

 An efficient acid catalyzed methodology has been employed to synthesize a variety of aza-flavanones and their α-glucosidase inhibitory activity is evaluated using acarbose, miglitol and voglibose as reference standards.
An efficient acid catalyzed methodology has been employed to synthesize a variety of aza-flavanones and their α-glucosidase inhibitory activity is evaluated using acarbose, miglitol and voglibose as reference standards.
Abstract
An efficient acid catalyzed methodology has been employed to synthesize a variety of aza-flavanones and their α-glucosidase inhibitory activity is evaluated using acarbose, miglitol and voglibose as reference standards. Molecular modeling studies were performed for all compounds to identify the important binding modes responsible for the inhibition activity of α-glucosidase which helped find key interactions between the enzyme and the active compounds. Among all the compounds 5g, 5r and 5w have shown high α-glucosidase inhibition activity compared to standard reference drugs and have been identified as promising potential antidiabetic agents. This study is the first biological evaluation of aza-flavanones as α-glucosidase inhibitors.
1. Introduction
Diabetes is one of the global chronic metabolic human diseases which results from inadequate insulin secretion or inefficient utilization of insulin. Between the two major types of diabetes,1 type 2 diabetes is the critical one which is characterized by hyperglycemia.2 Hyperglycemia causes serious damage to body parts like kidneys, heart, eyes and the nervous system. α-Glucosidase is an enzyme which catalyzes the conversion of carbohydrates to absorbable monosaccharides resulting in an increase in blood glucose levels. Inhibition of α-glucosidase can delay digestive processes leading to a decrease in sugar levels in the blood. α-Glucosidase inhibition is identified as a therapeutic target to cure diabetes. Recent standard glucosidase inhibitors in clinical practice viz., acarbose, miglitol, and voglibose, are associated with various side effects like bloating, flatulence, diarrhea and pain and their synthesis involves multiple steps. Hence, there is a strong demand for discovery of improved α-glucosidase inhibitors for the treatment of diabetes.
2-Aza-flavanones are an important class of building blocks in organic chemistry3 and have potent biological and pharmaceutical properties like anti-cancer4 and antibacterial.5 2-Aza-flavanones regulate the cell cycle by inhibiting microRNA which leads to the control of tumor progression.6 Furthermore, these scaffolds also play a vital role in drug development.7 Careful literature studies revealed that synthesis and applications of aza-flavanones are limited.8–11
Most reports in the literature involved either cyclization of amino chalcones by aza-Michael addition8–10 or by asymmetric synthesis from ortho-aminoacetophenone11 with substituted benzaldehydes. Recently Lee et al.12 have reported AgOTf catalyzed one-pot synthesis of ortho-aminoacetophenone with an aromatic aldehyde to accomplish aza-flavanones. Nevertheless, methods to synthesize aza-flavanones suffer from long reaction times, low yields and expensive catalysts.12 To overcome these challenges, we have developed an efficient metal-free, inexpensive TsOH catalytic system to synthesize a variety of aza-flavanones in good yields and evaluated their in vitro inhibitory activity against α-glucosidase.
2. Results and discussion
2.1. Chemistry
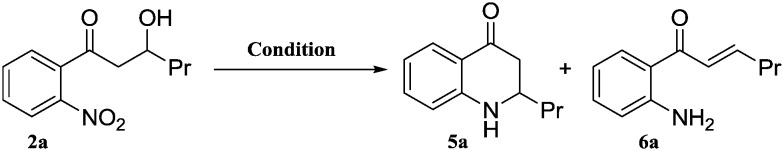
We have taken 1-(2-aminophenyl)-3-hydroxyhexan-1-one (4a) as a model substrate for this reaction, which was readily prepared from 2-nitroacetophenone (Scheme 1).13 Aldol reaction of 2-nitroacetophenone with various aldehydes furnished alcohols 2a–w in moderate yields (48–69%) along with the enones 3a–w (8–15%). Isolation of alcohols 2a–w and their subsequent reduction afforded suitable substrates 4a–w in good yield (78–91%).
Scheme 1. Reagents and conditions: (i) RCHO (1.1 equiv.), LDA (2.5 equiv.), THF, –78 °C, 1 h, 48–69% (ii) 10% Pd/C (20% wt), EtOH, 50 psi, 1 h, 78–91%.

Once the substrate 4a was synthesized, we carried out our attempts using several Lewis and Brønsted acids for the reaction, in which the desired aza-flavanone 5a and the enone 6a were formed in different ratios (Table 1). The reaction was unsuccessful (the starting material was recovered) in the absence of a catalyst, even after heating at 120 °C for 12 h in toluene. Initially we have screened with 10 mol% and 30 mol% catalyst. In both conditions we observed that the reactions were very sluggish and there was no progress after 16 h. It is observed that 50 mol% catalyst is necessary in order to achieve complete conversion. It was gratifying to note that the conversion is clean and the desired product 5a was isolated in 90% yield in the presence of TsOH (without formation of side product 6a). Surprisingly, Amberlyst and Dowex afforded the desired compound in almost quantitative yields. These catalysts can be easily recovered and reused without loss of activity.
Table 1. Optimization of reaction conditions for the synthesis of 5a a .
| |||
| Entry | Conditions | 5a (%) | 6a (%) |
| 1 | No catalyst | 0 | 0 |
| 2 | H3PO4 | 28 | 36 |
| 3 | AlCl3 | 37 | 48 |
| 4 | FeCl3 | 52 | 18 |
| 5 | TfOH | 78 | 10 |
| 6 | ZnCl2 | 74 | 5 |
| 7 | TsOH | 90 | 0 |
| 8 | Amberlyst | 97 | 0 |
| 9 | Dowex | 98 | 0 |
aCatalyst (0.5 equiv.), toluene, 120 °C, 1–2 h.
In addition, we extended the scope of the reaction to heterocycles attached to hydroxy bearing carbon (4m–u). Attempts to synthesize pyridine substituted aza-flavanones (5m–o) either using our catalytic system or Amberlyst and Dowex catalytic systems were futile, whereas the reaction was successful when 0.75 equiv. of ZnCl2 was employed.
Inspired by the results, we explored the reaction using various substrates and synthesized a library of aza-flavanones. To generalise the synthetic approach a long range of examples were synthesized adopting this methodology using simple nitroacetophenone with different aldehydes (aliphatic, aromatic (with electron donating & withdrawing groups), hetero aromatic (five membered and six membered rings)) and substituted nitroacetophenone (electron donating & withdrawing functionalities on the phenyl ring) with valeraldehyde, and the products are tabulated in Table 3.
Table 3. Substrate scope.
| Cpd no | Product | Conditions | Yield | Cpd no | Product | Conditions | Yield |
| 5a |
|
a | 90% | 5m |
|
b | 51% |
| 5b |
|
a | 85% | 5n |
|
b | 48% |
| 5c |
|
a | 85% | 5o |
|
b | 55% |
| 5d |
|
a | 80% | 5p |
|
a | 71% |
| 5e |
|
a | 74% | 5q |
|
a | 62% |
| 5f |
|
a | 54% | 5r |
|
a | 72% |
| 5g |
|
a | 60% | 5s |
|
a | 74% |
| 5h |
|
a | 68% | 5t |
|
a | 76% |
| 5i |
|
a | 74% | 5u |
|
a | 69% |
| 5j |
|
a | 70% | 5v |
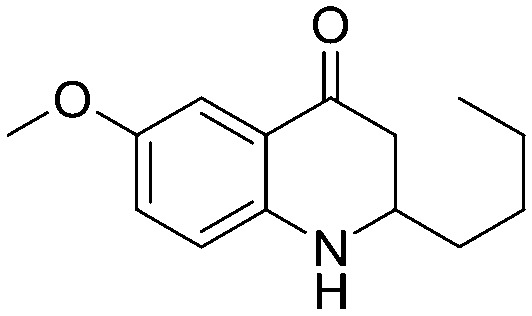
|
a | 76% |
| 5k |
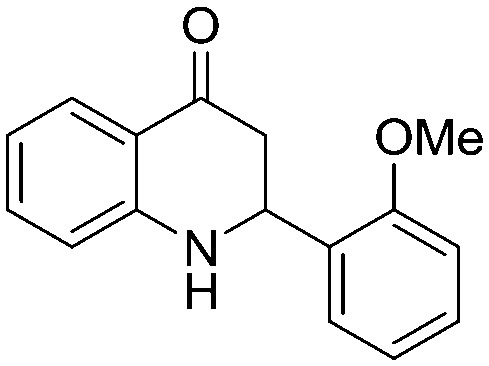
|
a | 72% | 5w |
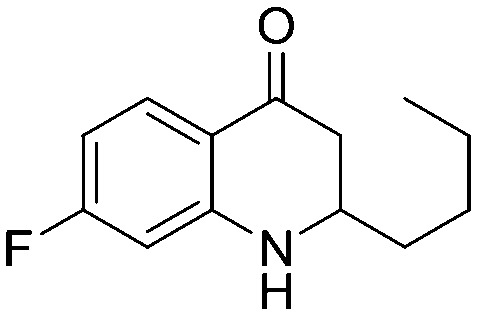
|
a | 73% |
| 5l |
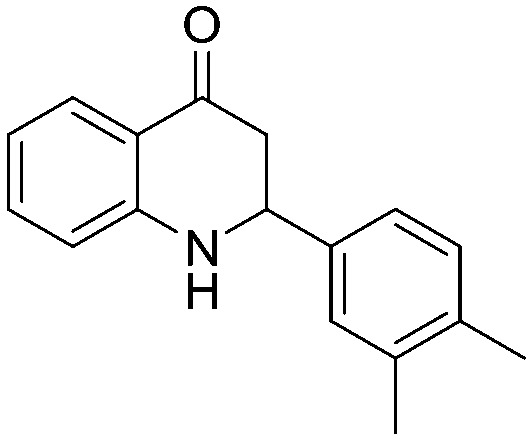
|
a | 65% |
aTsOH (0.5 equiv.), toluene, 120 °C, 1–2 h.
bZnCl2 (0.75 equiv.), toluene, 120 °C, 1–2 h.
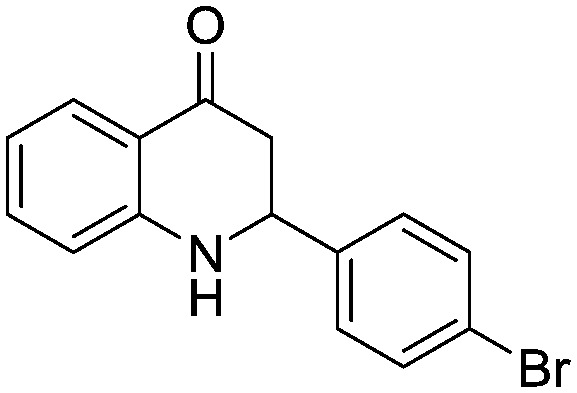
For the nitro reduction of 3-(4-bromophenyl)-3-hydroxy-1-(2-nitrophenyl)propan-1-one (2f), we avoided the use of Pd/C (because of the bromo group) and attempted to use Fe/AcOH. Surprisingly 56% of the 2-(4-bromophenyl)-2,3-dihydroquinolin-4(1H)-one (5f) final compound is isolated. The results were encouraging hence we have explored one pot cyclization of 2a using a variety of reducing agents. The best result was obtained when Fe/AcOH was employed which directly afforded 5a (58% yield) with enone 6a (12% yield) (Table 2). The enone side product increased when other reducing agents were used such as Fe/TsOH and Zn/AcOH. Eventually even with isolated 6a under optimized conditions the desired product 5a was obtained in good yields. We have attempted to use a metal (iron, Pd/cyclohexene) with polymer acid resins (Amberlyst and Dowex) but all attempts were unsuccessful.
