

 We report the discovery of an orally efficacious 3-aza-aminooxazoline-BACE1 inhibitor with 1 800 000× fold selectivity against CatD and no retinal effects in an early screening rat toxicology study.
We report the discovery of an orally efficacious 3-aza-aminooxazoline-BACE1 inhibitor with 1 800 000× fold selectivity against CatD and no retinal effects in an early screening rat toxicology study.
Abstract
As part of an ongoing effort at Amgen to develop a disease-modifying therapy for Alzheimer's disease, we have previously used the aminooxazoline xanthene (AOX) scaffold to generate potent and orally efficacious BACE1 inhibitors. While AOX-BACE1 inhibitors demonstrated acceptable cardiovascular safety margins, a retinal pathological finding in rat toxicological studies demanded further investigation. It has been widely postulated that such retinal toxicity might be related to off-target inhibition of Cathepsin D (CatD), a closely related aspartyl protease. We report the development of AOX-BACE1 inhibitors with improved selectivity against CatD by following a structure- and property-based approach. Our efforts culminated in the discovery of a picolinamide-substituted 3-aza-AOX-BACE1 inhibitor absent of retinal effects in an early screening rat toxicology study.
Introduction
A widely held hypothesis for the underlying pathogenesis of Alzheimer's disease (AD) proposes aggregation of amyloid β (Aβ) peptides and cerebral deposition of amyloid plaques as critical events.1 Aβ is produced via sequential endoproteolytic cleavage of amyloid precursor protein (APP) by the aspartyl protease β-site APP cleaving enzyme-1 (BACE1) and γ-secretase.2 Inhibition of BACE1 decreases production of all major forms of Aβ including the most pathogenic species Aβ42, suggesting that BACE1 is a prime therapeutic target for the development of disease modifying therapies for AD.3–6 The translation of changes in cerebrospinal fluid (CSF) Aβ levels, as an effect biomarker for brain Aβ lowering, in response to small molecule BACE1 inhibition is well established in rodents and higher preclinical animal species.7 The first prominent and long-lasting Aβ reduction in human lumbar cerebrospinal fluid was disclosed in 2011.8 Since then, several companies have advanced additional small molecule BACE1 inhibitors to human clinical trials for the treatment of AD.9–11 Potential safety liabilities of BACE1 inhibitors are less well studied. As an example, the development of the clinical candidate LY2811376 was terminated in phase 1 due to adverse retinal toxicity in a 3-month rat preclinical toxicology study.8 We reported a similar pathology in a 1-month preclinical toxicology study in Sprague Dawley rats administered with the lead compound, AMG-8718.12,13 Retinal toxicity in the rat was characterized by retinal thinning preceded by the accumulation of autofluorescent granules (AFG) in the retinal pigment epithelium (RPE), suggestive of disruption of phagolysosomal function in the RPE.14 These findings prompted us and others to evaluate additional BACE1 inhibitors for their potential to increase AFG in the RPE of preclinical species, as an early marker of incipient retinal thinning. To date, the results of several preclinical toxicology studies involving BACE1 inhibitors as they relate to retinal toxicity have been disclosed (Fig. 1): six BACE1 inhibitors tested “positive” for induction of AFG in the RPE (LY2811376,8AMG-8718,121,15PF-3195,16PF-1283,16 and PF-928316) whereas four BACE1 inhibitors tested “negative” (LY2886721,16,17 2,183,16 and NB-36019). We report herein the discovery of an aminooxazoline xanthene (AOX)-BACE1 inhibitor (Fig. 1, 30) that is devoid of early signs of retinal pathology.
Fig. 1. BACE1 inhibitors that tested “positive” (*) or “negative” (**) for induction of AFG in the RPE in preclinical species. Each study design varied with respect to preclinical species, compound dose and study length.

A current hypothesis is that the retinal toxicity in preclinical species is due to off-target inhibition of Cathepsin D (CatD), a structurally related aspartyl protease that plays an important role in phagolysosomal processing in the RPE.20 The scientific evidence for CatD as a possible mediator of the retinal toxicity is mounting as progress has been made linking the observed in vivo accumulation of AFG in the RPE to enzymatic (biophysical or biochemical) and cellular CatD inhibition, together with lysosomal accumulation.16
Our objective was to improve the selectivity of the AOX-BACE1 inhibitors against human CatD (CatD/BACE1 selectivity) by using a structure- and property-based approach. Our measurement of CatD/BACE1 selectivity was based on an enzymatic in vitro assay and we aimed for a large in vitro selectivity window.21 This in vitro data allowed us to quickly rank-order compounds for subsequent in vivo toxicological profiling in rats with an emphasis on retinal evaluation. We focused our efforts on the aminooxazoline 3-aza-xanthene (3-aza-AOX) core as the retinal toxicity profile of the lead molecule of this series, (compound 1 in Fig. 1) had been extensively studied in rats and cynomolgus monkeys.15
Our structure-based approach was based on exploiting the residue differences in the S3 sub-pocket (S3sp) – also referred to as the deep S3 pocket – of the BACE1 protein versus the CatD protein.18,22 Fig. 2A shows the co-crystal structure of rat CatD with BACE1 inhibitor 423 (PDB ; 5UX4) – a closely related analog of 1. The amino acid sequence of rat CatD shows 83% identity with human CatD and the residues within 5 Å of the co-crystallized ligand 4 are identical between the two species.24,25 To the best of our knowledge, this constitutes the first time a co-crystal of a BACE1 inhibitor with the CatD protein has been published. The catalytic aspartic acids and the flap tyrosine are conserved in both CatD (Fig. 2A) and BACE1 (Fig. 2B, PDB ; 4WTU)23 and drive the same alignment of AOX core in both proteins. The P326 group occupies the S3 pocket in CatD as well as in BACE1, but the protein surfaces reveal an opportunity to extend a vector from the 7 position of the xanthene core deeper into the S3 pocket of BACE1 and potentially increase selectivity over CatD (Fig. 2A and B). In particular, analysis of the aligned S3 regions of BACE1 and CatD (Fig. 2C) reveal a key difference between an alanine residue (Ala335) in BACE1 and the correspondingly larger and more polar Asp318 residue in the same binding side region of CatD. This key steric and polar residue mismatch in a relatively buried region of the S3 subsite afforded an attractive and compelling potential selectivity handle to pursue.
Fig. 2. (A) Co-crystal structure of rat CatD and AOX-BACE1 inhibitor 4 (yellow, PDB ; 5UX4). The mobile flap region is shown in pink and its surface is not depicted for clarity. (B) Co-crystal structure of human BACE1 and AOX-BACE1 inhibitor 4 (yellow; PDB ; 4WTU).23 Red spheres represent water molecules. The mobile flap region is shown in blue and its surface is not depicted for clarity. (C) Overlaid co-crystal structure of human BACE1 (blue) + compound 4 (yellow) overlaid with co-crystal structure of rat CatD (pink) + compound 4. Key residues are labelled in yellow for BACE1 and pink for CatD. Key hydrogen bonds are depicted as yellow dashed lines.

