

 Vascular endothelial growth factor receptor-2 (VEGFR-2) plays a crucial role in tumor angiogenesis, and inhibition of the VEGFR-2 signaling pathway has emerged as an attractive target for cancer therapy.
Vascular endothelial growth factor receptor-2 (VEGFR-2) plays a crucial role in tumor angiogenesis, and inhibition of the VEGFR-2 signaling pathway has emerged as an attractive target for cancer therapy.
Abstract
Vascular endothelial growth factor receptor-2 (VEGFR-2) plays a crucial role in tumor angiogenesis, and inhibition of the VEGFR-2 signaling pathway has emerged as an attractive target for cancer therapy. In our effort, a novel series of picolinamide-based derivatives were designed and synthesized as potent and effective VEGFR-2 inhibitors. All the newly prepared compounds were evaluated in vitro for their antiproliferative activity against A549 and HepG2 cell lines. Among the new compounds, 8j and 8l exhibited better activity against both A549 and HepG2 cell lines. Molecular docking was performed to investigate the binding capacity and binding mode with VEGFR-2 (PDB code: ; 4ASD).
Introduction
Cancer is the second leading cause of death worldwide. Cancer management is an extremely challenging field for medicinal chemists to discover effective and safer chemotherapeutic agents targeting various biochemical processes involved in the progression of different kinds of cancers.1 Among these targets, angiogenesis, the generation and growth of new blood vessels from the endothelium of an existing vascular network, is one of the critical processes that affect the growth and development of cancerous cells.2 Angiogenesis is also a complex process regulated by multiple growth factors that involves the migration, growth and differentiation of endothelial cells.3,4 Among these factors, vascular endothelial growth factor (VEGF) and its receptors (VEGFR-1/Flt-1, VEGFR-2/KDR/FLK-1 and VEGFR-3/Flt-4) are recognized as the major mediators of tumor angiogenesis.5–7 In particular, VEGFR-2, a type of receptor tyrosine kinase, mediates the major angiogenic function of VEGF,8 which is expressed primarily in endothelial cells and secreted by endothelial cells and various tumor cells. Over-activation of VEGFR-2 is correlated with poor prognosis and metastasis in the majority of solid tumor patients.9,10 Due to the vital roles of VEGFR-2 in tumor angiogenesis, inhibition of VEGFR-2 activation has become a compelling approach for developing targeted cancer therapies.11 Hence, several kinase inhibitors targeting VEGFR-2 are acknowledged as promising therapeutic drugs for treatment against a wide variety of human cancers. At present, many structurally diverse VEGFR-2 kinase inhibitors have been approved or in clinical trials for the treatment of cancers, such as Sorafenib,12 Regorafenib,13 Axitinib14 and ENMD-207615 (Fig. 1).
Fig. 1. VEGFR-2 inhibitors currently approved or in clinical trials.

As is well known, Sorafenib and Axitinib, known as tyrosine kinase inhibitors, could significantly inhibit the activity of VEGFR-2 and then cause the reduction of angiogenesis.12,16 The structure–activity relationships (SARs) demonstrate that Sorafenib binds to the ATP binding site of VEGFR-2. It presents a typical pharmacophore model where the central aryl moiety, occupying the linker region, is bound to the pyridine core occupying the adenine pocket via an ether linker, while the terminal aryl ring in the hydrophobic pocket is bound to the central aryl moiety via the urea group in the para position. A similar model between Axitinib and VEGFR-2 can also be discovered, where the central indazole moiety holds the linker region, while the aryl ring and the terminal pyridine ring are linked to the indazole.
In this paper, we report the design, synthesis and SARs of a novel series of picolinamide-based derivatives as potential VEGFR-2 inhibitors. In an attempt to develop new scaffolds for VEGFR-2 inhibitors, we hypothesized that a hybrid scaffold could be created by grafting the 2-ethenylpyridine fragment of Axitinib onto the 4-phenoxypicolinamide fragment of Sorafenib (Fig. 2). In order to further explore the SARs, a series of picolinamide-based derivatives 8 were designed and synthesized as VEGFR-2 inhibitors. As expected, these picolinamide derivatives displayed effective antiproliferative activity and could be considered as promising VEGFR-2 inhibitors.
Fig. 2. Design of picolinamide-based derivatives 8.