Table 2. One-pot cyclization a .
| |||
| Entry | Conditions | 5a (%) | 6a (%) |
| 1 | Fe/AcOH | 58 | 12 |
| 2 | Fe/TsOH | 26 | 32 |
| 3 | Zn/AcOH | 28 | 40 |
| 4 b | SnCl2/AcOH | 35 | 28 |
| 5 b | Pd/HCO2NH4 | 45 | 24 |
aMetal (1.0 equiv.), acid (3.0 equiv.), toluene, 120 °C, 6 h.
bEtOH used as solvent.
2.2. Biology
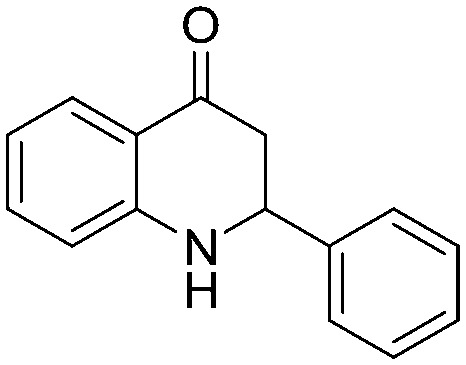
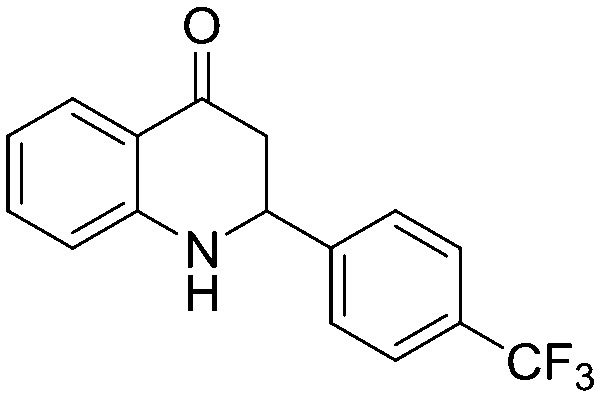
Next, we have evaluated the twenty compounds for their α-glucosidase inhibitory activity in comparison with acarbose, miglitol and voglibose as reference standards and Baker's yeast α-glucosidase inhibitory activity was assayed using the reported method.14 The IC50 values (best fit) were calculated using GraphPad Prism v6.0 where n = 3 and the results are tabulated in Table 4. It is evident from the results that many of the aza-flavanone derivatives showed promising inhibitory activity. Among the aliphatic derivatives the acyclic aza-flavanone derivatives (5a and 5c) exhibited high α-glucosidase activity compared to the alicyclic derivatives (5b and 5d). The simple phenyl ring-containing aza-flavanone derivative (5e) has shown negligible α-glucosidase activity. An enormous variation was observed when the phenyl ring is substituted with an electron withdrawing group like the phenyl ring of the 4-trifluoromethyl phenyl derivative (5g) that showed a 6–8 fold higher α-glucosidase activity with an IC50 value of 53.52 μM compared to standard references and that of the 2,4-difluoro phenyl derivative (5h) that also showed significant inhibition activity. The enzyme inhibition activity decreased when the phenyl ring was replaced with electron rich phenyl rings like that in the methoxyphenyl derivative (5i) or 3,4-dimethyl phenyl derivative (5l).
Table 4. α-Glucosidase inhibitory activity of aza-flavanones.
| Compound no | α-Glucosidase inhibition IC50 (μM) | Compound no | α-Glucosidase inhibition IC50 (μM) |
| 5a | 189.7 | 5p | 352.1 |
| 5b | 283.8 | 5q | 486.7 |
| 5c | 271.6 | 5r | 72.39 |
| 5d | 494.4 | 5s | 381.8 |
| 5e | 685.6 | 5t | 866.9 |
| 5f | SI | 5u | 667.1 |
| 5g | 53.52 | 5v | 683.2 |
| 5h | 210.6 | 5w | 133.8 |
| 5i | 302.8 | Acarbose | 360.2 |
| 5l | 606.2 | Voglibose | 324.7 |
| 5n | NI | Miglitol | 462.2 |
| 5o | 772.7 |
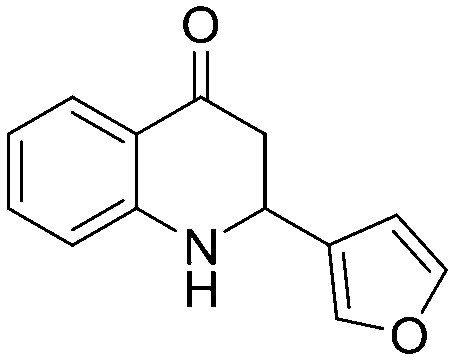
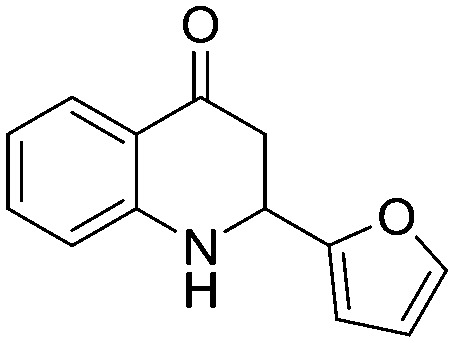
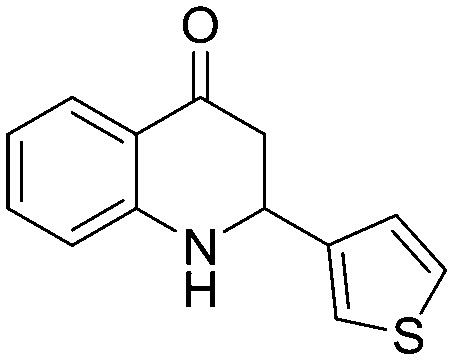
Among the heterocyclic derivatives, compounds containing pyridine scaffolds (5n and 5o) have shown low inhibition activity compared to furan and thiophene derivatives. The 3-furan derivative (5p) exhibited better activity compared to the corresponding reduced derivative 3-tetrahydrofuran (5t). Among all the heterocyclic derivatives promising activity was observed in the 3-thiophene derivative (5r) with an IC50 value of 72.39 μM. The results clearly indicated that the 3-substitueted aromatic five-membered heterocyclic rings (5p, 5r) are more potent than the 2-substituted derivatives (5q, 5s). The fluorodihydroquinolinone derivative (5w) with an IC50 value of 133.8 μM showed higher inhibition activity compared to the methoxydihydroquinolinone derivative (5v) with an IC50 value of 683.2 μM.
3. Molecular modeling
To gain some structural insight into the inhibitory mechanisms of the identified inhibitors of α-glucosidase, their binding modes in the active site were investigated using Maestro 9.7 in Schrödinger software.15
3.1. Homology modeling of α-glucosidase
To the best of our knowledge, the crystal structure for α-glucosidase of Saccharomyces cerevisiae (Baker's yeast) has not been reported yet. So, we developed a three dimensional (3D) model for the α-glucosidase of S. cerevisiae by a comparative modelling technique. The homology model of S. cerevisiae α-glucosidase was built with Prime 3.5 in Schrödinger software.
The protein sequence of S. cerevisiae α-glucosidase was downloaded from the universal protein resource (UniProt, entry Id: P53341). A template search was performed using Blast Homology-Search tools against the PDB databank implemented in Prime 3.5. The crystallographic structure of Saccharomyces cerevisiae isomaltase (PDB ID: ; 3A4A, 1.6 Å resolution)16 with a 72% sequence identity and an 85% sequence similarity with the target was selected as the template. The three dimensional (3D) model of yeast α-glucosidase was built using Prime homology modeling tools. The modeled 3D structure was evaluated with the help of the Ramachandran plot and protein reports.
Fig. 1 shows the homology-modeled structure of α-glucosidase and its Ramachandran plot. Fig. S1 (ESI†) displays the sequence alignment between α-glucosidase from S. cerevisiae and isomaltase from S. cerevisiae (PDB ID: ; 3A4A). Ramachandran plot analysis of the yeast α-glucosidase structure showed that >95% residues are favored and allowed φ, ψ backbone conformational regions (Fig. S2, ESI†). Visualization and characterization of the catalytic binding site was performed using the SiteMap module of Maestro 9.7.
Fig. 1. a) Graphic representation of the homology modeling structure of α-glucosidase from S. cerevisiae. b) Ramachandran plot for the modeled α-glucosidase of S. cerevisiae. The plot is organized as follows: glycine, proline and all other residues are plotted as triangles, squares, and circles respectively. The red, yellow and white regions represent the favoured, allowed and the disallowed regions respectively.

3.2. Molecular docking simulations and binding energy calculations
Molecular docking studies were carried out to explore the binding mode of the synthesized aza-flavanones within the binding pocket of α-glucosidase using Glide 6.2 in Schrödinger software. The docking procedure was followed using the standard protocol implemented in Maestro 9.7 and the compounds were docked against the three dimensional homology model of α-glucosidase. As a means of validation of the docking protocol, the known inhibitor acarbose was docked into the binding pocket of a developed homology model, and acarbose is accommodated well in the binding pocket and showed interactions with the important active site residues (Fig. S3, ESI†).
All the compounds were docked into the binding pocket of the developed homology model of the α-glucosidase enzyme. Table S5 (ESI†) demonstrates the result of the molecular docking along with hydrogen bonding as well as arene–arene interactions of compounds with the α-glucosidase enzyme. Based on the docking scores and the experimental IC50 values, we have performed MM/GBSA binding energy calculations of the complexes between the compounds and glucosidase enzyme (Table S6, ESI†). From these molecular modeling studies it was observed that the top ranked conformation of the most active compound 5g (Fig. 2) established four hydrogen bonds between the carbonyl group on the aza-flavanone ring of the compound and the active site residues (Lys 153, Arg 312, Tyr 313, Asn 412) with a binding energy of 48.60 kcal mol–1.
Fig. 2. a) Docking of the most active compound 5g (yellow colour stick) and b) its ligand–protein interactions in the binding site of modeled α-glucosidase. The red dashed lines represent hydrogen bonds. H-bond distances (in Å) between heteroatoms of the ligand and amino acid residues are as follows: Lys 153 (3.9), Arg 312 (2.2), Tyr 313 (3.8) and Asn 412 (3.7). The black dashed line indicates arene–arene interaction with His 237.

Additionally the aryl group of the compound formed a π–π stacking interaction (arene–arene interaction) with His 237. Furthermore, several hydrophobic interactions were observed between the compound 5g and the active site residues, e.g., Phe 155, Phe 156, Leu 174, Phe 175, Leu 216, Pro 238, Ala 276, Phe 300, Val 303, Pro 309, Phe 310, Phe 311 and Tyr 313 being the other residues that stabilized the binding of the compound 5g in the active site of α-glucosidase. Similar interactions were found in compound 5r (binding energy = –43.69 kcal mol–1) with residues Lys 153, Arg 312, Tyr 313 and Asn 412. These interactions made this compound the second most active compound in the series. A strong hydrogen bonding network was observed for compounds 5g, 5r, 5w, 5h and 5a where the carbonyl group is attached to the aza-flavanone ring. The good biological activity of compounds 5g, 5w and 5h might be due to the presence of fluoro groups; particularly the trifluoromethyl group in compound 5g exhibits a strong electron withdrawing inductive effect. This effect from the trifluoromethyl moiety might increase the ability of the compound to interact with catalytic site residues, which might be one of the reasons for the highest activity observed in the series. Similarly, compounds 5w and 5a also exhibited good biological activities against the enzyme, but upon comparing compound 5w with 5a, it is found that 5w has a higher biological activity and docking score and more protein–ligand interactions because of the presence of both an electron withdrawing group and an aliphatic side chain. These observations can be verified in the cases of compounds 5i to 5l as they have low biological activities as well as low docking scores and fewer interactions with the active site residues as compared to compound 5g. These compounds have groups that exhibit electron donating effects instead of the fluoro group present in compounds 5g, 5h and 5w. Similarly other less active compounds lacking electron withdrawing groups makes them slightly less active as compared to compound 5g. From the docking study it was observed that the presence of a carbonyl group is responsible for strong hydrogen bonding, the presence of an arene moiety in the basic scaffold of aza-flavanone is helpful in establishing π–π stacking and finally the presence of substituents at the 2-position of the aza-flavanone system is supportive of hydrophobic interactions. Thus, molecular modeling studies performed using azaflavanones had provided us with some good results, which best explain the compounds' in vitro α-glucosidase inhibitory activity.