The challenge was multifaceted regarding the property-based approach. The importance of choosing an amidine-type BACE1 inhibitor warhead with an optimal pKa has been extensively discussed in the context of: a) BACE1 cell potency;27–30 b) central nervous system (CNS) exposure and robust CNS pharmacodynamic (PD) activity;31,32 and c) potential lysosomal accumulation and polypharmacology.14,33–35 The pKa of the aminooxazoline warhead is influenced by the core,31 with the 3-aza-xanthene core maintaining the pKa of the 2-aminooxazoline (e.g.1 pKa = 5.7) in a desirable range (5.5 < pKa < 7.5); i.e., maximizing engagement with the catalytic aspartate residues within the BACE1 protein while minimizing the potential for lysosomal accumulation. Additionally, the overall properties of the molecule as they relate to CNS penetration/CNS drug-like space were closely monitored throughout the process of modifying the structure towards the goal of increasing selectivity over CatD.36–38
The aim of this work was to evaluate alkynylpyridines39–47 and picolinamides17,19,31,32,41,46,48–53 as P3 sub-pocket (P3sp) groups on the 3-aza-AOX core and test their influence on CatD selectivity. The strategy was to initially omit the P2′ substituent, in order to offset the increase in molecular weight (MW) upon extending into the S3sp and maximize lipophilic efficiency (Fig. 3).
Fig. 3. Strategy to improve selectivity of 3-aza-AOX-BACE1 inhibitors against CatD.

Results and discussion
Chemistry
The 3-aza-AOX BACE1 inhibitors profiled in Tables 1–3 were synthesized via several routes. The synthesis of the truncated BACE1 inhibitors 6 and 7 is shown in Scheme 1. The sequence for the synthesis of key intermediate 5 was disclosed in a previous publication15 and additional experimental details for each step including characterization of the intermediates can be found in the ESI‡ of this publication. Suzuki–Miyaura-coupling of the unprotected enantiomerically pure intermediate 5 with either 2-fluoro-3-pyridine boronic acid or 5-fluoro-3-pyridine boronic acid yielded compounds 6 and 7, respectively. The reaction proceeded with high regioselectivity for the bromide (96% (6) and 58% (7) respectively) using (Pd(AmPhos)2Cl2).54
Table 1. Different trajectories to access the S3 sub-pocket.

|
R1 = 
|

|

|

|

|
R2 = 
|
Cl | Cl | Cl | Cl | |
| Cmd # | 1 | 6 | 7 | 9 | 21 |
| BACE1 IC50 [μM] a | 0.00092 | 0.072 | 0.32 | 0.013 | 0.021 |
| CatD IC50 [μM] a | 0.66 | 15 | 130 | 72 | 1100 |
| HEK IC50 [μM] b | 0.021 | 0.83 | 4.7 | 0.12 | 0.19 |
| Selectivity c CatD/BACE1 | 660 | 210 | 400 | 5600 | 53 000 |
| MW [g mol–1] | 443 | 383 | 383 | 402 | 442 |
| clog P d /LipE e | 2.6/6.5 | 2.5/4.7 | 2.5/4.0 | 3.0/4.9 | 2.8/4.9 |
| PSA [Å2]/#HBD | 95/1 | 83/1 | 83/1 | 83/1 | 112/2 |
aIC50 values were averaged values determined by at least two independent experiments.
bHuman embryonic kidney cells.
cSelectivity ratio [IC50 CatD/IC50 BACE1].
dclog P values were calculated using ACD LogD Suite (version 10).
eLipE = –log 10(BACE1 IC50) – clog P.
Table 2. P2′ SAR of deep-P3 alkyne substituted 3-aza-AOX BACE1 inhibitors.

|
IC50
a
[μM] |
Selectivity c | P app d [nm s–1] | P-gp efflux ratio
e
|
Microsomal stability Clint
f
[μL min–1 mg–1] |
||||||
| Cpd | R1 | R2 | BACE1 | HEK b | CatD | CatD/BACE1 | rER | hER | RLM | HLM | |
| 9 | Me |

|
0.013 | 0.12 | 72 | 5600 | 22 | 1.7 | 2.8 | 21 | 27 |
| 13 | Me |

|
0.00031 | 0.0012 | 5.9 | 19 000 | 18 | 2.1 | 3.1 | 63 | 77 |
| 14 | Me |

|
0.00046 | 0.0022 | 2.7 | 5900 | 19 | 1.6 | 1.9 | 45 | 76 |
| 15 | Me |

|
0.00043 | 0.0041 | 4.5 | 10 000 | 15 | 1.8 | 2.4 | 20 | 40 |
| 16 |
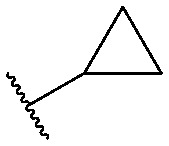
|

|
0.0018 | 0.087 | 3.6 | 2100 | 13 | 1.4 | 2.3 | 23 | 80 |
| 17 |

|

|
0.00058 | 0.0024 | 4.4 | 7500 | 15 | 44 | 50 | 46 | 84 |
| 18 |

|

|
0.0055 | 0.10 | 12 | 2200 | 18 | — | 5.6 | 73 | 420 |
aIC50 values were averaged values determined by at least two independent experiments.
bHuman embryonic kidney cells.
cSelectivity ratio [IC50 CatD/IC50 BACE1].
dApparent permeability measured in parental LLC-PK1 cells. Values are an average of apical to basolateral (A to B) and basolateral to apical (B to A) velocities and are reported as nm s–1.
eEfflux measured in LLC-PK1 cells transfected with human MDR1 and are reported as a ratio of (B to A)/(A to B).
fRat liver microsomal (RLM) and human liver microsomal (HLM) clearance reported as apparent intrinsic clearance with no correction for binding and not scaled. Compound concentration = 1 μM. Microsomal protein concentration = 0.25 mg mL–1.
Table 3. P2′ SAR of deep-P3 picolinamide substituted 3-aza-AOX BACE1 inhibitors.

|
IC50
a
[μM] |
Selectivity c | P app d [nm s–1] | P-gp efflux ratio
e
|
Microsomal stability CLint
f
[μL min–1 mg–1] |
LipE g | |||||
| Cpd | R | BACE1 | HEK b | CatD | CatD/BACE1 | hER | rER | RLM | HLM | ||
| 21 | Cl | 0.021 | 0.19 | 1100 | 53 000 | 28 | 2.8 | 2.9 | <14 | 37 | 4.9 |
| 32 | OMe | 0.0075 | 0.052 | 1200 | 160 000 | 25 | 2.0 | 3.1 | <14 | <14 | 5.4 |
| 27 | Me | 0.031 | 0.054 | 640 | 20 000 | 40 | 7.8 | 8.0 | <14 | 16 | 5.1 |
| 28 |

|
0.0036 | 0.013 | 310 | 85 000 | 29 | 2.6 | 2.5 | <14 | <14 | 5.5 |
| 29 |

|
0.00057 | 0.0025 | 1000 | 1 800 000 | 27 | 6.5 | 8.5 | 79 | 80 | 7.2 |
| 30 |

|
0.00062 | 0.0014 | 480 | 770 000 | 29 | 3.4 | 3.3 | 84 | 48 | 7.0 |
aIC50 values were averaged values determined by at least two independent experiments.
bHuman embryonic kidney cells.
cSelectivity ratio [IC50 CatD/IC50 BACE1].
dApparent permeability measured in parental LLC-PK1 cells. Values are an average of apical to basolateral (A to B) and basolateral to apical (B to A) velocities and are reported as nm s–1.
eEfflux measured in LLC-PK1 cells transfected with human MDR1 and are reported as a ratio of (B to A)/(A to B).
fRat liver microsomal (RLM) and human liver microsomal (HLM) clearance reported as apparent intrinsic clearance with no correction for binding and not scaled. Compound concentration = 1 μM. Microsomal protein concentration = 0.25 mg mL–1.
gLipE = –log 10(BACE1 IC50) – clog P (calculated).
Scheme 1. Reagents: (a) 2-fluoropyridin-3-ylboronic acid or 5-fluoropyridin-3-ylboronic acid, Pd(AmPhos)2Cl2 (cat.), K3PO4, dioxane/water, 100 °C, 96% (6) or 58% (7).