Results and discussion
Chemistry
The synthetic route to compounds 8a–8v is depicted in Scheme 1. Specifically, the synthesis was initiated by reacting picolines 1a and 1b with 4-hydroxybenzaldehyde 2 in refluxing acetic anhydride/acetic acid to furnish compounds 3a and 3b in 75% and 72% yield. Next, hydrolysis of compounds 3a and 3b was achieved via potassium hydroxide in ethanol to afford the corresponding compounds 4a and 4b in 92% and 94% yield, respectively. Furthermore, the starting material 2-picolinic acid 5 was treated with thionyl chloride to prepare 4-chloropicolinoyl chloride 6. Subsequently, 4-chloropicolinoyl chloride 6 was treated with the corresponding amine to form 4-chloropicolinamide derivatives 7a–7k in 71–81% yield. Finally, the target compounds 8a–8v were prepared through the reaction of 4-chloropicolinamide derivatives 7a–7k and compounds 4a and 4b in 54–70% yield. The final products were purified by column chromatography and their structures were characterized by 1H NMR, 13C NMR and HRMS. The crystal structures of 8b (CCDC number 1588889) and 8l (CCDC number 1588886) were confirmed by single crystal X-ray diffraction (Fig. 3 and 4).
Scheme 1. Synthetic route of target compounds 8a–8v. Reagents and conditions: a) acetic anhydride/acetic acid, reflux, 72–75%; b) KOH, EtOH, reflux, 92–94%; c) SOCl2/DMF, reflux, 100%; d) amines, dichloromethane, rt, 71–81%; e) Cu, Cs2CO3, DMF, N2, 110 °C, 54–70%.

Fig. 3. ORTEP diagram of the crystal structure of 8b (thermal ellipsoids drawn at the 50% probability level).

Fig. 4. ORTEP diagram of the crystal structure of 8l (thermal ellipsoids drawn at the 50% probability level).

Biological evaluation
The in vitro antiproliferative activity of the 22 picolinamide derivatives was evaluated against A549 and HepG2 cell lines by the CCK assay using Sorafenib and Axitinib as controls. Both cell lines were stimulated by VEGF and usually used as the tested cells in the discovery of VEGFR-2 kinase inhibitors. As shown in Table 1, most target compounds exhibited effective inhibitory activities against both cell lines with IC50 = 12.5–25.6 μM and 18.2–37.2 μM compared to Sorafenib (IC50 = 19.3 μM and 29.0 μM) and Axitinib (IC50 = 22.4 μM and 38.7 μM). The results indicated that the hybrid scaffold created by grafting the 2-ethenylpyridine fragment onto the 4-phenoxypicolinamide structure could maintain and even enhance the antiproliferative activity. Among them, 8d and 8k demonstrated better antiproliferative activities against A549 cells with IC50 = 16.2 μM and 15.6 μM, and 8a and 8u demonstrated better inhibitory activities against HepG2 cells with IC50 = 21.6 μM and 22.4 μM compared to other compounds. Moreover, 8j showed effective inhibitory activities against A549 and HepG2 cell lines with IC50 = 12.5 μM and 20.6 μM, and 8l showed similar inhibitory activities against both cell lines with IC50 = 13.2 μM and 18.2 μM, respectively.
Table 1. The structures and IC50 values of the target compounds against cancer cell lines.
| Entry | Substituent |
IC50
a
(μM) |
Selectivity b | |||
| R | X | Y | A549 | HepG2 | ||
| 8a |
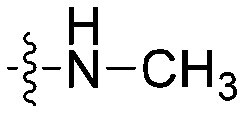
|
N | CH | 17.4 ± 1.1 | 21.6 ± 1.3 | 1.2 |
| 8b |