4. Experimental
4.1. Chemistry
Melting points were determined in capillaries, recorded on a Buchi Melting Point B-540 and are uncorrected. IR data was recorded on a Perkin Elmer Spectrum 100. 1H and 13CNMR spectra were recorded on Varian 400 MHz and 300 MHz spectrometers, using CDCl3 and DMSO-d6 as solvents. Chemical shifts are given in ppm with TMS as an internal reference. J values are given in Hertz. Reactions were monitored by thin-layer chromatography (TLC) coated with silica gel. Column chromatography was performed using 100–200 mesh silica.
4.1.1. General procedure for the preparation of compounds 2a–w
To a cooled solution of diisopropylamine (3.0 eq.) at –78 °C in THF (20 vol) was added n-BuLi (2.5 eq., 1.0 M solution in hexane). After addition of n-BuLi, the reaction mixture was stirred at 0 °C for 30 min and then cooled to –78 °C. To this solution was added dropwise nitroacetophenone (1.0 eq.). The reaction mixture was stirred at –78 °C for 20 min and to this solution was added a solution of an aldehyde derivative (1.1 eq.) in THF (5 vol). The reaction mixture was stirred for 1 h at –78 °C and was quenched with a saturated NH4Cl solution. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate. The organic phase was combined and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash silica gel chromatography to give compounds 2a–w (48–69%).
4.1.1.1. 3-Hydroxy-1-(2-nitrophenyl)hexan-1-one (2a)
Yield: 63%; IR (KBr, cm–1): 3436 (OH), 1654 (CO), 1530 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.13 (d, J = 8.2 Hz, 1H), 7.74 (t, J = 7.6 Hz, 1H), 7.62 (t, J = 7.6 Hz, 1H), 7.44 (dd, J = 1.2, 7.6 Hz, 1H), 4.32–4.28 (m, br, 1H), 3.16 (dd, J = 2.6, 17.2 Hz, 1H), 2.89–2.83 (m, 1H), 1.59–1.38 (m, 4H), 0.95 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 203.23, 137.62, 134.38, 130.67, 127.37, 124.41, 124.02, 67.79, 49.62, 38.61, 18.62, 13.92; LC-MS (ESI): m/z calcd. for C12H15NO4: 237.10, found 238.01 [M + H]+.
4.1.1.2. 3-Cyclopropyl-3-hydroxy-1-(2-nitrophenyl)propan-1-one (2b)
Yield: 68%; IR (KBr, cm–1): 3431 (OH), 1689 (CO), 1560 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.12 (d, J = 8.4 Hz, 1H), 7.74 (t, J = 7.6 Hz, 1H), 7.62 (t, J = 9.0 Hz, 1H), 7.47 (dd, J = 1.2, 7.6 Hz, 1H), 3.54 (t, J = 8.4 Hz, 1H), 3.19–3.02 (m, 2H), 2.80 (s, br, OH, 1H), 1.04–0.95 (m, 1H), 0.63–0.49 (m, 2H), 0.46–0.40 (m, 1H), 0.30–0.24 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 202.71, 145.54, 137.68, 134.33, 130.63, 127.51, 124.32, 72.85, 49.52, 16.99, 3.35, 2.24.
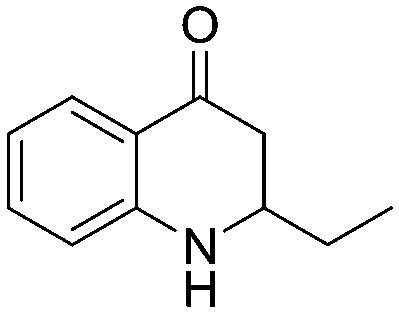
4.1.1.3. 3-Hydroxy-1-(2-nitrophenyl)pentan-1-one (2c)
Yield: 62%; IR (KBr, cm–1): 3432 (OH), 1689 (CO), 1525 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.12 (dd, J = 0.8, 8.4 Hz, 1H), 7.74 (t, J = 8.0 Hz, 1H), 7.62 (t, J = 8.4 Hz, 1H), 7.45 (dd, J = 1.6, 8.0 Hz, 1H), 4.22–4.18 (m, br, 1H), 3.01 (dd, J = 2.4, 17.2 Hz, 1H), 2.90–2.83 (m, 2H), 1.60–1.53 (m, 2H), 0.98 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 203.15, 145.54, 137.64, 134.37, 130.67, 127.41, 124.40, 69.38, 49.20, 29.45, 9.76; LC-MS (ESI): m/z calcd. for C11H13NO4: 223.08, found 223.91 [M + H]+.
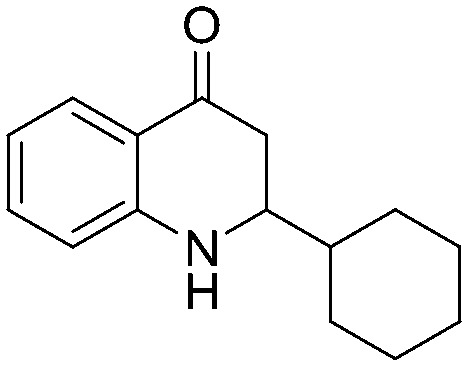
4.1.1.4. 3-Cyclohexyl-3-hydroxy-1-(2-nitrophenyl)propan-1-one (2d)
Yield: 59%; IR (KBr, cm–1): 3434 (OH), 1703 (CO), 1538 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.12 (dd, J = 0.8, 10.8 Hz, 1H), 7.73 (td, J = 1.2, 10.0 Hz, 1H), 7.61 (td, J = 1.6, 10.8 Hz, 1H), 7.45 (dd, J = 1.6, 10.0 Hz, 1H), 4.07–4.03 (m, 1H), 3.05–2.99 (m, 1H), 2.91–2.56 (m, 2H), 1.87–1.66 (m, 4H), 1.48–1.01 (m, 6H); 13C NMR (100 MHz, CDCl3): δ 203.63, 145.48, 137.79, 134.34, 130.59, 127.45, 124.34, 72.13, 46.93, 43.11, 28.81, 28.04, 26.35, 26.09, 25.99; LC-MS (ESI): m/z calcd. for C15H19NO4: 277.13, found 278.22 [M + H]+.
4.1.1.5. 3-Hydroxy-1-(2-nitrophenyl)-3-phenylpropan-1-one (2e)
Yield: 64%; MR: 85–88 °C; IR (KBr, cm–1): 3445 (OH), 1703 (CO), 1560 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.13 (d, J = 7.2 Hz, 1H), 7.72 (t, J = 8.0 Hz, 1H), 7.61 (t, J = 8.0 Hz, 1H), 7.41–7.26 (m, 6H), 5.42–5.39 (m, 1H), 3.24–3.20 (m, 2H); LC-MS (ESI): m/z calcd. for C15H13NO4: 271.08, found NO mass ionization.
4.1.1.6. 3-(4-Bromophenyl)-3-hydroxy-1-(2-nitrophenyl)propan-1-one (2f)
Yield: 54%; IR (KBr, cm–1): 3476 (OH), 1688 (CO), 1539 (NO2); 1H NMR (300 MHz, CDCl3): δ 8.14 (dd, J = 1.2, 8.1 Hz, 1H), 7.74 (t, J = 7.8 Hz, 1H), 7.63 (t, J = 8.1 Hz, 1H), 7.49–7.39 (m, 3H), 7.32–7.26 (m, 2H), 5.40–5.37 (m br, 1H), 3.26 (d, J = 3.0 Hz, 1H), 3.15–3.13 (m, 2H); LC-MS (ESI): m/z calcd. for C15H12BrNO4: 348.99, found 374.5 [M + Na]+.
4.1.1.7. 3-Hydroxy-1-(2-nitrophenyl)-3-(4-(trifluoromethyl)phenyl)propan-1-one (2g)
Yield: 52%; IR (KBr, cm–1): 3427 (OH), 1701 (CO), 1530 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.14 (dd, J = 0.8, 8.0 Hz, 1H), 7.73 (td, J = 1.2, 7.8 Hz, 1H), 7.65–7.26 (m, 3H), 7.54–7.47 (m, 2H), 7.40 (dd, J = 1.2, 7.2 Hz, 1H), 5.48 (dd, J = 3.2, 8.8 Hz, 1H), 3.38 (s, OH, 1H), 3.18–3.15 (m, 2H); LC-MS (ESI): m/z calcd. for C16H12F3NO4: 339.07, found NO mass ionization.
4.1.1.8. 3-(2,4-Difluorophenyl)-3-hydroxy-1-(2-nitrophenyl)propan-1-one (2h)
Yield: 54%; MR: 77–75 °C; IR (KBr, cm–1): 3368 (OH), 1655 (CO), 1525 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.13 (dd, J = 1.2, 8.4 Hz, 1H), 7.74 (td, J = 1.2, 7.4 Hz, 1H), 7.63 (td, J = 1.2, 8.4 Hz, 1H), 7.58–7.52 (m, 1H), 7.43 (dd, J = 1.6, 8.0 Hz, 1H), 6.90 (td, J = 2.0, 8.0 Hz, 1H), 6.78 (td, J = 1.2, 8.4 Hz, 1H), 5.62 (dd, J = 2.0, 8.8 Hz, 1H), 3.28–3.12 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 202.22, 162.35 (dd), 159.36 (dd), 145.50, 137.05, 134.42, 130.93, 128.43–128.37 (m), 127.31, 125.33 (d), 124.47, 111.44 (dd), 103.71 (t), 64.31, 49.94; LC-MS (ESI): m/z calcd. for C15H11F2NO4: 299.12, found NO mass ionization.
4.1.1.9. 3-Hydroxy-3-(4-methoxyphenyl)-1-(2-nitrophenyl)propan-1-one (2i)
Yield: 61%; IR (KBr, cm–1): 3458 (OH), 1615 (CO), 1542 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.12 (dd, J = 1.2, 10.8 Hz, 1H), 7.72 (t, J = 8.0 Hz, 1H), 7.61 (t, J = 8.4 Hz, 1H), 7.41 (dd, J = 1.2, 8.8 Hz, 1H), 7.32 (d, J = 8.4 Hz, 2H), 6.88 (d, J = 12.0 Hz, 2H), 5.35–5.32 (m, 1H), 3.79 (s, 3H), 3.19–3.16 (m, 2H), 3.06 (s, br, OH, 1H); LC-MS (ESI): m/z calcd. for C16H15NO5: 301.10, found 324 [M + Na]+.
4.1.1.10. 3-Hydroxy-3-(3-methoxyphenyl)-1-(2-nitrophenyl)propan-1-one (2j)
Yield: 60%; IR (KBr, cm–1): 3467 (OH), 1703 (CO), 1545 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.12 (d, J = 7.6 Hz, 1H), 7.72 (d, J = 8.4 Hz, 1H), 7.61 (t, J = 8.0 Hz, 1H), 7.41 (d, J = 8.0 Hz, 1H), 7.26 (t, J = 6.0 Hz, 1H), 6.96 (d, J = 7.6 Hz, 2H), 6.81 (d, J = 8.4 Hz, 1H), 5.36 (t, J = 5.6 Hz, 1H), 3.81 (s, 3H), 3.18 (d, J = 6.0 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 202.29, 159.85, 145.45, 144.14, 137.47, 134.38, 130.77, 129.64, 127.45, 124.38, 117.88, 113.48, 111.04, 70.33, 55.25, 51.68; LC-MS (ESI): m/z calcd. for C16H15NO5: 301.10, found 300.4 [M – H]–.