The synthesis of deep-P326 alkyne containing 3-aza-AOX compounds is illustrated in Scheme 2.55 Miyaura borylation of intermediate 5 under standard reaction conditions afforded boronic ester 8 in 91% yield. Subsequent palladium-catalyzed Suzuki-cross coupling of 8 with the corresponding 3-bromo-5-alkyne-pyridines using Pd(AmPhos)2Cl2 provided compounds 9–12 in moderate yields. The chloro-pyridine of these alkyne-substituted 3-aza-AOX analogs (9–12) was further converted into more elaborate P2′ substituents via Suzuki-cross coupling. Three privileged P2′ substituents from formerly published lead AOX-BACE1 inhibitors15,23,56 were installed by using either the corresponding boronic acid or pinacol boronic ester affording compounds 13–18.
Scheme 2. Reagents: (a) B2Pin2, Pd(dppf)Cl2 (cat.), KOAc, dioxane, 91%; (b) 3-bromo-5-alkyne-pyridines, Pd(AmPhos)2Cl2 (cat.), K3PO4, dioxane/water, 100 °C, 26–58%; (c) 2-(5,6-dihydro-2H-pyran-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (for 13, 16, 17 and 18), 2-(3,6-dihydro-2H-pyran-4-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (for 14), or (2-fluoropyridin-4-yl)boronic acid (for 15) with catalyst system A (Pd(PPh3)4 (cat.), K2CO3, dioxane/water, 120 °C for synthesis of 13 and 15) or catalyst system B (Pd(AmPhos)2Cl2 (cat.), K3PO4, dioxane/water, 100 °C for synthesis of 14, 16–18).

All deep-P3 amide-substituted 3-aza-AOX compounds were synthesized via amide coupling of the corresponding amino-(phenyl)-substituted 3-aza-AOX intermediates (20, 23–26 and 31) and 5-chloropicolinic acid but with slight variations, as depicted in Scheme 3. For ease of discussion “protected” and “unprotected” refers to the aminooxazoline warhead functional group. Intermediate 5 was mono-benzoyl protected to give bromide 19, which was converted to the amino-substituted-benzoyl-protected 3-aza-AOX 20via Pd-catalyzed coupling with LiHMDS as the ammonia surrogate.57 Intermediate 20 was reacted with 5-chloropicolinic acid under 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) amide coupling conditions, followed by cleavage of the benzoyl protecting group using sodium hydroxide, to yield the final compound 21.
Scheme 3. Reagents: (a) (i) NaN3, CuI (cat.), trans-N,N′-dimethylcyclohexane-1,2-diamine, sodium ascorbate, EtOH/water, 90 °C, (ii) NaBH4, 80%; (b) benzoic anhydride, 90%; (c) Pd2(dba)3 (cat.), CyJohnPhos, LiHMDS, 69%; (d) (i) EDCI, HOBt, 5-chloropicolinic acid; (ii) NaOH; 36% overall yield. (e) Trimethyl borate (for 23), cyclopropyl boronic acid (for 24) or corresponding pinacol boranes (for 25 and 26), Pd(AmPhos)2Cl2 (cat.), K3PO4, dioxane/water, 110 °C, 69–99%; (f) 5-chloropicolinic acid, DMTMM, 26–65%. (g) (i) NaOMe, 55%; (ii) Pd2(dba)3 (cat.), CyJohnPhos, LiHMDS, 71%; (iii) NaOH, 90%.

Alternatively, the unprotected amino-substituted 3-aza-AOX 22 was obtained from the unprotected bromide 5via a Cu-catalyzed azide coupling, followed by reduction with sodium borohydride.58 Precursor 22 was elaborated to intermediates 23–26 through Suzuki coupling without any additional protecting of either the warhead or the aminophenyl group. 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM)59,60 promoted the coupling of 5-chloropicolinic acid with intermediates 23–26 giving compounds 27–30. Analog 32 was synthesized by nucleophilic aromatic substitution of chloro-pyridine 19 with sodium methoxide, introduction of an amino-group via LiHMDS coupling, warhead deprotection, and amide coupling with 5-chloropicolinic acid.
Biological activity
As previously discussed, our strategy was to initially minimize MW by reducing the size of the P2′-substituent on the 3-aza-AOX core while fully exploiting CatD and BACE1 protein residue differences in the S3 sub-pocket. To this end P2′-Cl-substituted compound 6, a truncated version of compound 1, was chosen as a reference compound to monitor progress (Table 1). Despite replacing the 2-fluoro-4-pyridinyl substituent with a chlorine, compound 6 retained high potency in the BACE1 enzymatic assay (IC50 = 72 nM) and moderate potency in the cell assay (IC50 = 830 nM). The cellular potency of the compounds was assessed by measuring the inhibition of Aβ40 production in HEK293 cells expressing APP. The CatD/BACE1 selectivity of 6 was calculated to be 210× based on measured enzymatic CatD potency. Based on the co-crystal structures shown in Fig. 2A, the P3 group (2-fluoropyridin-3-yl) in 6 was replaced by a structural isomer. It was predicted that a 5-fluoro-3-pyridyl P3 group would position the fluorine in a trajectory towards the S3 sub-pocket. The affinity of the 5-fluoro-3-pyridyl analog 7 towards CatD decreased by 8.5×-fold compared to compound 6, while the affinity towards BACE1 dropped only by 4.5×-fold (Table 1). This result suggested that the fluorine in 5-fluoro-3-pyridyl was a potential vector into the S3 sub-pocket which could be further exploited. As a follow-up, a 5-propynyl-3-pyridyl analog 9 was prepared to further probe this vector.39,40 Compound 9 showed improved BACE1 enzyme (13 nM) and cell potency (120 nM), as well as increased CatD/BACE1 selectivity (∼5600×). Additionally, the physicochemical properties of 9 were in an acceptable range for CNS penetration (MW = 402 g mol–1, clog P = 3.0, PSA = 83 Å2).36
As an alternative approach, we replaced the 5-substituted-3-pyridyl P3 group with an elongated picolinamide, which has been a privileged moiety heavily utilized in several previously disclosed BACE1 inhibitor designs and initially described by Shionogi.17,19,31,32,41,46,48–52 The 5-chloropicolinamide was well tolerated on the 3-aza-AOX core with compound 21 showing high enzymatic BACE1 activity (21 nM), a 10× cell shift (190 nM) and very high CatD/BACE1 selectivity (∼53 000×). Of particular note, the absolute CatD IC50 value exceeded 1 mM.61 The superior CatD/BACE1 selectivity obtained with the 5-chloropicolinamide-substituted compound 21 (Table 1) came at the cost of increased MW, PSA (442 g mol–1, and 112 Å2, respectively) and the addition of another hydrogen bond donor. In order to assess if CNS penetration, and ultimately in vivo Aβ40 lowering was attainable for these 3-aza-AOX BACE1 inhibitors, data on permeability, P-gp transporter recognition and microsomal stability was collected. The results shown in Table 2 (entry 1, 9) and Table 3 (entry 1, 21) confirmed that both compounds were highly permeable, had low efflux ratios and low turnover in human and rat liver microsomes. These results warranted further work on both types of deep P3 groups with the goal of improving BACE1 potency without eroding selectivity towards CatD.
We replaced the chlorine in compound 9 with P2′ substituents that were successfully used in former lead AOX-BACE1 inhibitors.15,23,56 Three alkyne analogs 13–15 with these privileged P2′ substituents were tested in the BACE1 enzymatic and cellular assays and showed picomolar BACE1 affinity with low cell shift (Table 2). High passive permeability and low P-gp efflux ratios were maintained for all three analogs. Compound 15 was metabolically stable in rat and human liver microsomal studies, whereas increased microsomal turnover was observed for the dihydropyrans 13 and 14. Importantly, high CatD/BACE1 selectivity was maintained for all three compounds, with the 5,6-dihydro-2H-pyran-3-yl analog 13 displaying the highest CatD/BACE1 selectivity (∼19 000×). A X-ray co-crystal structure of compound 13 bound to BACE1 was obtained to confirm the hypothesized orientation of the propyne into the S3 sub-pocket (Fig. 4, PDB ; 5UYU). As observed in previously reported AOX-BACE1 X-ray co-crystal structure, the aminooxazoline moiety interacts with the two catalytic aspartic acids (Asp228, Asp32) and Tyr 71 engages in the π-stacking interaction with the 3-aza-AOX core. The nitrogen of the 3-aza-AOX core nitrogen forms a hydrogen bond interaction with Trp 76 through a water bridge and the dihydropyran unit resides in the S2′ pocket of the protein where the oxygen atom is within hydrogen bond distance to Arg128. As proposed, the propynyl group occupies the S3 sub-pocket and engages in a lipophilic interaction with Ala335. The high CatD selectivity could be attributed to the residue difference in the S3 sub-pocket as highlighted in the CatD protein overlay in Fig. 4b (Ala335 in BACE1 vs. Asp318 in CatD): whereas the methyl terminus of the propynyl moiety engages in a van der Waals interaction (3.8 Å) with the methyl group of Ala335; structural alignment of the CatD protein clearly demonstrates the repulsive steric interaction which would ensue with the Asp318 side chain of CatD.
Fig. 4. Left: Co-crystal structure of human BACE1 + compound 13 (turquoise, PDB ; 5UYU). The mobile flap region is shown in orange. Key hydrogen bonds are depicted as black dashed lines. Right: Co-crystal structure of human BACE1 (blue) + compound 13 (turquoise) overlaid with rat CatD (pink). Key residues are labelled in yellow for BACE1 and pink for CatD.

