same way in principle, we tested the most active piperidine carbamate 32 for inhibition of the serine hydrolases monoacylglycerol lipase (MAGL) and cytosolic phospholipase A2α (cPLA2α). MAGL is also involved in the process of endocannabinoid inactivation,1,2 cPLA2α is the key enzyme of the arachidonic acid cascade.33 Like FAAH, both enzymes hydrolyze very lipophilic substrates. At the highest test concentration of 10 μM, MAGL34 and cPLA2α35 were not affected by compound 32. These data provide evidence that the compound class investigated exhibits some specificity for FAAH.
Conclusions
In conclusion, integration of the amino group and the α-, β- and γ-carbons of the alkyl chain of the indolylalkylcarbamates 5–8 into a piperidine ring system does not increase inhibitory potency against FAAH. However, metabolic stability in rat liver S9 fractions and in porcine plasma can be enhanced by this structural variation. With the piperidine carbamate 32 a FAAH inhibitor was obtained, which possesses similar activity and metabolic stability as the known FAAH standard inhibitor URB597.
Supplementary Material
Footnotes
†The authors declare no competing interests.
‡Electronic supplementary information (ESI) available: Syntheses and analytical data of the test compounds. See DOI: 10.1039/c6md00683c
References
- Lutz B., Marsicano G., Maldonado R., Hillard C. J. Nat. Rev. Neurosci. 2015;16:705. doi: 10.1038/nrn4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parolaro D. and Rubino T., The endocannabinoid system in General and Molecular Pharmacology, ed. F. Clementi and G. Fumagalli, John Wiley & Sons, Hoboken, New Jersey, 2015. [Google Scholar]
- Kaur R., Ambwani S. R., Singh S. Curr. Clin. Pharmacol. 2016;11:110. doi: 10.2174/1574884711666160418105339. [DOI] [PubMed] [Google Scholar]
- McKinney M. K., Cravatt B. F. Annu. Rev. Biochem. 2005;74:411. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- Bisogno T., Maccarrone M. Expert Opin. Drug Discovery. 2013;8:509. doi: 10.1517/17460441.2013.780021. [DOI] [PubMed] [Google Scholar]
- Lodola A., Castelli R., Mor M., Rivara S. Expert Opin. Ther. Pat. 2015;25:1247. doi: 10.1517/13543776.2015.1067683. [DOI] [PubMed] [Google Scholar]
- Boger D. L., Sato H., Lerner A. E., Hedrick M. P., Fecik R. A., Miyauchi H., Wilkie G. D., Austin B. J., Patricelli M. P., Cravatt B. F. Proc. Natl. Acad. Sci. U. S. A. 2000;97:5044. doi: 10.1073/pnas.97.10.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahov S., Drews A., Hess M., Schulze Elfringhoff A., Lehr M. ChemMedChem. 2011;6:544. doi: 10.1002/cmdc.201000473. [DOI] [PubMed] [Google Scholar]
- Johnson D. S., Stiff C., Lazerwith S. E., Kesten S. R., Fay L. K., Morris M., Beidler D., Liimatta M. B., Smith S. E., Dudley D. T., Sadagopan N., Bhattachar S. N., Kesten S. J., Nomanbhoy T. K., Cravatt B. F., Ahn K. ACS Med. Chem. Lett. 2011;2:91. doi: 10.1021/ml100190t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria S., Gaetani S., Fegley D., Valino F., Duranti A., Tontini A., Mor M., Tarzia G., La Rana G., Calignano A., Giustino A., Tattoli M., Palmery M., Cuomo V., Piomelli D. Nat. Med. 2003;9:76. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Huggins J. P., Smart T. S., Langman S., Taylor L., Young T. Pain. 2012;153:1837. doi: 10.1016/j.pain.2012.04.020. [DOI] [PubMed] [Google Scholar]
- von Schaper E. Nat. Biotechnol. 2016;34:223. doi: 10.1038/nbt0316-223a. [DOI] [PubMed] [Google Scholar]
- Celorrio M., Fernández-Suárez D., Rojo-Bustamante E., Echeverry-Alzate V., Ramírez M. J., Hillard C. J., López-Moreno J. A., Maldonado R., Oyarzábal J., Franco R., Aymerich M. S. Brain, Behav., Immun. 2016;57:94. doi: 10.1016/j.bbi.2016.06.010. [DOI] [PubMed] [Google Scholar]
- Marco E. M., Rapino C., Caprioli A., Borsini F., Laviola G., Maccarrone M. PLoS One. 2015;10:e0137034. doi: 10.1371/journal.pone.0137034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki S., Munakata R., Kawano N., Samizu K., Oka H., Ishii T. and Sugane T., WO2010007966, 2010.