4.1.1.11. 3-Hydroxy-3-(2-methoxyphenyl)-1-(2-nitrophenyl)propan-1-one (2k)
Yield: 58%; MR: 85–88 °C; IR (KBr, cm–1): 3492 (OH), 1688 (CO), 1518 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 8.4 Hz, 1H), 7.70 (t, J = 8.4 Hz, 1H), 7.59 (t, J = 8.4 Hz, 1H), 7.44 (t, J = 8.0 Hz, 2H), 7.25 (t, J = 8.4 Hz, 1H), 6.97 (t, J = 7.2 Hz, 1H), 6.85 (d, J = 8.4 Hz, 1H), 5.57–5.52 (m, 1H), 3.83 (s, 3H), 3.36–3.10 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 202.45, 155.93, 145.76, 137.66, 134.21, 130.58, 130.44, 128.62, 127.72, 126.47, 124.24, 120.82, 110.28, 66.56, 55.24, 49.79; LC-MS (ESI): m/z calcd. for C16H15NO5: 301.10, found 324.61 [M + Na]+.
4.1.1.12. 3-(3,4-Dimethylphenyl)-3-hydroxy-1-(2-nitrophenyl)propan-1-one (2l)
Yield: 63%; MR: 82–85 °C; IR (KBr, cm–1): 3429 (OH), 1689 (CO), 1529 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.12 (dd, J = 0.9, 8.1 Hz, 1H), 7.72 (t, J = 6.6 Hz, 1H), 7.62 (t, J = 9.0 Hz, 1H), 7.42 (dd, J = 1.5, 7.5 Hz, 1H), 7.17 (s, br, 1H), 7.11 (s, br, 2H), 5.35–5.31 (m, 1H), 3.40–3.17 (m, 2H), 2.25 (d, J = 3.9 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 202.38, 145.55, 139.93, 137.58, 136.82, 136.21, 134.33, 130.68, 129.78, 127.50, 126.91, 124.34, 123.05, 70.31, 51.66, 19.74, 19.40; LC-MS (ESI): m/z calcd. for C17H17NO4: 299.12, found NO mass ionization.
4.1.1.13. 3-Hydroxy-1-(2-nitrophenyl)-3-(pyridin-2-yl)propan-1-one (2m)
Yield: 48%; IR (KBr, cm–1): 3392 (OH), 1704 (CO), 1528 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.53–8.51 (m, 1H), 8.10 (d, J = 8.4 Hz, 1H), 7.72 (t, J = 7.6 Hz, 2H), 7.60 (t, J = 8.0 Hz, 1H), 7.53–7.47 (m, 2H), 7.24–7.20 (m, 1H), 5.38 (dd, J = 3.6, 8.8 Hz, 1H), 3.71–3.58 (m, 1H), 3.44 (dd, J = 4.0, 12.6 Hz, 1H), 3.28–3.22 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 201.74, 160.38, 148.40, 136.98, 134.30, 130.59, 127.79, 124.23, 123.19, 122.99, 122.67, 120.60, 69.85, 50.45; LC-MS (ESI): m/z calcd. for C14H12N2O4: 272.08, found 273.31 [M + H]+.
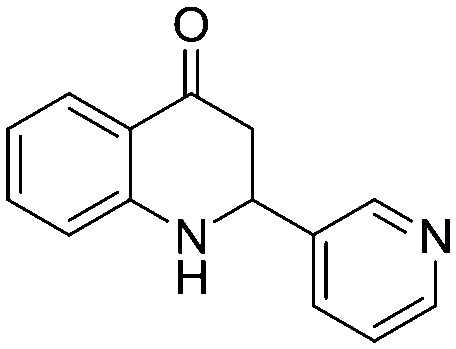
4.1.1.14. 3-Hydroxy-1-(2-nitrophenyl)-3-(pyridin-3-yl)propan-1-one (2n)
Yield: 51%; IR (KBr, cm–1): 3435 (OH), 1663 (CO), 1531 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.65 (d, J = 2.0 Hz, 1H), 8.54 (dd, J = 1.2, 4.8 Hz, 1H), 8.15 (d, J = 8.0 Hz, 1H), 7.78–7.73 (m, 2H), 7.64 (t, J = 8.4 Hz, 1H), 7.42 (dd, J = 1.2, 7.2 Hz, 1H), 7.31–7.27 (m, 1H), 5.47 (t, J = 6.8 Hz, 1H), 3.20 (d, J = 6.8 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 202.04, 150.08, 149.09, 147.81, 137.98, 137.19, 134.48, 133.61, 130.95, 127.38, 124.50, 123.51, 68.22, 51.32; LC-MS (ESI): m/z calcd. for C14H12N2O4: 272.08, found 273.06 [M + H]+.
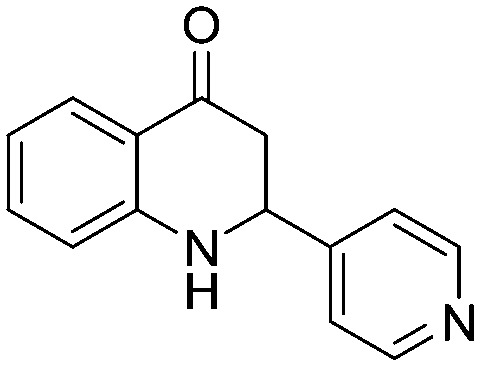
4.1.1.15. 3-Hydroxy-1-(2-nitrophenyl)-3-(pyridin-4-yl)propan-1-one (2o)
Yield: 55%; IR (KBr, cm–1): 3392 (OH), 1704 (CO), 1528 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.53 (d, J = 5.2 Hz, 2H), 8.14 (d, J = 8.0 Hz, 1H), 7.75 (t, J = 7.6 Hz, 1H), 7.64 (t, J = 7.2 Hz, 1H), 7.43 (dd, J = 1.2, 7.6 Hz, 1H), 7.34 (d, J = 6.0 Hz, 2H), 5.42 (dd, J = 3.2, 8.8 Hz, 1H), 3.23–3.10 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 201.72, 151.66, 149.77, 145.39, 137.13, 134.55, 130.96, 127.31, 124.48, 120.61, 68.75, 51.12; LC-MS (ESI): m/z calcd. for C14H12N2O4: 272.08, found 273.13.
4.1.1.16. 3-(Furan-3-yl)-3-hydroxy-1-(2-nitrophenyl)propan-1-one (2p)
Yield: 65%; IR (KBr, cm–1): 3421 (OH), 1703 (CO), 1527 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.13 (d, J = 8.0 Hz, 1H), 7.72 (t, J = 8.4 Hz, 1H), 7.62 (t, J = 8.4 Hz, 1H), 7.44–7.42 (m, 2H), 7.38–7.37 (m, 1H), 6.40 (s, 1H), 5.38–5.34 (m, br, 1H), 3.22–3.21 (m, 2H), 3.07 (d, J = 3.6 Hz, 1H); 13CNMR (100 MHz, CDCl3): δ 202.01, 145.56, 143.45, 139.10, 137.37, 134.36, 130.78, 127.42, 127.27, 124.40, 108.34, 63.45, 50.33; LC-MS (ESI): m/z calcd. for C13H11NO5: 261.06, found 284.00 [M + Na]+.
4.1.1.17. 3-(Furan-2-yl)-3-hydroxy-1-(2-nitrophenyl)propan-1-one (2q)
Yield: 62%; IR (KBr, cm–1): 3429 (OH), 1702 (CO), 1526 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.12 (dd, J = 1.2, 8.0 Hz, 1H), 7.73 (t, J = 8.0 Hz, 1H), 7.62 (t, J = 8.0 Hz, 1H), 7.43 (dd, J = 1.2, 7.6 Hz, 1H), 7.35 (s, 1H), 6.34–6.31 (m, 2H), 5.39–5.36 (m, 1H), 3.39–3.34 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 202.49, 154.51, 145.49, 142.24, 137.35, 134.36, 130.80, 127.49, 124.40, 110.34, 106.63, 64.16, 47.91; LC-MS (ESI): m/z calcd. for C13H11NO5: 261.06, found 262.27 [M + H]+.
4.1.1.18. 3-Hydroxy-1-(2-nitrophenyl)-3-(thiophen-3-yl)propan-1-one (2r)
Yield: 65%; IR (KBr, cm–1): 3491 (OH), 1711 (CO), 1525 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.13 (dd, J = 1.2, 8.4 Hz, 1H), 7.73 (t, J = 7.6 Hz, 1H), 7.62 (t, J = 8.4 Hz, 1H), 7.40 (dd, J = 1.6, 7.6 Hz, 1H), 7.31–7.29 (m, 1H), 7.27–7.26 (m, 1H), 7.08 (dd, J = 1.6, 5.2 Hz, 1H), 5.48 (t, J = 5.2 Hz, 1H), 3.24 (d, J = 7.6 Hz, 2H), 3.20 (s, 1H); 13C NMR (100 MHz, CDCl3): δ 202.24, 145.45, 143.71, 137.40, 134.44, 130.79, 127.41, 126.40, 125.36, 124.40, 121.09, 66.83, 50.85; LC-MS (ESI): m/z calcd. for C13H11NO4S: 277.04, found, no ionization.
4.1.1.19. 3-Hydroxy-1-(2-nitrophenyl)-3-(thiophen-2-yl)propan-1-one (2s)
Yield: 67%; IR (KBr, cm–1): 3445 (OH), 1703 (CO), 1527 (NO2); 1H NMR (400 MHz, CDCl3): δ 8.13 (dd, J = 0.8, 8.0 Hz, 1H), 7.73 (t, J = 1.2, 7.8 Hz, 1H), 7.62 (t, J = 1.2, 8.0 Hz, 1H), 7.41 (dd, J = 1.2, 7.8 Hz, 1H), 7.25 (dd, J = 1.2, 7.8 Hz, 1H), 7.01 (d, J = 3.2 Hz, 1H), 6.96 (t, J = 5.2 Hz, 1H), 5.64 (t, J = 5.6 Hz, 1H), 3.33 (d, J = 6.0 Hz, 2H), 3.30 (s, 1H); 13C NMR (100 MHz, CDCl3): δ 201.59, 146.09, 145.54, 137.30, 134.37, 130.81, 127.43, 126.71, 124.90, 124.40, 123.84, 66.64, 51.53; LC-MS (ESI): m/z calcd. for C13H11NO4S: 277.04, found, no ionization.
4.1.1.20. 3-Hydroxy-1-(5-methoxy-2-nitrophenyl)heptan-1-one (2v)
Yield: 59%; IR (KBr, cm–1): 3288 (OH), 1647 (CO), 1503 (NO2); 1H NMR (400 MHz, CDCl3): δ 7.46 (d, J = 8.4 Hz, 1H), 7.42 (d, J = 2.8 Hz, 1H), 7.15 (dd, J = 2.4, 8.8 Hz, 1H), 6.61 (s, 1H), 4.18–4.10 (m, br, 1H), 3.91 (s, 3H), 3.24–3.20 (m, 2H), 1.54–1.25 (m, 6H), 0.94–0.89 (m, 3H); LC-MS (ESI): m/z calcd. for C14H19NO5: 281.13, found 281.95 [M + H]+.
4.1.1.21. 1-(4-Fluoro-2-nitrophenyl)-3-hydroxyheptan-1-one (2w)
Yield: 52%; IR (KBr, cm–1): 3098 (OH), 1708 (CO), 1539 (NO2); 1H NMR (400 MHz, CDCl3): δ 7.80 (dd, J = 2.4, 8.0 Hz, 1H), 7.50–7.41 (m, 2H), 4.23 (s, br, 1H), 2.97 (dd, J = 2.4, 17.2 Hz, 1H), 2.89–2.82 (m, 1H), 2.67 (s, OH, 1H), 1.63–1.30 (m, 6H), 0.91 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 201.66, 162.64 (d), 146.92, 133.62, 129.52 (d), 121.34 (d), 112.16 (d), 68.14, 49.57, 36.31, 27.53, 22.52, 13.93; LC-MS (ESI): m/z calcd. for C13H16FNO4: 269.11, found 269.96 [M + H]+.