Additional alkyne analogs (Table 2, 16–18) were investigated in an attempt to improve the microsomal stability of 13 and explore the impact on CatD/BACE1 selectivity. A larger cell shift and moderate CatD/BACE1 selectivity was observed for the cyclopropylethynyl analog 16 and the 3-methyl-3-oxetan-ethynyl analog 18. In addition, the hydroxymethyl analog 17 demonstrated high P-gp mediated efflux. An attempt to mask the hydroxy group in form of a 3-methyl-oxetan-3-yl group (18) resulted in reduced P-gp efflux.13 However, all three analogs 16–18 suffered from high in vitro metabolism.
We continued our investigation on the deep-P3 amide-substituted compound 21 in order to improve cell potency (Tables 1 and 3). Cognizant of the physicochemical properties, small substituents were chosen as alternative P2′ groups (Table 3, 27, 28 and 32). The methoxy (32) and the cyclopropyl (28) P2′ groups not only improved BACE1 enzyme and cell potency (52 nM and 13 nM, respectively) but also maintained the high CatD/BACE1 selectivity relative to chloro analog 21. Both compounds were highly permeable with favourable P-gp efflux ratios and were stable in human and rat liver microsomes. Replacement of the chlorine by a methyl group (27) increased P-gp efflux ratios. To further test the limits of the CNS drug-like space as it relates to the deep-P3 amide-substituted 3-aza-AOX scaffold, the occupancy of S2′ pocket was maximized via implementation of dihydropyran P2′ substituents (Table 3, 29 and 30). The additional contact with the BACE1 protein due to the P2′ substituents led to BACE1 affinities in the picomolar range. A low cell shift (2–4 fold) was maintained and CatD selectivity values were extremely high, ranging from ∼770 000× to ∼1 800 000×. Despite the high molecular weight (MW = 490 g mol–1 for 29 and 30), both analogs remained highly permeable. The increase in PSA (121 Å2 for 29 and 30) led only in the case of compound 29 to increased recognition by P-gp efflux transporter (h/rER = 6.5/8.5). Elevated turnover in microsomes was measured for both dihydropyran analogs 29 and 30.
On the basis of the overall in vitro profiles and lipophilic efficiency (LipE) values (Table 3), the PD response of compounds 28, 29 and 30 was assessed. Naïve Sprague Dawley (SD) rats were administered a single low oral dose (10 mg kg–1) of the corresponding test compound and the Aβ40 levels in CSF and brain as well as exposure levels in plasma and CSF were measured after 4 h (Table 4). While all three compounds showed comparable reduction (>58%) of Aβ40 in the CSF compartment after a 10 mg kg–1 single oral dose, various degrees of Aβ40 reduction in the brain were obtained. The compounds were rank-ordered using estimated unbound plasma concentration to achieve 50% Aβ40 lowering in the brain from the single-point experiment.62 The estimated unbound plasma drug concentration corresponding to 50% Aβ40 reduction in the brain (EC50,unbound) for compound 29 was 12-fold shifted relative to the cell IC50 consistent with the previous observation that BACE1 inhibitors with large efflux ratios require higher unbound plasma concentrations relative to IC50 values to generate robust Aβ40 reduction.15 Compound 30 had the lowest estimated unbound plasma drug concentration corresponding to 50% Aβ40 reduction in the CSF (EC50,unbound = 5 nM) and in the brain (EC50,unbound = 11 nM) and was subsequently evaluated in a time-course PD study in rats (Fig. 5). Aβ40 levels in the CSF and brain as well as drug plasma levels were measured at multiple timepoints in the same study. The Aβ40 reduction in the CSF and brain compartments of rats after a single 30 mg kg–1 oral dose reached its maximum after 4 hours and returned to baseline after 24 h.
Table 4. Reduction of Aβ40 levels in CSF and brain in wild-type Sprague-Dawley rats and additional profiling.
| Cpd a | Aβ40 reduction
b
[%] |
Estimated EC50,unbound
c
[μM] |
Cplasma,unbound d [μM] | CCSF e [μM] | Shift | pKa | ||
| CSF | Brain | CSF | Brain | Brain EC50/cell IC50 | ||||
| 1 | 74 | 65 | 0.011 | 0.017 | 0.032 | 0.06 | 0.81 | 5.6 f |
| 28 | 66 | 52 | 0.012 | 0.021 | 0.022 | 0.02 | 1.5 | NA g |
| 29 | 58 | 25 | 0.011 | 0.030 | 0.015 | 0.005 | 12 | NA g |
| 30 | 62 | 41 | 0.0049 | 0.011 | 0.0078 | 0.003 | 7.9 | 6.1 h |
aCompounds were dosed orally 10 mg kg–1 as a solution (2% HMPC, 1% Tween80) to male Sprague-Dawley rats. n = 5 animals per group.
bCSF and brain Aβ40 measured at t = 4 h.
cEstimated unbound plasma EC50 for CSF or brain effect from a single dose experiment at 4 h timepoint or unbound plasma concentration corresponding to 50% Aβ40 in CSF or brain, respectively.
dUnbound plasma drug concentration measured at t = 4 h.
eCSF drug concentration measured at t = 4 h.
fExperimental pKa determined by potentiometry at T = 25 °C.
gNA = not available.
hExperimental pKa determined by UV spectrophotometry at T = 25 °C.
Fig. 5. Time dependent Aβ40 reduction in CSF and brain in rat after 30 mg kg–1 oral dose of 30 and total plasma concentrations of compound 30 measured in the same time-course study.