|
N | CH | 19.1 ± 0.9 | 30.2 ± 1.4 | 1.6 |
| 8c |
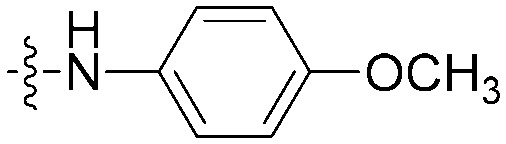
|
N | CH | 20.0 ± 1.5 | 30.0 ± 2.0 | 1.5 |
| 8d |

|
N | CH | 16.2 ± 1.7 | 31.3 ± 1.3 | 1.9 |
| 8e |

|
N | CH | 18.8 ± 0.8 | 27.9 ± 1.5 | 1.5 |
| 8f |

|
N | CH | 63.6 ± 2.1 | 26.0 ± 1.0 | 0.4 |
| 8g |

|
N | CH | >100 | 45.1 ± 2.6 | <0.5 |
| 8h |

|
N | CH | 21.4 ± 0.9 | >100 | >4.7 |
| 8i |
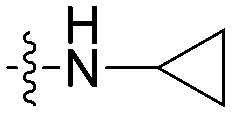
|
N | CH | 17.3 ± 1.3 | 31.0 ± 2.4 | 1.8 |
| 8j |

|
N | CH | 12.5 ± 1.0 | 20.6 ± 1.9 | 1.6 |
| 8k |
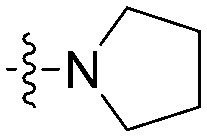
|
N | CH | 15.6 ± 2.2 | 27.0 ± 2.1 | 1.7 |
| 8l |

|
CH | N | 13.2 ± 1.6 | 18.2 ± 0.8 | 1.4 |
| 8m |

|
CH | N | 79.7 ± 0.8 | >100 | >1.2 |
| 8n |

|
CH | N | 25.6 ± 2.2 | >100 | >3.9 |
| 8o |

|
CH | N | 62.1 ± 2.8 | 56.2 ± 1.4 | 0.9 |
| 8p |

|
CH | N | 17.0 ± 0.7 | 28.9 ± 2.5 | 1.7 |
| 8q |
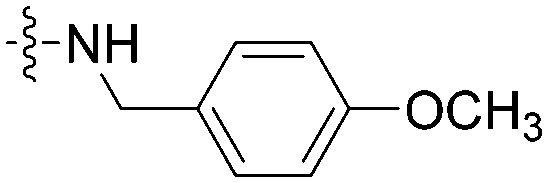
|
CH | N | 19.3 ± 1.5 | >100 | >5.2 |
| 8r |

|
CH | N | 18.2 ± 1.7 | 37.2 ± 1.6 | 2.0 |
| 8s |
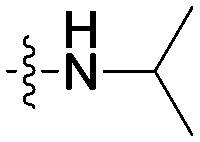
|
CH | N | 20.6 ± 2.3 | 33.7 ± 0.6 | 1.6 |
| 8t |
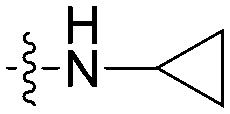
|
CH | N | 35.5 ± 0.9 | 77.5 ± 2.7 | 2.2 |
| 8u |