4.1.2. General procedure for the preparation of compounds 4a–w
Compound 2 (1.0 eq.) was added to EtOH (10.0 vol) and hydrogenated (3.5 bar H2) over 10% palladium on charcoal (20% wt). The reaction mixture was filtered through a pad of Celite and the filtrate was evaporated under reduced pressure. The residue was purified by silica gel column chromatography to give compounds 4a–w (78–91%).
4.1.2.1. 1-(2-Aminophenyl)-3-hydroxyhexan-1-one (4a)
Yield: 90%; IR (KBr, cm–1): 3453 (OH), 3345 (NH2), 1615 (CO); 1H NMR (300 MHz, CDCl3): δ 7.70 (dd, J = 1.5, 8.4 Hz, 1H), 7.30–7.24 (m, 1H), 6.67–6.62 (m, 2H), 6.28 (s br, NH2, 2H), 4.23–4.15 (m, br, 1H), 3.39 (s, OH, 1H), 3.15 (dd, J = 2.4, 17.1 Hz, 1H), 3.02–2.93 (m, 1H), 1.64–1.41 (m, 4H), 0.96 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 203.05, 150.47, 134.72, 131.09, 117.82, 117.42, 115.87, 67.71, 45.25, 38.64, 18.79, 14.05; LC-MS (ESI): m/z calcd. for C12H17NO2: 207.13, found 208.20 [M + H]+.
4.1.2.2. 1-(2-Aminophenyl)-3-cyclopropyl-3-hydroxypropan-1-one (4b)
Yield: 86%; IR (KBr, cm–1): 3445 (OH), 3343 (NH2), 1616 (CO); 1H NMR (400 MHz, CDCl3): δ 7.74 (d, J = 8.0 Hz, 1H), 7.29–7.25 (m, 1H), 6.67–6.63 (m, 2H), 6.28 (s, br, NH2, 2H), 3.45 (t, J = 8.8 Hz, 1H), 3.33–3.16 (m, 2H), 1.03–0.99 (m, 1H), 0.63–0.42 (m, 3H), 0.24–0.19 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 202.68, 150.51, 134.71, 131.19, 117.84, 117.40, 115.80, 72.94, 45.36, 16.78, 3.43, 2.15; LC-MS (ESI): m/z calcd. for C12H15NO2: 205.11, found 206.13 [M + H]+.
4.1.2.3. 1-(2-Aminophenyl)-3-hydroxypentan-1-one (4c)
Yield: 88%; IR (KBr, cm–1): 3433 (OH), 3385 (NH2), 1660 (CO); 1H NMR (400 MHz, CDCl3): δ 7.70 (dd, J = 1.2, 8.4 Hz, 1H), 7.29–7.27 (m, 1H), 6.66–6.62 (m, 2H), 6.27 (s br, NH2, 2H), 4.20–4.05 (m, br, 1H), 3.35 (s, OH, 1H), 3.16 (dd, J = 2.8, 17.2 Hz, 1H), 3.00–2.94 (m, 1H), 1.69–1.54 (m, 2H), 1.01 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 203.02, 150.51, 134.71, 131.12, 117.94, 117.44, 115.90, 64.42, 44.86, 29.41, 9.97.
4.1.2.4. 1-(2-Aminophenyl)-3-cyclohexyl-3-hydroxypropan-1-one (4d)
Yield: 85%; IR (KBr, cm–1): 3468 (OH), 3886 (NH2), 1614 (CO); 1H NMR (400 MHz, CDCl3): δ 7.71 (dd, J = 1.6, 8.0 Hz, 1H), 7.28–7.24 (m, 1H), 6.66–6.62 (m, 2H), 6.27 (s, br, NH2, 2H), 3.97–3.93 (m, 1H), 3.17 (dd, J = 2.0, 17.2 Hz, 1H), 3.01–2.94 (m, 1H), 1.94–1.91 (m, 1H), 1.80–1.67 (m, 4H), 1.50–1.07 (m, 6H); 13C NMR (100 MHz, CDCl3): δ 203.41, 150.48, 134.65, 131.12, 118.04, 117.44, 115.87, 72.11, 43.14, 42.38, 29.05, 28.42, 26.53, 26.29, 26.18; LC-MS (ESI): m/z calcd. for C15H21NO2: 247.33, found 248.21 [M + H]+.
4.1.2.5. 1-(2-Aminophenyl)-3-hydroxy-3-phenylpropan-1-one (4e)
Yield: 87%; IR (KBr, cm–1): 3469 (OH), 3349 (NH2), 1616 (CO); 1H NMR (400 MHz, CDCl3): δ 7.64 (dd, J = 1.2, 8.1 Hz, 1H), 7.45–7.24 (m, 6H), 6.66–6.58 (m, 2H), 6.32 (s br, NH2, 2H), 5.31 (dd, J = 3.6, 8.1 Hz, 1H), 3.34–3.31 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 202.09, 150.58, 143.07, 134.87, 131.08, 128.51, 127.56, 125.77, 117.53, 117.42, 115.90, 70.28, 47.62; LC-MS (ESI): m/z calcd. for C15H15NO2: 241.11, found 264.18 [M + Na]+.
4.1.2.6. 1-(2-Aminophenyl)-3-hydroxy-3-(4-(trifluoromethyl)phenyl)propan-1-one (4g)
Yield: 79%; IR (KBr, cm–1): 3391 (OH), 3326 (NH2), 1619 (CO); 1H NMR (400 MHz, CDCl3): δ 7.60–7.49 (m, 4H), 7.08 (d, J = 9.6 Hz, 2H), 6.75–6.63 (m, 2H), 5.18–5.02 (m, 2H), 2.52–2.46 (m, 1H), 2.12–2.04 (m, 1H); LC-MS (ESI): m/z calcd. for C16H14F3NO2: 309.10, found 310.11 [M + H]+.
4.1.2.7. 1-(2-Aminophenyl)-3-(2,4-difluorophenyl)-3-hydroxypropan-1-one (4h)
Yield: 82%; MR: 73–75 °C; IR (KBr, cm–1): 3454 (OH), 3350 (NH2), 1617 (CO); 1H NMR (400 MHz, CDCl3): δ 7.64–7.56 (m, 2H), 7.29–7.25 (m, 1H), 6.91 (td, J = 1.6, 8.0 Hz, 1H), 6.79 (td, J = 2.4, 8.4 Hz, 1H), 6.66–6.59 (m, 2H), 6.30 (s, br, NH2, 2H), 5.53 (d, J = 9.2 Hz, 1H), 3.98 (s, br, OH, 1H), 3.42 (dd, J = 2.4, 17.6 Hz, 1H), 3.24–3.17 (m, 1H); LC-MS (ESI): m/z calcd. for C15H13F2NO2: 277.09, found 278.53 [M + H]+.
4.1.2.8. 1-(2-Aminophenyl)-3-hydroxy-3-(4-methoxyphenyl)propan-1-one (4i)
Yield: 85%; IR (KBr, cm–1): 3444 (OH), 3278 (NH2), 1614 (CO); 1H NMR (300 MHz, CDCl3): δ 7.66 (dd, J = 1.6, 10.8 Hz, 1H), 7.37–7.26 (m, 3H), 6.92–6.87 (m, 2H), 6.66–6.61 (m, 2H), 6.31 (s, NH2, 2H), 5.29–5.24 (m, 1H), 3.81 (s, 3H), 3.33–3.30 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 202.20, 159.07, 150.59, 135.32, 134.83, 131.11, 127.38, 117.64, 115.92, 114.05, 113.90, 69.95, 56.56, 47.60; LC-MS (ESI): m/z calcd. for C16H17NO3: 271.12, found 294.16 [M + Na]+.
4.1.2.9. 1-(2-Aminophenyl)-3-hydroxy-3-(3-methoxyphenyl)propan-1-one (4j)
Yield: 81%; IR (KBr, cm–1): 3468 (OH), 3976 (NH2), 1614 (CO); 1H NMR (400 MHz, CDCl3): δ 7.64 (dd, J = 1.6, 8.4 Hz, 1H), 7.30–7.24 (m, 2H), 7.01–6.98 (m, 2H), 6.83 (dd, J = 1.6, 8.4 Hz, 1H), 6.63–6.60 (m, 2H), 6.31 (s, NH2, 2H), 5.28 (dd, J = 3.2, 8.8 Hz, 1H), 3.82 (s, 3H), 3.33–3.28 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 202.04, 159.81, 150.60, 144.83, 134.86, 131.09, 129.52, 118.03, 117.57, 117.42, 115.91, 113.14, 111.22, 70.21, 55.23, 47.64; LC-MS (ESI): m/z calcd. for C16H17NO3: 271.12, found 272.24 [M + H]+.
4.1.2.10. 1-(2-Aminophenyl)-3-hydroxy-3-(2-methoxyphenyl)propan-1-one (4k)
Yield: 84%; IR (KBr, cm–1): 3465 (OH), 3382 (NH2), 1614 (CO); 1H NMR (300 MHz, CDCl3): δ 7.69 (dd, J = 1.2, 8.1 Hz, 1H), 7.56 (dd, J = 1.5, 7.2 Hz, 1H), 7.29–7.24 (m, 2H), 7.01 (t, J = 6.9 Hz, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.67–6.58 (m, 2H), 5.57 (dd, J = 2.1, 9.0 Hz, 1H), 3.84 (s, 3H), 3.53–3.47 (m, 1H), 3.21–3.12 (m, 1H); 13C NMR (75 MHz, CDCl3): δ 202.66, 155.67, 150.52, 134.66, 131.28, 128.23, 126.47, 120.84, 117.81, 117.35, 115.82, 110.12, 109.95, 65.68, 55.24, 46.11; LC-MS (ESI): m/z calcd. for C16H17NO3: 271.12, found 294.68 [M + Na]+.
4.1.2.11. 1-(2-Aminophenyl)-3-(3,4-dimethylphenyl)-3-hydroxypropan-1-one (4l)
Yield: 90%; IR (KBr, cm–1): 3459 (OH), 1615 (CO); 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 7.6 Hz, 1H), 7.28–7.24 (m, 1H), 7.21 (s, 1H), 7.16–7.11 (m, 2H), 6.61–6.58 (m, 2H), 6.31 (s br, NH2, 2H), 5.24 (dd, J = 3.6, 7.6 Hz, 1H), 3.69 (s, OH, 1H), 3.32–3.30 (m, 2H), 2.27 (d, J = 7.6 Hz, 6H); 13CNMR (100 MHz, CDCl3): δ 202.22, 150.58, 140.60, 136.73, 135.88, 134.79, 131.12, 129.72, 127.06, 123.20, 117.66, 117.41, 115.90, 70.16, 47.70, 19.81, 19.43; LC-MS (ESI): m/z calcd. for C17H19NO2: 269.14, found 270.23 [M + H]+.
4.1.2.12. 1-(2-Aminophenyl)-3-hydroxy-3-(pyridin-2-yl)propan-1-one (4m)
Yield: 83%; MR: 94–99 °C; IR (KBr, cm–1): 3482 (OH), 3345 (NH2), 1665 (CO); 1H NMR (400 MHz, CDCl3): δ 8.54 (d, J = 4.8 Hz, 1H), 7.72–7.67 (m, 2H), 7.53 (d, J = 8.0 Hz, 1H), 7.27–7.17 (m, 2H), 6.64–6.58 (m, 2H), 6.29 (s NH2, 2H), 5.36 (dd, J = 2.8, 8.4 Hz, 1H), 4.38 (s OH, 1H), 3.58 (dd, J = 3.2, 16.8 Hz, 1H), 3.42–3.35 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 201.72, 161.84, 150.53, 148.54, 136.73, 134.67, 131.35, 122.28, 120.41, 117.85, 117.30, 115.85, 70.49, 46.32; LC-MS (ESI): m/z calcd. for C14H14N2O2: 242.11, found, NO mass ionization.