An indirect response model was used to model the PK–PD relationship which accounts for the formation and clearance of Aβ40 in the CSF.63 Using this model in vivo unbound plasma IC50 values for CSF and brain effect were generated (15 nM and 17 nM, respectively). These values were in good agreement with the estimated unbound plasma EC50 values obtained in the single dose, single time point PD study (5 nM and 11 nM, respectively).
The pharmacokinetic (PK) profile of 30 was evaluated in rat and dog (Table 5). Consistent with the in vitro rat microsomal stability data, 30 showed moderate clearance in rat. Moderate clearance and similar Vdss was also maintained in dog. Taking into account the different protein binding of 30 in rat versus dog, unbound clearance was lower in dog (Clunbound = 15.6 L h–1 kg–1) compared to rat (Clunbound = 80 L h–1 kg–1). The bioavailability was calculated to be moderate in both species (%F = 30).
Table 5. Pharmacokinetic profiles and plasma protein binding of 30.
| Species | iv
a
|
po
b
|
PPB Fu | |||
| Cl [L h–1 kg–1] | V dss [L kg–1] | C max [μM] | t 1/2 [h] | %F | ||
| Rat | 0.61 | 1.2 | 0.7 | 2.4 | 28 | 0.0076 |
| Dog | 0.36 | 1.3 | 0.7 | 4.5 | 30 | 0.023 |
aRat: 2 mg kg–1 dose as a solution in DMSO; dog: 1 mg kg–1 dose as a solution in 25% Captisol/75% water at pH = 4.
b5 mg kg–1 dose (rat) or 2 mg kg–1 dose (dog) as a solution in 1% Tween80/2% HMPC/97% water at pH = 2 (rat) or pH = 5 (dog).
A X-ray co-crystal structure of 30 in BACE1 overlaid with CatD protein is shown in Fig. 6 (PDB ; 5I3W).64,65 Every interaction of the BACE1 protein with the amino-oxazoline-warhead, the 3-aza-xanthene backbone and the P2′ substituent is conserved compared to AOX-BACE1 inhibitors absent of a deep-P3 substituent. Fig. 5 highlights the hydrogen-bonding between the NH of the amide and the backbone carbonyl of Gly230 as well as the near co-planarity of the amide linker between the 3-aza-AOX core and the 2-pyridyl. The picolinamide extends into the S3 sub-pocket and the chlorine in para-position engages in a repulsive steric interaction with the Asp318 resulting in the very high CatD selectivity of this compound. When tested against a panel of other aspartyl proteases, compound 30 showed high selectivity for BACE1 over Cathepsin E, pepsin and renin, but no selectivity over homologue BACE2.
Fig. 6. Co-crystal structure of human BACE1 (blue) + compound 30 (green, PDB ; 5I3W) overlaid with CatD (pink). Key residues are labeled in yellow for BACE1 and pink for CatD.