|
CH | N | 16.8 ± 1.2 | 22.4 ± 2.2 | 1.3 |
| 8v |

|
CH | N | 18.6 ± 1.7 | 41.6 ± 1.5 | 2.2 |
| Sorafenib | — | — | — | 19.3 ± 0.9 | 29.0 ± 2.0 | 1.5 |
| Axitinib | — | — | — | 22.4 ± 1.4 | 38.7 ± 1.9 | 1.7 |
aIC50 values are the mean ± S.D. of three separate experiments.
bSelectivity = IC50 value (HepG2)/IC50 value (A549).
In the following, the impacts of the substituent R on the carbonyl group and the substituents X/Y on the terminal pyridine were investigated, which may influence the selectivity on A549 and HepG2 cell lines. As shown in Table 1, compound 8g bearing a 4-fluorobenzylamino group provided a significant reduction of inhibition potency on A549 (IC50 > 100 μM) and selectivity (<0.5-fold vs. HepG2). This trait was also obvious in compound 8f containing 4-methoxybenzylamino. In contrast, compound 8q containing a 4-methoxybenzylamino group showed good antiproliferative activity to A549 with IC50 = 19.3 μM and >5.2-fold selectivity against HepG2. Moreover, it was notable that compound 8h bearing an isopropylamino group and compound 8n bearing a 4-methoxyanilino group showed good inhibitory activities against A549 (IC50 = 21.4 μM and 25.6 μM) and selectivity (>3.9-fold and >5.2-fold vs. HepG2).
To assess the inhibition for VEGFR-2 of the target compounds, the most active compounds 8a, 8j, 8l and 8u were selected to be further evaluated for their inhibitory activity against VEGFR-2 kinase with Sorafenib as the positive control. The results indicated that these compounds displayed good inhibitory activity on VEGFR-2 kinase with IC50 values ranging from 0.29 to 1.22 μM (Table 2). Among them, compound 8l potently inhibited VEGFR-2 at a low IC50 value of 0.29 μM. Moreover, compounds 8a and 8j possessed IC50 values of 0.87 and 0.53 μM, respectively. The result of the VEGFR-2 kinase assay corresponded to that of the in vitro inhibitory activity on A549 and HepG2 cell lines, which might indicate that the target compounds acted as VEGFR-2 kinase inhibitors.
Table 2. The VEGFR-2 inhibitory activity of the target compounds.
| Entry | VEGFR-2 IC50 a (μM) |
| 8a | 0.87 ± 0.06 |
| 8j | 0.53 ± 0.09 |
| 8l | 0.29 ± 0.02 |
| 8u | 1.22 ± 0.12 |
| Sorafenib | 0.95 ± 0.09 |
aIC50 values are the mean ± S.D. of three separate experiments.
Molecular docking