4.1.2.13. 1-(2-Aminophenyl)-3-hydroxy-3-(pyridin-3-yl)propan-1-one (4n)
Yield: 80%; IR (KBr, cm–1): 3465 (OH), 3387 (NH2), 1608 (CO); 1H NMR (400 MHz, CDCl3): δ 8.66 (s, 1H), 8.55 (d, J = 3.2 Hz, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.63 (d, J = 7.6 Hz, 1H), 7.32–7.28 (m, 2H), 6.68–6.60 (m, 2H), 6.31 (s, NH2, 2H), 3.93 (d, J = 2.8 Hz, 1H), 3.41–3.37 (m, 2H).
4.1.2.14. 1-(2-Aminophenyl)-3-hydroxy-3-(pyridin-4-yl)propan-1-one (4o)
Yield: 82%; MR: 132.136 °C; IR (KBr, cm–1): 3492 (OH), 3426 (NH2), 1624 (CO); 1H NMR (400 MHz, CDCl3): δ 8.48 (s, 2H), 7.62 (d, J = 8.0 Hz, 1H), 7.36 (s, 2H), 7.30–7.26 (s, 1H), 6.67–6.60 (m, 2H), 6.21 (s NH2, 2H), 5.30 (d, J = 8.0 Hz, 1H), 4.08 (s OH, 1H), 3.38–3.23 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 201.20, 152.07, 150.72, 149.90, 135.11, 130.93, 120.73, 117.52, 117.32, 116.02, 68.96, 46.88; LC-MS (ESI): m/z calcd. for C14H14N2O2: 242.11, found 243.18 [M + H]+.
4.1.2.15. 1-(2-Aminophenyl)-3-(furan-3-yl)-3-hydroxypropan-1-one (4p)
Yield: 66%; IR (KBr, cm–1): 3435 (OH), 2920, 1615 (CO); 1H NMR (400 MHz, CDCl3): δ 7.68 (dd, J = 1.2, 8.0 Hz, 1H), 7.46–7.40 (m, 2H), 7.30–7.25 (m, 1H), 6.66–6.62 (m, 2H), 6.45 (s, 1H), 6.30 (s, br, NH2, 2H), 5.28 (t, J = 6.0 Hz, 1H), 3.35 (2d, J = 6.4 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 201.84, 150.57, 143.25, 138.96, 134.87, 130.99, 120.69, 117.54, 117.43, 115.90, 108.58, 63.46, 46.06; LC-MS (ESI): m/z calcd. for C13H13NO3: 231.090, found 255.12 [M + Na]+ (note: during the nitro reduction of 3-(furan-3-yl)-3-hydroxy-1-(2-nitrophenyl)propan-1-one (2p), along with 1-(2-aminophenyl)-3-(furan-3-yl)-3-hydroxypropan-1-one (4p), the furan ring reduced product 1-(2-aminophenyl)-3-hydroxy-3-(tetrahydrofuran-3-yl)propan-1-one (4q) was isolated and characterized).
4.1.2.16. 1-(2-Aminophenyl)-3-(furan-2-yl)-3-hydroxypropan-1-one (4q)
Yield: 60%; IR (KBr, cm–1): 3460 (OH), 3006 (NH2), 1615 (CO); 1H NMR (400 MHz, CDCl3): δ 7.71 (d, J = 7.6 Hz, 1H), 7.39 (s, 1H), 7.28 (t, J = 8.0 Hz, 1H), 6.64 (t, J = 8.0 Hz, 2H), 6.36–6.29 (m, 4H), 3.65–3.64 (m, 1H), 3.53–3.41 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 201.37, 155.38, 150.62, 142.02, 134.91, 131.08, 117.61, 117.45, 115.97, 110.25, 106.68, 64.36, 43.67; LC-MS (ESI): m/z calcd. for C13H13NO3: 231.09, found, NO mass ionization (note: during the nitro reduction of 3-(furan-2-yl)-3-hydroxy-1-(2-nitrophenyl)propan-1-one (2r), along with 1-(2-aminophenyl)-3-(furan-2-yl)-3-hydroxypropan-1-one (4r), the furan ring reduced product 1-(2-aminophenyl)-3-hydroxy-3-(tetrahydrofuran-2-yl)propan-1-one (4s) was isolated and characterized).
4.1.2.17. 1-(2-Aminophenyl)-3-hydroxy-3-(thiophen-3-yl)propan-1-one (4r)
Yield: 87%; IR (KBr, cm–1): 3491 (OH), 2903, 1711 (CO); 1H NMR (400 MHz, CDCl3): δ 7.66 (d, J = 8.0 Hz, 1H), 7.34–7.21 (m, 3H), 7.06 (d, J = 5.2 Hz, 1H), 6.65–6.59 (m, 2H), 6.29 (s, NH2, 2H), 5.38 (dd, J = 3.2, 7.6 Hz, 1H), 3.77 (s, 1H), 3.37 (d, J = 4.0 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 201.85, 150.60, 144.45, 134.85, 131.05, 126.06, 125.63, 123.27, 120.74, 117.44, 115.91, 66.86, 46.69; LC-MS (ESI): m/z calcd. for C13H13NO2S: 247.07, found 248.11 [M + H]+.
4.1.2.18. 1-(2-Aminophenyl)-3-hydroxy-3-(thiophen-2-yl)propan-1-one (4s)
Yield: 82%; IR (KBr, cm–1): 3466 (OH), 3423 (NH2), 1615 (CO); 1H NMR (400 MHz, CDCl3): δ 7.67 (dd, J = 1.2, 7.6 Hz, 1H), 7.30–7.23 (m, 2H), 7.02–6.89 (m, 2H), 6.66–6.63 (m, 2H), 6.32–6.28 (s, NH2, 2H), 5.55 (dd, J = 4.4, 7.6 Hz, 1H), 3.97 (s, OH, 1H), 3.47–3.45 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 201.42, 150.61, 146.79, 134.96, 131.96, 126.63, 124.59, 119.93, 117.73, 115.94, 114.85, 66.68, 47.28; LC-MS (ESI): m/z calcd. for C13H13NO2S: 247.07, found 248.06 [M + H]+.
4.1.2.19. 1-(2-Aminophenyl)-3-hydroxy-3-(tetrahydrofuran-3-yl)propan-1-one (4t)
Yield: 32%; IR (KBr, cm–1): 3446 (OH), 3223 (NH2), 1615 (CO); 1H NMR (400 MHz, CDCl3): δ 7.70–7.60 (m, 1H), 7.31–7.26 (m, 1H), 6.67–6.63 (m, 2H), 6.29 (s, br, NH2, 2H), 4.11–3.73 (m, 4H), 3.72–3.56 (m, 1H), 3.26–2.95 (m, 2H), 2.45–2.34 (m, 1H), 2.10–1.62 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 150.58, 134.91, 131.00, 118.52, 117.50, 115.94, 70.87 & 70.65, 69.85 & 69.45, 68.40 & 68.20, 44.64, 43.87, 29.00 & 28.40; LC-MS (ESI): m/z calcd. for C13H17NO3: 235.12, found 236.16 [M + H]+.
4.1.2.20. 1-(2-Aminophenyl)-3-hydroxy-3-(tetrahydrofuran-2-yl)propan-1-one (4u)
Yield: 35%; IR (KBr, cm–1): 3436 (OH), 3365 (NH2), 1616 (CO); 1H NMR (400 MHz, CDCl3): δ 7.75–7.72 (m, 1H), 7.26 (t, J = 8.0 Hz, 1H), 6.65–6.62 (m, 2H), 6.26 (s, br, NH2, 2H), 4.19–4.08 (m, 1H), 3.93–3.74 (m, 4H), 3.37–3.32 (m, 1H), 3.26–3.19 (m, 1H), 3.10–3.01 (m, 2H), 1.46–1.44 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 202.66 & 201.56, 150.50, 134.70 & 134.59, 131.35 & 131.29, 117.36, 115.91, 115.85, 81.42 & 81.17, 70.32 & 70.05, 68.56 & 68.51, 42.71 & 42.21, 29.57 & 27.53, 26.00 & 25.76; LC-MS (ESI): m/z calcd. for C13H17NO3: 235.12, found 236.00 [M + H]+.
4.1.2.21. 1-(2-Amino-5-methoxyphenyl)-3-hydroxyheptan-1-one (4v)
Yield: 85%; IR (KBr, cm–1): 3433 (OH), 3368 (NH2), 1610 (CO); 1H NMR (400 MHz, CDCl3): δ 7.62 (d, J = 8.0 Hz, 1H), 6.43 (s, br, NH2, 2H), 6.22 (d, J = 2.4, 8.8 Hz, 1H), 6.06 (d, J = 2.4 Hz, 1H), 4.17–4.13 (m, 1H), 3.80 (s, 3H), 3.57 (s, OH, 1H), 3.09 (dd, J = 2.4, 17.2 Hz, 1H), 2.92–2.86 (m, 1H), 1.64–1.24 (m, 6H), 0.92 (t, J = 10.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 201.19, 164.57, 153.03, 133.20, 112.45, 104.81, 99.15, 68.15, 55.23, 44.80, 36.25, 27.77, 22.70, 14.06; LC-MS (ESI): m/z calcd. for C14H21NO3: 251.15, found 252.14 [M + H]+.
4.1.2.22. 1-(2-Amino-4-fluorophenyl)-3-hydroxyheptan-1-one (4w)
Yield: 81%; IR (KBr, cm–1): 3438 (OH), 3340 (NH2), 1634 (CO); 1H NMR (400 MHz, CDCl3): δ 7.70 (dd, J = 6.3, 8.7 Hz, 1H), 6.40 (s, br, NH2, 2H), 6.38–6.28 (m, 2H), 4.17–4.13 (s, br, 1H), 3.31 (s, OH, 1H), 3.09 (dd, J = 2.7, 17.1 Hz, 1H), 2.98–2.89 (m, 1H), 1.65–1.25 (m, 6H), 0.92 (t, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 201.71, 166.65 (d), 152.77 (d), 133.98 (d), 115.02, 104.20 (d), 102.46 (d), 67.90, 45.33, 36.19, 27.74, 22.66, 14.00; LC-MS (ESI): m/z calcd. for C13H18FNO2: 239.13, found 240.08 [M + H]+.
4.1.3. General procedure for the preparation of compounds 5a–w
To a solution of compound 4 (1.0 eq.) in toluene (10 vol) was added TsOH (0.5 eq.) under an argon atmosphere. The reaction mixture was stirred at 120 °C for 1–2 h and then cooled to RT, toluene was removed under reduced pressure and the residue was diluted with DCM (30 vol) and washed with saturated NaHCO3 solution (20 vol) followed by saturated brine solution (20 vol). The organic phase was dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash silica gel chromatography to give compounds 5a–w (70–98%) (note: ZnCl2 (0.75 eq.) was used for preparation of pyridine derivatives (5k–m)).
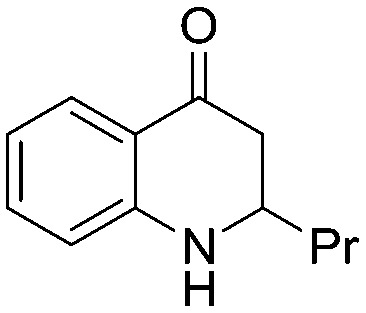
4.1.3.1. 2-Propyl-2,3-dihydroquinolin-4(1H)-one (5a)
MR: 101–104 °C; IR (KBr, cm–1): 3437 (NH), 1653 (CO); 1H NMR (400 MHz, CDCl3): δ 7.82 (dd, J = 1.6, 8.4 Hz, 1H), 7.31–7.26 (m, 1H), 6.72 (t, J = 8.0 Hz, 1H), 6.65 (d, J = 8.4 Hz, 1H), 4.29 (m, br, NH, 1H), 3.67–3.60 (m, 1H), 2.69–2.65 (m, 1H), 2.51–2.44 (m, 1H), 1.67–1.54 (m, 2H), 1.44–1.33 (m, 2H), 0.97 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 194.06, 151.41, 135.14, 127.49, 119.13, 117.92, 115.71, 53.07, 43.94, 37.32, 18.54, 13.95; LC-MS (ESI): m/z calcd. for C12H15NO: 189.12, found 190.18 [M + H]+.