Compound 30 demonstrated a significant pharmacodynamic response and an acceptable PK profile in rat and dog. To further evaluate the likelihood for retinal toxicity of compound 30, a 4 day repeat dose toxicology study was conducted in male Sprague Dawley rats.66 Animals were administered daily oral doses of vehicle, 100, 300 and 900 mg kg–1 (n = 3/dose level) of compound 30 for 4 days and necropsied 24 hours after the last dose. After the last dose, the mean exposure (area under the curve; AUC0–24) was 985 μg h mL–1 at 900 mg kg–1. No test-article related increases in AFG in the RPE were observed at any dose level when evaluated by fluorescence microscopy.12 Thus, at high multiples (127×) over the exposure necessary for 50% Aβ40 reduction in the CSF (AUC0–24 = 7.72 μg h mL–1 based on estimated total plasma drug concentration corresponding to 50% Aβ40 reduction in the CSF EC50,total = 0.658 nM) no microscopic changes in the retina were observed for 30 after 4 days. In contrast, we observed accumulation of AFG in the RPE after only 4 days after a daily oral 30 mg kg–1 dose of compound 1, which corresponded to a 10× multiple.67
During the evolution of AOX-BACE1 inhibitors great attention has been paid to the activity at the hERG cardiac ion channel.15,56 Consequently, the cardiovascular (CV) effects of compound 30 were evaluated. Compound 30 was found to block hERG-mediated current in vitro (IC50 = 0.9 μM). The risk of QTc prolongation was further assessed in an anesthetized dog cardiovascular model. Two male Beagle dogs were anesthetized and instrumented for cardiovascular monitoring before and after intravenous infusion of 30 at doses of 3.0, 7.5 and 25 mg kg–1 (30 min per dose). A blood sample was taken every 10 min at the end of each 30 min infusion to determine plasma drug concentration. After each dose of 30, the average unbound plasma concentration was 0.11 μM, 0.35 μM and 0.85 μM, respectively. These drug levels corresponded to unbound plasma concentrations of 9×, 25× and 59× fold over the unbound BACE IC50 in the rat as determined by the indirect response model. Based on exposure-QTc prolongation response analysis (18 total PK-QT points; 9 per dog), plasma drug levels >0.3 μM (unbound) caused QTc interval prolongation (≥10% relative to baseline) in a concentration-dependent manner. We concluded, that the QTc prolongation observed for 30 in the dog was consistent with its hERG channel inhibition, and decided not to further pursue compound 30 due to the low cardiovascular safety margin.
Conclusions
In summary, a series of highly selective 2-aminooxazoline 3-aza-xanthene BACE1 inhibitors versus CatD were synthesized by installing substituents reaching into the S3 sub-pocket of the BACE1 protein. A combined structure- and property-based approach led to the identification of picolinamide-substituted 3-aza-AOX BACE1 inhibitors with suitable in vitro profiles, warranting further in vivo studies. Despite pushing the limits of physicochemical properties as they relate to CNS drug-likeness, the picolinamide-substituted 3-aza-AOX BACE1 inhibitors showed robust reduction of brain and CSF Aβ. The highly BACE1 over CatD selective compound 30 was further evaluated in a 4 day repeat toxicology study in rats and did not show any retinal effects, including AFG in the RPE, at doses up to 900 mg kg–1. Despite our efforts culminating in the discovery of a picolinamide-substituted 3-aza-AOX-BACE1 inhibitor (30) with significantly greater selectivity over CatD, which appeared to have reduced the likelihood of retinal toxicity, a low safety margin for the elongation of the QTc interval in the dog cardiovascular model prevented the further assessment of compound 30.
Supplementary Material
Acknowledgments
The authors thank Jennifer Allen for helpful discussions and editing of the manuscript, Kyung-Hyun Gahm, Wes Barnhart, Samuel Thomas, and Larry Miller for chiral separation; Patricia Lopez for scale-up of compound 30; Alexander Long for purification of the BACE protein and Opas Nuanmanee for purification of the CatD protein; Steven Louie, Joel Esmay, Leeanne Zalameda and Yi Luo for performing in vitro screening of compounds; Paul H. Wen and Li Zhu for performing in vivo studies. The Advanced Light Source is supported by the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
†The authors declare no competing interests.
‡Electronic supplementary information (ESI) available. See DOI: 10.1039/c7md00106a
References
- Karran E., Mercken M., De Strooper B. Nat. Rev. Drug Discovery. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- Munro K. M., Nash A., Pigoni M., Lichtenthaler S. F., Gunnersen J. M. J. Mol. Neurosci. 2016;60:305–315. doi: 10.1007/s12031-016-0800-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T., Atwal J. K., Steinberg S., Snaedal J., Jonsson P. V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J., Hoyte K., Gustafson A., Liu Y., Lu Y., Bhangale T., Graham R. R., Huttenlocher J., Bjornsdottir G., Andreassen O. A., Joensson E. G., Palotie A., Behrens T. W., Magnusson O. T., Kong A., Thorsteinsdottir U., Watts R. J., Stefansson K. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- Yuan J., Venkatraman S., Zheng Y., McKeever B. M., Dillard L. W., Singh S. B. J. Med. Chem. 2013;56:4156–4180. doi: 10.1021/jm301659n. [DOI] [PubMed] [Google Scholar]
- Oehlrich D., Prokopcova H., Gijsen H. J. M. Bioorg. Med. Chem. Lett. 2014;24:2033–2045. doi: 10.1016/j.bmcl.2014.03.025. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K., Osswald H. L. Chem. Soc. Rev. 2014;43:6765–6813. doi: 10.1039/c3cs60460h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Riddell D., Hajos-Korcsok E., Bales K., Wood K. M., Nolan C. E., Robshaw A. E., Zhang L., Leung L., Becker S. L., Tseng E., Barricklow J., Miller E. H., Osgood S., O'Neill B. T., Brodney M. A., Johnson D. S., Pettersson M. J. Pharmacol. Exp. Ther. 2012;342:366–375. doi: 10.1124/jpet.112.192625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May P. C., Dean R. A., Lowe S. L., Martenyi F., Sheehan S. M., Boggs L. N., Monk S. A., Mathes B. M., Mergott D. J., Watson B. M., Stout S. L., Timm D. E., LaBell E. S., Gonzales C. R., Nakano M., Jhee S. S., Yen M., Ereshefsky L., Lindstrom T. D., Calligaro D. O., Cocke P. J., Hall D. G., Friedrich S., Citron M., Audia J. E. J. Neurosci. 2011;31:16507–16516. doi: 10.1523/JNEUROSCI.3647-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R. Alzheimer's Res. Ther. 2014;6:1–14. doi: 10.1186/s13195-014-0089-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan C. Nat. Biotechnol. 2015;33:115–116. doi: 10.1038/nbt0215-115. [DOI] [PubMed] [Google Scholar]
- Kennedy Matthew E., Chen X., Hodgson Robert A., Hyde Lynn A., Kuvelkar R., Parker Eric M., Stamford Andrew W., Cumming Jared N., Li W., Scott Jack D., Cox K., Dockendorf Marissa F., Kleijn Huub J., Mei H., Stone Julie A., Egan M., Ereshefsky L., Jhee S., Mattson Britta A., Palcza J., Tanen M., Troyer Matthew D., Tseng Jack L., Forman Mark S. Sci. Transl. Med. 2016;8:363ra150. doi: 10.1126/scitranslmed.aad9704. [DOI] [PubMed] [Google Scholar]
- Fielden M. R., Werner J., Jamison J. A., Coppi A., Hickman D., Dunn, II R. T., Trueblood E., Zhou L., Afshari C. A., Lightfoot-Dunn R. Toxicol.Toxicol. Pathol.Pathol. 2015;43:581–592. doi: 10.1177/0192623314553804. [DOI] [PubMed] [Google Scholar]
- Dineen T. A., Chen K., Cheng A. C., Derakhchan K., Epstein O., Esmay J., Hickman D., Kreiman C. E., Marx I. E., Wahl R. C., Wen P. H., Weiss M. M., Whittington D. A., Wood S., Fremeau R. T., White R. D., Patel V. F. J. Med. Chem. 2014;57:9811–9831. doi: 10.1021/jm5012676. [DOI] [PubMed] [Google Scholar]