To understand the SARs between inhibitors and VEGFR-2, molecular docking simulations were performed using AutoDock 4.2 software.17 The VEGFR-2 kinase domain crystal structure of Sorafenib (PDB ID: ; 4ASD) was selected as the receptor and Sorafenib and Axitinib would be considered as the reference compounds in the docking study.18 All the target compounds were docked into the binding site, and the binding affinities were reported as the binding free energy (ΔG) and inhibition constant (Ki). The docking results are presented in Table 3. Compound 8 fitted well into the binding site of VEGFR-2, with ΔG ranging from –10.52 to –8.31 kcal mol–1 and Ki ranging from 0.019 to 0.815 μM. Among the target compounds, 8l (ΔG = –10.52 kcal mol–1, Ki = 0.019 μM) exhibited a better binding capacity with VEGFR-2 owing to the introduction of methylamino in suitable positions. The introduction of cyclohexylamino in 8j (ΔG = –10.28 kcal mol–1, Ki = 0.029 μM) and 8u (ΔG = –10.27 kcal mol–1, Ki = 0.030 μM) also resulted in better binding capacity than other compounds. It was obvious that the docking results of 8j, 8l and 8u were consistent with their better antiproliferative activities.
Table 3. The docking results of the target compounds.
| Entry | ΔG a | K i b | Entry | ΔG a | K i b |
| 8a | –9.53 | 0.104 | 8l | –10.52 | 0.019 |
| 8b | –8.53 | 0.561 | 8m | –9.65 | 0.084 |
| 8c | –9.00 | 0.253 | 8n | –9.20 | 0.179 |
| 8d | –8.95 | 0.274 | 8o | –9.56 | 0.098 |
| 8e | –9.66 | 0.082 | 8p | –9.10 | 0.214 |
| 8f | –9.14 | 0.198 | 8q | –8.59 | 0.503 |
| 8g | –9.32 | 0.147 | 8r | –9.76 | 0.069 |
| 8h | –9.37 | 0.135 | 8s | –8.59 | 0.505 |
| 8i | –9.61 | 0.090 | 8t | –8.31 | 0.815 |
| 8j | –10.28 | 0.029 | 8u | –10.27 | 0.030 |
| 8k | –9.42 | 0.125 | 8v | –9.45 | 0.119 |
| Sorafenib | –11.03 | 0.008 | |||
| Axitinib | –9.15 | 0.195 |
aBinding free energy (kcal mol–1).
bInhibition constant (μM).
To further evaluate the SARs, compound 8l was chosen as a representative compound and docked into the binding site of VEGFR-2 in comparison with Sorafenib and Axitinib. As displayed in Fig. 5 and 6, compound 8l almost totally overlapped with Sorafenib and Axitinib, and showed a good binding mode with VEGFR-2. On the moiety of the pyridine core, there were two important hydrogen bonds formed with Cys919. The N1-nitrogen of the pyridine core was hydrogen bonded to the backbone NH of hinge residue Cys919, and the NH group of picolinamide was hydrogen bonded to the backbone carbonyl of hinge residue Cys919. Furthermore, the pyridine core had some hydrophobic interactions and van der Waals contacts with Leu840, Leu1035, Ala866 and Glu917. The central aryl moiety is located in the linker region and in contact with Ala866, Cys1045, Lys868, Val848, Val916 and Phe1047. The terminal pyridine ring that connected the central aryl moiety via ethenyl penetrated into the hydrophobic pocket created by the hinge residues Asp1046, Glu885, Leu889, Leu1019 and Val914.
Fig. 5. Comparison of the binding site between 8l (magenta), Sorafenib (yellow) and Axitinib (blue).