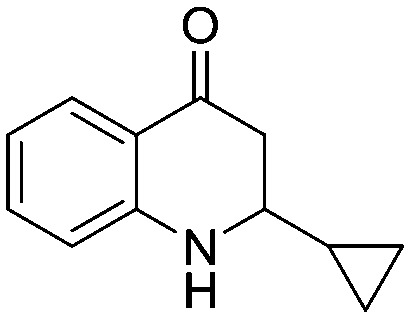
4.1.3.2. 2-Cyclopropyl-2,3-dihydroquinolin-4(1H)-one (5b)
MR: 122–125 °C; IR (KBr, cm–1): 3325 (NH), 1656 (CO); 1H NMR (400 MHz, CDCl3): δ 7.80 (dd, J = 1.6, 8.0 Hz, 1H), 7.30–7.27 (m, 1H), 6.72–6.65 (m, 2H), 4.48 (s, br, NH, 1H), 2.78–2.60 (m, 3H), 1.09–1.03 (m, 1H), 0.62–0.56 (m, 2H), 0.30–0.21 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 194.03, 151.44, 135.19, 127.53, 119.15, 117.95, 115.68, 59.32, 44.26, 15.96, 3.18, 3.12; LC-MS (ESI): m/z calcd. for C12H13NO: 187.10, found 188.16 [M + H]+.
4.1.3.3. 2-Ethyl-2,3-dihydroquinolin-4(1H)-one (5c)
IR (KBr, cm–1): 3445 (NH), 1651 (CO); 1H NMR (400 MHz, CDCl3): δ 7.81 (dd, J = 1.6, 8.0 Hz, 1H), 7.30–7.26 (m, 1H), 6.72 (t, J = 7.6 Hz, 1H), 6.66 (d, J = 8.4 Hz, 1H), 4.32 (s, br, NH, 1H), 3.58–3.53 (m, 1H), 2.67 (dd, J = 3.6, 16.0 Hz, 1H), 2.51–2.43 (m, 1H), 1.70–1.63 (m, 2H), 1.01 (t, J = 7.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 194.05, 151.44, 135.13, 127.47, 119.12, 117.91, 115.71, 54.66, 43.48, 28.05, 9.63; LC-MS (ESI): m/z calcd. for C11H13NO: 175.10, found 176.13 [M + H]+.
4.1.3.4. 2-Cyclohexyl-2,3-dihydroquinolin-4(1H)-one (5d)
MR: 121–125 °C; IR (KBr, cm–1): 3444 (NH), 1650 (CO); 1H NMR (400 MHz, CDCl3): δ 7.80 (dd, J = 1.2, 8.0 Hz, 1H), 7.28 (t, J = 8.4 Hz, 1H), 6.71 (t, J = 6.8 Hz, 1H), 6.66 (d, J = 8.0 Hz, 1H), 4.34 (s, NH, 1H), 3.44–3.39 (m, 1H), 2.66–2.51 (m, 2H), 1.83–1.69 (m, 5H), 1.57–1.49 (m, 6H); 13C NMR (100 MHz, CDCl3): δ 194.53, 151.62, 135.13, 127.38, 119.01, 117.73, 115.74, 58.12, 41.53, 40.98, 28.87, 28.56, 26.29, 26.07; LC-MS (ESI): m/z calcd. for C15H19NO: 229.14, found 230.67 [M + H]+.
4.1.3.5. 2-Phenyl-2,3-dihydroquinolin-4(1H)-one (5e)
MR: 133–136 °C; IR (KBr, cm–1): 3436 (NH), 1650 (OH); 1H NMR (400 MHz, CDCl3): δ 7.87 (dd, J = 1.2, 8.4 Hz, 1H), 7.46 (dd, J = 1.6, 8.0 Hz, 2H), 7.42–7.32 (m, 4H), 6.79 (t, J = 8.0 Hz, 1H), 6.71 (d, J = 8.0 Hz, 1H), 4.75 (dd, J = 4.0, 13.6 Hz, 1H), 4.50 (m, br, NH, 1H), 2.89–2.79 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 193.24, 151.53, 141.00, 135.38, 128.99, 128.47, 127.62, 126.61, 119.05, 118.45, 115.88, 58.50, 46.44; LC-MS (ESI): m/z calcd. for C15H13NO: 223.10, found: 224.62 [M + H]+.
4.1.3.6. 2-(4-Bromophenyl)-2,3-dihydroquinolin-4(1H)-one (5f)
MR: 165–169 °C; IR (KBr, cm–1): 3467 (NH), 1644 (CO); 1H NMR (400 MHz, CDCl3): δ 7.87 (d, J = 8.0 Hz, 1H), 7.52 (d, J = 8.4 Hz, 2H), 7.40–7.33 (m, 3H), 6.81 (t, J = 8.0 Hz, 1H), 6.72 (d, J = 7.6 Hz, 1H), 4.72 (dd, J = 4.0, 12.8 Hz, 1H), 4.47 (s, NH, 1H), 2.84–2.76 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 192.77, 151.28, 140.05, 135.49, 132.13, 128.30, 127.62, 122.25, 119.07, 118.74, 115.94, 57.96, 46.36; LC-MS (ESI): m/z calcd. for C15H12BrNO: 302.01, found 304.12 [M + 2]+2 (note: during the reduction of 3-(4-bromophenyl)-3-hydroxy-1-(2-nitrophenyl)propan-1-one (2f) using iron (1.0 eq.) and acetic acid (3.0 eq.) in toluene under reflux for 6 h, the directly isolated cyclized 2-(4-bromophenyl)-2,3-dihydroquinolin-4(1H)-one (5f) was obtained in 56% yield).
4.1.3.7. 2-(4-(Trifluoromethyl)phenyl)-2,3-dihydroquinolin-4(1H)-one (5g)
IR (KBr, cm–1): 3337 (NH), 1610 (CO); 1H NMR (300 MHz, CDCl3): δ 7.88 (d, J = 8.1 Hz, 1H), 7.68–7.51 (m, 4H), 7.37 (t, J = 6.9 Hz, 1H), 6.83 (t, J = 7.5 Hz, 1H), 6.74 (d, J = 8.1 Hz, 1H), 4.84 (dd, J = 5.1, 12.6 Hz, 1H), 4.49 (s, NH, 1H), 2.87–2.76 (m, 2H); LC-MS (ESI): m/z calcd. for C16H12F3NO: 291.09, found 292.08 [M + H]+.
4.1.3.8. 2-(2,4-Difluorophenyl)-2,3-dihydroquinolin-4(1H)-one (5h)
IR (KBr, cm–1): 3287 (NH), 1647 (CO); 1H NMR (400 MHz, CDCl3): δ 7.87 (dd, J = 8.4, 1.2 Hz, 1H), 7.57–7.50 (m, 1H), 7.35 (t, J = 8.4 Hz, 1H), 6.95–6.79 (m, 3H), 6.72 (d, J = 8.4 Hz, 1H), 5.09 (t, J = 8.4 Hz, 1H), 2.87 (d, J = 8.1 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 192.49, 151.17, 135.45, 134.44, 128.54, 127.62, 119.16, 118.80, 117.30, 115.96, 111.89 (d), 111.68, 104.30 (t), 50.64, 44.34; LC-MS (ESI): m/z calcd. for C15H11F2NO: 259.08, found 260.54 [M + H]+.
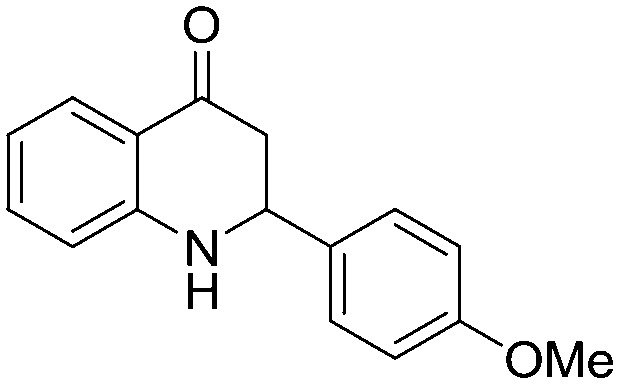
4.1.3.9. 2-(4-Methoxyphenyl)-2,3-dihydroquinolin-4(1H)-one (5i)
IR (KBr, cm–1): 3436 (NH), 1660 (CO); 1H NMR (400 MHz, CDCl3): δ 7.87 (d, J = 10.4 Hz, 1H), 7.40–7.30 (m, 3H), 6.93 (d, J = 11.6 Hz, 2H), 6.78 (t, J = 10.0 Hz, 1H), 6.69 (d, J = 11.2 Hz, 1H), 4.70 (dd, J = 4.8, 18.0 Hz, 1H), 4.43 (s, NH, 1H), 3.83 (s, 3H), 2.93–2.76 (m, 2H); LC–MS (ESI): m/z calcd. for C16H15NO2: 253.11, found 254.31 [M + H]+.
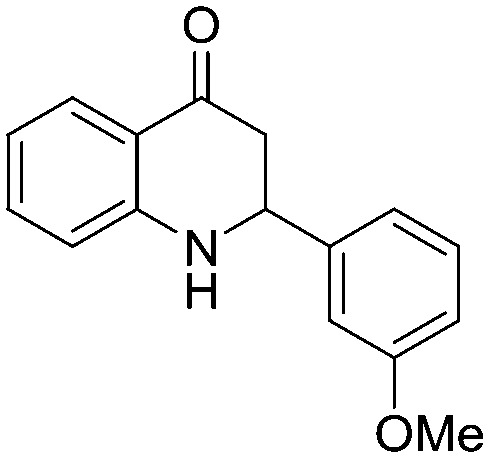
4.1.3.10. 2-(3-Methoxyphenyl)-2,3-dihydroquinolin-4(1H)-one (5j)
MR: 109–112 °C; IR (KBr, cm–1): 3467 (NH), 1655 (CO); 1H NMR (400 MHz, CDCl3): δ 7.87 (dd, J = 1.6, 10.8 Hz, 1H), 7.36–7.25 (m, 2H), 7.04–7.01 (m, 2H), 6.88 (dd, J = 2.0, 9.6 Hz, 1H), 6.79 (t, J = 9.2 Hz, 1H), 6.71 (t, J = 11.2 Hz, 1H), 4.72 (dd, J = 6.0, 17.6 Hz, 1H), 4.51 (s, NH, 1H), 3.82 (s, 3H), 2.92–2.73 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 193.20, 160.01, 151.47, 142.65, 135.38, 130.06, 127.61, 119.02, 118.82, 118.46, 115.89, 113.71, 112.23, 58.46, 55.29, 46.44; LC-MS (ESI): m/z calcd. for C16H15NO2: 253.11, found 254.22 [M + H]+.
4.1.3.11. 2-(2-Methoxyphenyl)-2,3-dihydroquinolin-4(1H)-one (5k)
IR (KBr, cm–1): 3436 (NH), 1660 (CO); 1H NMR (400 MHz, CDCl3): δ 7.85 (dd, J = 1.6, 8.0 Hz, 1H), 7.48 (dd, J = 1.2, 7.6 Hz, 1H), 7.33–7.25 (m, 2H), 6.98 (t, J = 8.0 Hz, 1H), 6.90 (d, J = 8.0 Hz, 1H), 6.76–6.69 (m, 2H), 5.16 (dd, J = 4.4, 7.6 Hz, 1H), 3.84 (s, 3H), 2.93–2.78 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 193.88, 156.57, 151.93, 135.18, 131.91, 129.01, 127.46, 126.41, 120.83, 118.96, 118.02, 116.05, 110.52, 55.32, 51.24, 43.67; LC-MS (ESI): m/z calcd. for C16H15NO2: 253.11, found 254.28 [M + H]+.