- Khoh-Reiter S., Sokolowski S. A., Jessen B., Evans M., Dalvie D., Lu S. Toxicol. Sci. 2015;145:383–395. doi: 10.1093/toxsci/kfv059. [DOI] [PubMed] [Google Scholar]
- Chen J. J., Liu Q., Yuan C., Gore V., Lopez P., Ma V., Amegadzie A., Qian W., Judd T. C., Minatti A. E., Brown J., Cheng Y., Xue M., Zhong W., Dineen T. A., Epstein O., Human J., Kreiman C., Marx I., Weiss M. M., Hitchcock S. A., Powers T. S., Chen K., Wen P. H., Whittington D. A., Cheng A. C., Bartberger M. D., Hickman D., Werner J. A., Vargas H. M., Everds N. E., Vonderfecht S. L., Dunn, II R. T., Wood S., Fremeau, Jr R. T., White R. D., Patel V. F. Bioorg. Med. Chem. Lett. 2015;25:767–774. doi: 10.1016/j.bmcl.2014.12.092. [DOI] [PubMed] [Google Scholar]
- Zuhl A. M., Nolan C. E., Brodney M. A., Niessen S., Atchison K., Houle C., Karanian D. A., Ambroise C., Brulet J. W., Beck E. M., Doran S. D., O'Neill B. T., am Ende C. W., Chang C., Geoghegan K. F., West G. M., Judkins J. C., Hou X., Riddell D. R., Johnson D. S. Nat. Commun. 2016;7:13042. doi: 10.1038/ncomms13042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May P. C., Willis B. A., Lowe S. L., Dean R. A., Monk S. A., Cocke P. J., Audia J. E., Boggs L. N., Borders A. R., Brier R. A., Calligaro D. O., Day T. A., Ereshefsky L., Erickso J. A., Gevorkyan H., Gonzales C. R., James D. E., Jhee S. S., Komjathy S. F., Li L., Lindstrom T. D., Mathe B. M., Martenyi F., Sheehan S. M., Stout S. L., Timm D. E., Vaught G. M., Watson B. M., Winneroski L. L., Yang Z., Mergott D. J. J. Neurosci. 2015;35:1199–1210. doi: 10.1523/JNEUROSCI.4129-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler C. R., Brodney M. A., Beck E. M., Barreiro G., Nolan C. E., Pan F., Vajdos F., Parris K., Varghese A. H., Helal C. J., Lira R., Doran S. D., Riddell D. R., Buzon L. M., Dutra J. K., Martinez-Alsina L. A., Ogilvie K., Murray J. C., Young J. M., Atchison K., Robshaw A., Gonzales C., Wang J., Zhang Y., O'Neill B. T. J. Med. Chem. 2015;58:2678–2702. doi: 10.1021/jm501833t. [DOI] [PubMed] [Google Scholar]
- Neumann U., Rueeger H., Machauer R., Veenstra S. J., Lueoend R. M., Tintelnot-Blomley M., Laue G., Beltz K., Vogg B., Schmid P., Frieauff W., Shimshek D. R., Staufenbiel M., Jacobson L. H. Mol. Neurodegener. 2015;10:1–15. doi: 10.1186/s13024-015-0033-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinfeld R., Reinhardt K., Schreiber K., Hillebrand M., Kraetzner R., Brueck W., Saftig P., Gaertner J. Am. J. Hum. Genet. 2006;78:988–998. doi: 10.1086/504159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standard deviations for the in vitro CatD enzymatic assay can be found in the ESI
- Baldwin E. T., Bhat T. N., Gulnik S., Hosur M. V., Sowder R. C., Cachau R. E., Collins J., Silva A. M., Erickson J. W. Proc. Natl. Acad. Sci. U. S. A. 1993;90:6796–6800. doi: 10.1073/pnas.90.14.6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Brown J., Judd T. C., Lopez P., Qian W., Powers T. S., Chen J. J., Bartberger M. D., Chen K., Dunn R. T., Epstein O., Fremeau R. T., Harried S., Hickman D., Hitchcock S. A., Luo Y., Minatti A. E., Patel V. F., Vargas H. M., Wahl R. C., Weiss M. M., Wen P. H., White R. D., Whittington D. A., Zheng X. M., Wood S. ACS Med. Chem. Lett. 2015;6:210–215. doi: 10.1021/ml500458t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonezawa S., Takahashi T., Wang X. J., Wong R. N. S., Hartsuck J. A., Tang J. J. Biol. Chem. 1988;263:16504–16511. [PubMed] [Google Scholar]
- Birch N. P., Loh Y. P. Nucleic Acids Res. 1990;18:6445–6446. doi: 10.1093/nar/18.21.6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In this publication, substituents on the aminooxazoline xanthene core were named in accordance with the space they occupy in the catalytic domain of the BACE1 enzyme as it was determined by X-ray cocrystal structure of previous AOX BACE1 inhibitors with BACE1 protein. For example, the substituents binding in the S3 region of the enzyme are named P3 substituents, those binding in the S3 sp pocket are named P3 sp or deep P3 substituents, those binding in the S2′ pocket are named P2′ substituents
- Stachel S. J., Coburn C. A., Rush D., Jones K. L. G., Zhu H., Rajapakse H., Graham S. L., Simon A., Katharine Holloway M., Allison T. J., Munshi S. K., Espeseth A. S., Zuck P., Colussi D., Wolfe A., Pietrak B. L., Lai M.-T., Vacca J. P. Bioorg. Med. Chem. Lett. 2009;19:2977–2980. doi: 10.1016/j.bmcl.2009.04.033. [DOI] [PubMed] [Google Scholar]
- Rajapakse H. A., Nantermet P. G., Selnick H. G., Barrow J. C., McGaughey G. B., Munshi S., Lindsley S. R., Young M. B., Ngo P. L., Katherine Holloway M., Lai M.-T., Espeseth A. S., Shi X.-P., Colussi D., Pietrak B., Crouthamel M.-C., Tugusheva K., Huang Q., Xu M., Simon A. J., Kuo L., Hazuda D. J., Graham S., Vacca J. P. Bioorg. Med. Chem. Lett. 2010;20:1885–1889. doi: 10.1016/j.bmcl.2010.01.137. [DOI] [PubMed] [Google Scholar]
- Ellis C. R., Shen J. J. Am. Chem. Soc. 2015;137:9543–9546. doi: 10.1021/jacs.5b05891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis C. R., Tsai C.-C., Hou X., Shen J. J. Phys. Chem. Lett. 2016;7:944–949. doi: 10.1021/acs.jpclett.6b00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woltering T. J., Wostl W., Hilpert H., Rogers-Evans M., Pinard E., Mayweg A., Gobel M., Banner D. W., Benz J., Travagli M., Pollastrini M., Marconi G., Gabellieri E., Guba W., Mauser H., Andreini M., Jacobsen H., Power E., Narquizian R. Bioorg. Med. Chem. Lett. 2013;23:4239–4243. doi: 10.1016/j.bmcl.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Hilpert H., Guba W., Woltering T. J., Wostl W., Pinard E., Mauser H., Mayweg A. V., Rogers-Evans M., Humm R., Krummenacher D., Muser T., Schnider C., Jacobsen H., Ozmen L., Bergadano A., Banner D. W., Hochstrasser R., Kuglstatter A., David-Pierson P., Fischer H., Polara A., Narquizian R. J. Med. Chem. 2013;56:3980–3995. doi: 10.1021/jm400225m. [DOI] [PubMed] [Google Scholar]
- Anderson N., Borlak J. FEBS Lett. 2006;580:5533–5540. doi: 10.1016/j.febslet.2006.08.061. [DOI] [PubMed] [Google Scholar]
- Price D. A., Blagg J., Jones L., Greene N., Wager T. Expert Opin. Drug Metab. Toxicol. 2009;5:921–931. doi: 10.1517/17425250903042318. [DOI] [PubMed] [Google Scholar]
- Peters J.-U., Hert J., Bissantz C., Hillebrecht A., Gerebtzoff G., Bendels S., Tillier F., Migeon J., Fischer H., Guba W., Kansy M. Drug Discovery Today. 2012;17:325–335. doi: 10.1016/j.drudis.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Hitchcock S. A., Pennington L. D. J. Med. Chem. 2006;49:7559–7583. doi: 10.1021/jm060642i. [DOI] [PubMed] [Google Scholar]
- Wager T. T., Chandrasekaran R. Y., Hou X., Troutman M. D., Verhoest P. R., Villalobos A., Will Y. ACS Chem. Neurosci. 2010;1:420–434. doi: 10.1021/cn100007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager T. T., Hou X., Verhoest P. R., Villalobos A. ACS Chem. Neurosci. 2010;1:435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming J. N., Smith E. M., Wang L., Misiaszek J., Durkin J., Pan J., Iserloh U., Wu Y., Zhu Z., Strickland C., Voigt J., Chen X., Kennedy M. E., Kuvelkar R., Hyde L. A., Cox K., Favreau L., Czarniecki M. F., Greenlee W. J., McKittrick B. A., Parker E. M., Stamford A. W. Bioorg. Med. Chem. Lett. 2012;22:2444–2449. doi: 10.1016/j.bmcl.2012.02.013. [DOI] [PubMed] [Google Scholar]