- Makriyannis A., Shukla V. G. and Alapafuja S. O., WO2016014975, 2016.
- Pember S. O., Mejia G. L., Price T. J., Pasteris R. J. Bioorg. Med. Chem. Lett. 2016;26:2965. doi: 10.1016/j.bmcl.2016.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor M., Rivara S., Lodola A., Plazzi P. V., Tarzia G., Duranti A., Tontini A., Piersanti C., Kathuria S., Piomelli D. J. Med. Chem. 2004;47:4998. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
- Sit S. Y., Conway C., Bertekap R., Xie K., Bourin C., Burris K., Deng H. Bioorg. Med. Chem. Lett. 2007;17:3287. doi: 10.1016/j.bmcl.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Micoli A., De Simone A., Russo D., Ottonello G., Colombano G., Ruda G. F., Bandiera T., Cavalli A., Bottegoni G. Med. Chem. Commun. 2016;7:537. [Google Scholar]
- Terwege T., Dahlhaus H., Hanekamp W., Lehr M. Med. Chem. Commun. 2014;5:932. [Google Scholar]
- Pippel D. J., Mapes C. M., Neelakandha S. M. J. Org. Chem. 2007;72:5828. doi: 10.1021/jo070646a. [DOI] [PubMed] [Google Scholar]
- Sugimoto H., Negoro N., Murata T. and Rikimaru K., WO2011055770, 2011.
- Barluenga J., Bayon A. M., Asensio G. J. Chem. Soc., Chem. Commun. 1984:1334. [Google Scholar]
- Holtfrerich A., Hanekamp W., Lehr M. Eur. J. Med. Chem. 2013;63:64. doi: 10.1016/j.ejmech.2013.01.050. [DOI] [PubMed] [Google Scholar]
- Forster L., Schulze Elfringhoff A., Lehr M. Anal. Bioanal. Chem. 2009;394:1679. doi: 10.1007/s00216-009-2850-5. [DOI] [PubMed] [Google Scholar]
- Shoichet B. K. Drug Discovery Today. 2006;11:607. doi: 10.1016/j.drudis.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B. Y., Shoichet B. K. Nat. Protoc. 2006;1:550. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegard A., Hanekamp W., Griessbach K., Fabian J., Lehr M. Eur. J. Med. Chem. 2012;48:153. doi: 10.1016/j.ejmech.2011.12.009. [DOI] [PubMed] [Google Scholar]
- Terwege T., Hanekamp W., Garzinsky D., König S., Koch O., Lehr M. ChemMedChem. 2016;11:429. doi: 10.1002/cmdc.201500445. [DOI] [PubMed] [Google Scholar]
- Bachovchin D. A., Cravatt B. F. Nat. Rev. Drug Discovery. 2012;11:52. doi: 10.1038/nrd3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander J. P., Cravatt B. F. Chem. Biol. 2005;12:1179. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie C. C. J. Lipid Res. 2015;56:1386. doi: 10.1194/jlr.R057588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtfrerich A., Makharadze T., Lehr M. Anal. Biochem. 2010;399:218. doi: 10.1016/j.ab.2009.12.015. [DOI] [PubMed] [Google Scholar]
- Hanekamp W., Lehr M. J. Chromatogr., B. 2012;900:79. doi: 10.1016/j.jchromb.2012.05.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.