Fig. 6. Docking simulations for 8l in the active site of VEGFR-2.

Conclusions
In summary, a novel series of picolinamide-based derivatives were designed and synthesized as potential VEGFR-2 inhibitors, and their structures were characterized by 1H NMR, 13C NMR, HRMS and single crystal X-ray diffraction. The antiproliferative activities were evaluated against A549 and HepG2 cell lines with Sorafenib (IC50 = 19.3 μM and 29.0 μM) and Axitinib (IC50 = 22.4 μM and 38.7 μM) as references. Of these derivatives, 8j (IC50 = 12.5 μM and 20.6 μM) and 8l (IC50 = 13.2 μM and 18.2 μM) emerged as the most active members against both cell lines. Also, 8d and 8k displayed good inhibitory activities against A549 with IC50 = 16.2 μM and 15.6 μM, and 8a and 8u exhibited effective inhibitory activities against HepG2 with IC50 = 21.6 μM and 22.4 μM. The inhibitory activities of the most active compounds 8a, 8j, 8l and 8u against VEGFR-2 were further evaluated. Compound 8l was found to be the most active compound against VEGFR-2 with an IC50 value of 0.29 μM. Molecular docking was performed to investigate the binding capacity and binding mode with VEGFR-2. The docking results indicated that 8j (ΔG = –10.28 kcal mol–1, Ki = 0.029 μM) and 8l (ΔG = –10.52 kcal mol–1, Ki = 0.019 μM) exhibited better binding capacities, corresponding to their antiproliferative activities. The representative compound 8l showed a good binding mode with VEGFR-2. The N1-nitrogen of pyridine and the NH group of picolinamide formed two essential hydrogen bonds with Cys919. The central aryl moiety and the terminal pyridine ring were involved in hydrophobic interactions with residues. In conclusion, these compounds have strong potential to be further developed as novel VEGFR-2 inhibitors. Further structural optimization of the most active candidates may lead to the discovery of better inhibitors, and the studies are currently in progress.
Conflicts of interest
All the authors declare that there is no conflict of interest.
Supplementary Material
Acknowledgments
This work was financially supported by the Key Projects in the Beijing Municipal Natural Science Foundation (No. KZ201510005007).
Footnotes
†Electronic supplementary information (ESI) available: 1588889 and 1588886. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8md00057c
References
- Yang Y., Shi L., Zhou Y., Li H.-Q., Zhu Z.-W., Zhu H.-L. Bioorg. Med. Chem. Lett. 2010;20:6653–6656. doi: 10.1016/j.bmcl.2010.09.014. [DOI] [PubMed] [Google Scholar]
- Patel H. M., Bari P., Karpoormath R., Noolvi M., Thapliyal N., Surana S., Jain P. RSC Adv. 2015;5:56724–56771. [Google Scholar]
- Folkman J., D'Amore P. A. Cell. 1996;87:1153–1155. doi: 10.1016/s0092-8674(00)81810-3. [DOI] [PubMed] [Google Scholar]
- Risau W. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- Olsson A.-K., Dimberg A., Kreuger J., Claesson-Welsh L. Nat. Rev. Mol. Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- Robinson C. J., Stringer S. E. J. Cell Sci. 2001;114:853–865. doi: 10.1242/jcs.114.5.853. [DOI] [PubMed] [Google Scholar]
- Shibuya M. Genes Cancer. 2011;2:1097–1105. doi: 10.1177/1947601911423031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N. Endocr. Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- Poon R. T.-P., Fan S.-T., Wong J. J. Clin. Oncol. 2001;19:1207–1225. doi: 10.1200/JCO.2001.19.4.1207. [DOI] [PubMed] [Google Scholar]
- Takahashi Y., Kitadai Y., Bucana C. D., Cleary K. R., Ellis L. M. Cancer Res. 1995;55:3964–3968. [PubMed] [Google Scholar]
- Huang L., Huang Z., Bai Z., Xie R., Sun L., Lin K. Future Med. Chem. 2012;4:1839–1852. doi: 10.4155/fmc.12.121. [DOI] [PubMed] [Google Scholar]
- Wilhelm S., Carter C., Lynch M., Lowinger T., Dumas J., Smith R. A., Schwartz B., Simantov R., Kelley S. Nat. Rev. Drug Discovery. 2006;5:835–844. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- Strumberg D., Scheulen M., Schultheis B., Richly H., Frost A., Büchert M., Christensen O., Jeffers M., Heinig R., Boix O. Br. J. Cancer. 2012;106:1722–1727. doi: 10.1038/bjc.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okaniwa M., Hirose M., Imada T., Ohashi T., Hayashi Y., Miyazaki T., Arita T., Yabuki M., Kakoi K., Kato J. J. Med. Chem. 2012;55:3452–3478. doi: 10.1021/jm300126x. [DOI] [PubMed] [Google Scholar]
- Tentler J. J., Bradshaw-Pierce E. L., Serkova N. J., Hasebroock K. M., Pitts T. M., Diamond J. R., Fletcher G. C., Bray M. R., Eckhardt S. G. Clin. Cancer Res. 2010;16:2989–2998. doi: 10.1158/1078-0432.CCR-10-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu-Lowe D. D., Zou H. Y., Grazzini M. L., Hallin M. E., Wickman G. R., Amundson K., Chen J. H., Rewolinski D. A., Yamazaki S., Wu E. Y. Clin. Cancer Res. 2008;14:7272–7283. doi: 10.1158/1078-0432.CCR-08-0652. [DOI] [PubMed] [Google Scholar]
- Sanner M. F. J. Mol. Graphics Modell. 1999;17:57–61. [PubMed] [Google Scholar]
- McTigue M., Murray B. W., Chen J. H., Deng Y. L., Solowiej J., Kania R. S. Proc. Natl. Acad. Sci. U. S. A. 2012;109:18281–18289. doi: 10.1073/pnas.1207759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.