- Stamford A. W., Scott J. D., Li S. W., Babu S., Tadesse D., Hunter R., Wu Y., Misiaszek J., Cumming J. N., Gilbert E. J., Huang C., McKittrick B. A., Hong L., Guo T., Zhu Z., Strickland C., Orth P., Voigt J. H., Kennedy M. E., Chen X., Kuvelkar R., Hodgson R., Hyde L. A., Cox K., Favreau L., Parker E. M., Greenlee W. J. ACS Med. Chem. Lett. 2012;3:897–902. doi: 10.1021/ml3001165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swahn B.-M., Kolmodin K., Karlstroem S., von Berg S., Soederman P., Holenz J., Berg S., Lindstroem J., Sundstroem M., Turek D., Kihlstroem J., Slivo C., Andersson L., Pyring D., Rotticci D., Oehberg L., Kers A., Bogar K., Bergh M., Olsson L.-L., Janson J., Eketjaell S., Georgievska B., Jeppsson F., Faelting J. J. Med. Chem. 2012;55:9346–9361. doi: 10.1021/jm3009025. [DOI] [PubMed] [Google Scholar]
- Eketjaell S., Janson J., Jeppsson F., Svanhagen A., Kolmodin K., Gustavsson S., Radesaeter A.-C., Eliason K., Briem S., Appelkvist P., Niva C., Berg A.-L., Karlstroem S., Swahn B.-M., Faelting J. J. Neurosci. 2013;33:10075–10084. doi: 10.1523/JNEUROSCI.1165-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eketjaell S., Janson J., Kaspersson K., Bogstedt A., Jeppsson F., Faelting J., Haeberlein S. B., Kugler A. R., Alexander R. C., Cebers G., Ho P. J. Alzheimer's Dis. 2016;50:1109–1123. doi: 10.3233/JAD-150834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt K. W., Cook A. W., Watts R. J., Clark C. T., Vigers G., Smith D., Metcalf A. T., Gunawardana I. W., Burkard M., Cox A. A., Geck Do M. K., Dutcher D., Thomas A. A., Rana S., Kallan N. C., DeLisle R. K., Rizzi J. P., Regal K., Sammond D., Groneberg R., Siu M., Purkey H., Lyssikatos J. P., Marlow A., Liu X., Tang T. P. J. Med. Chem. 2013;56:3379–3403. doi: 10.1021/jm4002154. [DOI] [PubMed] [Google Scholar]
- Gravenfors Y., Viklund J., Blid J., Ginman T., Karlstroem S., Kihlstroem J., Kolmodin K., Lindstroem J., von Berg S., von Kieseritzky F., Bogar K., Slivo C., Swahn B.-M., Olsson L.-L., Johansson P., Eketjaell S., Faelting J., Jeppsson F., Stroemberg K., Janson J., Rahm F. J. Med. Chem. 2012;55:9297–9311. doi: 10.1021/jm300991n. [DOI] [PubMed] [Google Scholar]
- Wu Y.-J., Guernon J., Rajamani R., Toyn J. H., Ahlijanian M. K., Albright C. F., Muckelbauer J., Chang C., Camac D., Macor J. E., Thompson L. A. Bioorg. Med. Chem. Lett. 2016;26:5729–5731. doi: 10.1016/j.bmcl.2016.10.055. [DOI] [PubMed] [Google Scholar]
- Ciordia M., Perez-Benito L., Delgado F., Trabanco A. A., Tresadern G. J. Chem. Inf. Model. 2016;56:1856–1871. doi: 10.1021/acs.jcim.6b00220. [DOI] [PubMed] [Google Scholar]
- Huestis M. P., Liu W., Volgraf M., Purkey H. E., Yu C., Wang W., Smith D., Vigers G., Dutcher D., Hunt K. W., Siu M. Tetrahedron Lett. 2013;54:5802–5807. [Google Scholar]
- Wu Y.-J., Guernon J., Yang F., Snyder L., Shi J., McClure A., Rajamani R., Park H., Ng A., Lewis H., Chang C. Y., Camac D., Toyn J. H., Ahlijanian M. K., Albright C. F., Macor J. E., Thompson L. A. ACS Med. Chem. Lett. 2016;7:271–276. doi: 10.1021/acsmedchemlett.5b00432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rombouts F. J. R., Tresadern G., Delgado O., Martinez-Lamenca C., Van Gool M., Garcia-Molina A., Alonso de Diego S. A., Oehlrich D., Prokopcova H., Alonso J. M., Austin N., Borghys H., Van Brandt S., Surkyn M., De Cleyn M., Vos A., Alexander R., Macdonald G., Moechars D., Gijsen H., Trabanco A. A. J. Med. Chem. 2015;58:8216–8235. doi: 10.1021/acs.jmedchem.5b01101. [DOI] [PubMed] [Google Scholar]
- Tresadern G., Delgado F., Delgado O., Gijsen H., Macdonald G. J., Moechars D., Rombouts F., Alexander R., Spurlino J., Van Gool M., Vega J. A., Trabanco A. A. Bioorg. Med. Chem. Lett. 2011;21:7255–7260. doi: 10.1016/j.bmcl.2011.10.050. [DOI] [PubMed] [Google Scholar]
- Kobayashi N., Ueda K., Itoh N., Suzuki S., Sakaguchi G., Kato A., Yukimasa A., Hori A., Koriyama Y., Haraguchi H., Yasui K. and Kanda Y., Shionogi & Co., Ltd., WO2007049532, 2007.
- Scott J. D., Li S. W., Brunskill A. P. J., Chen X., Cox K., Cumming J. N., Forman M., Gilbert E. J., Hodgson R. A., Hyde L. A., Jiang Q., Iserloh U., Kazakevich I., Kuvelkar R., Mei H., Meredith J., Misiaszek J., Orth P., Rossiter L. M., Slater M., Stone J., Strickland C. O., Voigt J. H., Wang G., Wang H., Wu Y., Greenlee W. J., Parker E. M., Kennedy M. E., Stamford A. W. J. Med. Chem. 2016;59:10435–10450. doi: 10.1021/acs.jmedchem.6b00307. [DOI] [PubMed] [Google Scholar]
- Guram A. S., Wang X., Bunel E. E., Faul M. M., Larsen R. D., Martinelli M. J. J. Org. Chem. 2007;72:5104–5112. doi: 10.1021/jo070341w. [DOI] [PubMed] [Google Scholar]
- Chen Y., White R., Amegadzie A., Brown J., Cheng A. C., Dimauro E. F., Dineen T., Gore V. K., Human J. B., Judd T., Kreiman C., Liu Q., Lopez P., Ma V. V., Marx I., Minatti A. E., Nguyen H. N., Paras N. A., Patel V. F., Qian W., Weiss M., Xue Q., Zheng X. M., Zhong W. and Amgen Inc., WO2011115938, 2011.
- Epstein O., Bryan M. C., Cheng A. C., Derakhchan K., Dineen T. A., Hickman D., Hua Z., Human J. B., Kreiman C., Marx I. E., Weiss M. M., Wahl R. C., Wen P. H., Whittington D. A., Wood S., Zheng X. M., Fremeau, Jr. R. T., White R. D., Patel V. F. J. Med. Chem. 2014;57:9796–9810. doi: 10.1021/jm501266w. [DOI] [PubMed] [Google Scholar]
- Lee D.-Y., Hartwig J. F. Org. Lett. 2005;7:1169–1172. doi: 10.1021/ol050141b. [DOI] [PubMed] [Google Scholar]
- Andersen J., Madsen U., Bjorkling F., Liang X. Synlett. 2005:2209–2213. doi: 10.1055/s-2005-872248. [DOI] [Google Scholar]
- Kaminski Z. J., Paneth P., Rudzinski J. J. Org. Chem. 1998;63:4248–4255. [Google Scholar]
- Falchi A., Giacomelli G., Porcheddu A., Taddei M. Synlett. 2000:275–277. doi: 10.1055/s-2000-6490. [DOI] [Google Scholar]
- As suggestedAs suggested in a previous publication (in a previous publication (ref. 19), attention should not be limited to achieveing a high numerical CatD/BACE1 selectivity ratio but also to the absolute CatD IC50 value
- An estimate of unbound EC50 for Ab lowering was calculated from a single dose, single time point (4 h) experiment assuming a standard sigmoid dose response (Hill slope of 1). This approach was initially validated by comparing EC50 values from a dose response experiment to the single dose, single time point estimate of unbound EC50. Subsequently, estimated EC50s have been shown to be a good approximation of the IC50 determined from a time course experiment using an indirect response PK/PD model
- Felmlee M. A., Morris M. E., Mager D. E. Methods Mol. Biol. 2012;929:583–600. doi: 10.1007/978-1-62703-050-2_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A X-ray co-crystal structure of compound 30 in BACE1 protein has previously been published in a different context (PDB ID ; 5I3W)
- Jordan J. B., Whittington D. A., Bartberger M. D., Sickmier E. A., Chen K., White R. D., Cheng Y., Judd T. J. Med. Chem. 2016;59:3732–3749. doi: 10.1021/acs.jmedchem.5b01917. [DOI] [PubMed] [Google Scholar]
- Male Sprague Dawley rats (Rattus norvegicus, Crl:CD®(SD); Charles River Laboratories; 10 weeks of age) were group-housed at an AAALAC, international-accredited facility. Animals were cared for in accordance with the Guide for the Care and Use of Laboratory Animals, 8th edition. All research protocols were reviewed and approved by the Amgen Institutional Animal Care and Use Committee. Rats were housed in an individual ventilated caging system on an irradiated corncob bedding (Envigo Teklad 7097). Lighting in animal holding rooms was maintained on 12:12 h light:dark cycle, and the ambient temperature and humidity range was at 68 °F to 79 °F and 30% to 70%, respectively. Animals had ad libitum access to irradiated pelleted feed (Envigo Teklad Global Rodent Diet; soy protein free extruded 2020×) and reverse-osmosis chlorinated (0.3 to 0.5 ppm) water via an automatic watering system. Cages were changed weekly inside an engineered cage changing station
- unpublished results.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.