4.1.3.12. 2-(3,4-Dimethylphenyl)-2,3-dihydroquinolin-4(1H)-one (5l)
IR (KBr, cm–1): 3327 (NH), 1663 (CO); 1H NMR (400 MHz, CDCl3): δ 7.87 (d, J = 8.0 Hz, 1H), 7.32 (t, J = 8.8 Hz, 1H), 7.22 (s, 1H), 7.17–7.14 (m, 2H), 6.77 (t, J = 7.6 Hz, 1H), 6.69 (d, J = 8.4 Hz, 1H), 4.68 (dd, J = 3.6, 13.6 Hz, 1H), 4.45 (s, br, NH, 1H), 2.92–2.71 (m, 2H), 2.28 (d, J = 4.4 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 193.50, 151.60, 138.46, 137.27, 136.91, 135.31, 130.12, 127.83, 127.61, 123.98, 119.00, 118.32, 115.85, 58.23, 46.248, 19.83, 19.46; LC-MS (ESI): m/z calcd. for C17H17NO: 251.13, found 252.30 [M + H]+.
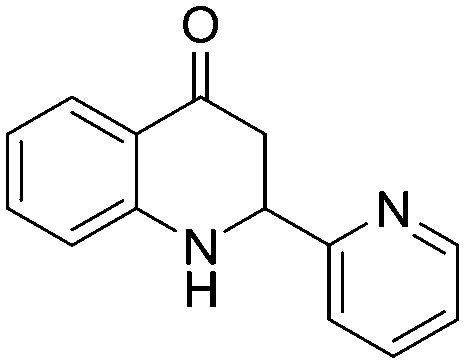
4.1.3.13. 2-(Pyridin-2-yl)-2,3-dihydroquinolin-4(1H)-one (5m)
IR (KBr, cm–1): 3435 (NH), 1611 (CO); 1H NMR (400 MHz, CDCl3): δ 8.60 (d, J = 4.8 Hz, 1H), 7.84 (dd, J = 1.6, 8.0 Hz, 1H), 7.73–7.69 (m, 1H), 7.36–7.30 (m, 2H), 7.24–7.18 (m, 1H), 6.78–6.73 (m, 2H), 5.37–5.34 (s, NH, 1H), 4.86 (dd, J = 4.4, 12.4 Hz, 1H), 3.04–2.89 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 193.06, 158.80, 151.06, 149.44, 137.19, 135.46, 131.34, 127.47, 122.99, 120.44, 118.17, 116.14, 57.35, 43.46; LC-MS (ESI): m/z calcd. for C14H12N2O: 224.09, found 225.11 [M + H]+.
4.1.3.14. 2-(Pyridin-3-yl)-2,3-dihydroquinolin-4(1H)-one (5n)
MR: 152–155 °C; IR (KBr, cm–1): 3344 (NH), 1607 (CO); 1H NMR (400 MHz, CDCl3): δ 8.69 (s, 1H), 8.60 (d, J = 3.6 Hz, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.82 (d, J = 7.6 Hz, 1H), 7.38–7.32 (m, 2H), 6.82 (t, J = 7.6 Hz, 1H), 6.74 (d, J = 8.0 Hz, 1H), 4.80 (dd, J = 3.2, 12.8 Hz, 1H), 4.57 (s NH, 1H), 2.92–2.77 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 192.30, 151.19, 150.00, 148.52, 136.52, 135.54, 134.21, 127.64, 123.80, 119.19, 118.95, 116.01, 56.17, 46.02; LC-MS (ESI): m/z calcd. for C14H12N2O: 224.09, found 225.27 [M + H]+.
4.1.3.15. 2-(Pyridin-4-yl)-2,3-dihydroquinolin-4(1H)-one (5o)
MR: 174–178 °C; IR (KBr, cm–1): 3435 (NH), 1670 (CO); 1H NMR (400 MHz, CDCl3): δ 8.64 (d, J = 5.6 Hz, 2H), 7.87 (d, J = 8.0 Hz, 1H), 7.39–7.35 (m, 3H), 6.83 (t, J = 8.0 Hz, 1H), 6.76 (d, J = 8.4 Hz, 1H), 4.78 (t, J = 8.4 Hz, 1H), 4.57 (s, 1H), 2.83 (d, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 191.97, 150.95, 150.55, 149.81, 135.63, 127.62, 121.41, 119.18, 119.02, 115.99, 57.35, 45.69; LC-MS (ESI): m/z calcd. for C14H12N2O: 224.09, found 224.96 [M + H]+.
4.1.3.16. 2-(Furan-3-yl)-2,3-dihydroquinolin-4(1H)-one (5p)
IR (KBr, cm–1): 3365 (NH), 1645 (CO); 1H NMR (400 MHz, CDCl3): δ 7.86 (dd, J = 1.6, 8.0 Hz, 1H), 7.47–7.42 (m, 2H), 7.32 (t, J = 8.4 Hz, 1H), 6.78 (t, J = 8.0 Hz, 1H), 6.69 (d, J = 8.4 Hz, 1H), 6.46 (s, 1H), 4.75–4.71 (m, 1H), 4.45 (s, NH, 1H), 2.86–2.81 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 193.04, 151.13, 143.79, 139.55, 135.34, 127.59, 125.96, 119.23, 118.54, 115.85, 108.60, 49.72, 45.26; LC-MS (ESI): m/z calcd. for C13H11NO2: 213.079, found 214.05 [M + H]+.
4.1.3.17. 2-(Furan-2-yl)-2,3-dihydroquinolin-4(1H)-one (5q)
MR: 78–82 °C; IR (KBr, cm–1): 3393 (NH), 1663 (CO); 1H NMR (400 MHz, CDCl3): δ 7.85 (dd, J = 8.0 Hz, 1H), 7.39 (s, 1H), 7.32 (t, J = 8.4 Hz, 1H), 6.78 (t, J = 8.0 Hz, 1H), 6.70 (d, J = 8.0 Hz, 1H), 6.34–6.33 (m, 1H), 6.26 (d, J = 3.2 Hz, 1H), 4.85–4.81 (m, 1H), 4.69 (s, NH, 1H), 3.04–2.92 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 192.56, 153.30, 150.41, 142.48, 135.42, 127.47, 119.24, 118.62, 115.98, 110.36, 106.84, 50.80, 49.93; LC-MS (ESI): m/z calcd. for C13H11NO2: 213.07, found 214.22 [M + H]+.
4.1.3.18. 2-(Thiophen-3-yl)-2,3-dihydroquinolin-4(1H)-one (5r)
MR: 131–133 °C; IR (KBr, cm–1): 3336 (NH), 1658 (CO); 1H NMR (400 MHz, CDCl3): δ 7.86 (d, J = 8.0 Hz, 1H), 7.36–7.25 (m, 3H), 7.15 (d, J = 4.8 Hz, 1H), 6.78 (t, J = 7.6 Hz, 1H), 6.70 (d, J = 8.4 Hz, 1H), 4.87 (dd, J = 4.8, 11.6 Hz, 1H), 4.55 (s, NH, 1H), 2.92–2.81 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 193.07, 151.19, 142.25, 135.36, 127.60, 126.89, 125.76, 121.91, 119.20, 118.51, 115.83, 53.79, 45.80; LC-MS (ESI): m/z calcd. for C13H11NOS: 229.06, found 229.88 [M + H]+.
4.1.3.19. 2-(Thiophen-2-yl)-2,3-dihydroquinolin-4(1H)-one (5s)
MR: 142–145 °C; IR (KBr, cm–1): 3331 (NH), 1665 (CO); 1H NMR (300 MHz, CDCl3): δ 7.87 (dd, J = 10.4 Hz, 1H), 7.34 (t, J = 9.6 Hz, 1H), 7.29–7.25 (m, 1H), 7.06 (s, 1H), 7.00–6.97 (m, 1H), 6.80 (t, J = 10.4 Hz, 1H), 6.71 (d, J = 10.8 Hz, 1H), 5.05 (dd, J = 5.7, 8.4 Hz, 1H), 4.64 (s, NH, 1H), 3.01–2.88 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 192.58, 150.75, 144.48, 135.44, 127.56, 126.87, 125.12, 124.98, 119.28, 118.81, 115.95, 53.73, 47.02; LC-MS (ESI): m/z calcd. for C13H11NOS: 229.06, found 230.03 [M + H]+.
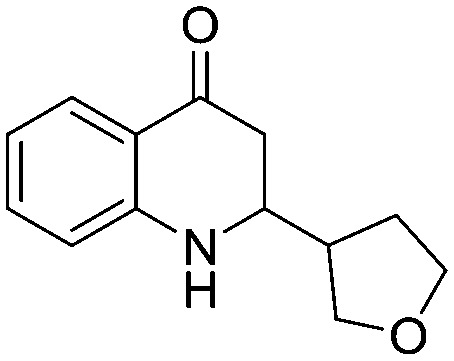
4.1.3.20. 2-(Tetrahydrofuran-3-yl)-2,3-dihydroquinolin-4(1H)-one (5t)
MR:114–117 °C; IR (KBr, cm–1): 3466 (NH), 1665 (CO); 1H NMR (400 MHz, CDCl3): δ 7.81 (d, J = 8.0 Hz, 1H), 7.30 (t, J = 8.0 Hz, 1H), 6.76–6.65 (m, 2H), 4.01–3.56 (m, 5H), 2.59–2.46 (m, 2H), 2.10–2.06 (m, 1H), 2.03–1.65 (m, 2H); 13C NMR (CDCl3-d3, 100 MHz): δ 193.32 & 193.26, 150.86 & 150.34, 135.34 & 135.28, 127.48 & 127.42, 119.20 & 119.00, 118.23 & 118.12, 115.88 & 115.78, 70.81 & 70.12, 68.26 & 68.14, 56.53 & 56.12, 43.16 & 42.95, 42.66 & 42.61, 29.65 & 29.31; LC-MS (ESI): m/z calcd. for C13H15NO2: 217.110, found 218.08 [M + H]+.
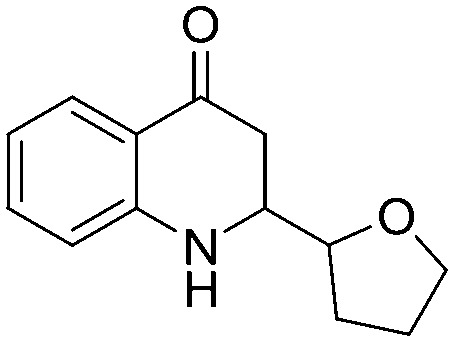
4.1.3.21. 2-(Tetrahydrofuran-2-yl)-2,3-dihydroquinolin-4(1H)-one (5u)
IR (KBr, cm–1): 3435 (NH), 1662 (CO); 1H NMR (400 MHz, CDCl3): δ 7.82–7.79 (m, 1H), 7.33–7.27 (m, 1H), 6.74–6.67 (m, 2H), 3.98–3.56 (m, 4H), 2.55–2.17 (m, 2H), 2.08–1.65 (m, 4H); LC-MS (ESI): m/z calcd. for C13H15NO2: 217.110, found 218.13 [M + H]+.
4.1.3.22. 2-Butyl-6-methoxy-2,3-dihydroquinolin-4(1H)-one (5v)
IR (KBr, cm–1): 3317 (NH), 1636 (CO); 1H NMR (400 MHz, CDCl3): δ 7.77 (d, J = 8.4 Hz, 1H), 6.32 (dd, J = 2.0, 8.4 Hz, 1H), 6.08 (d, J = 2.0 Hz, 1H), 3.77 (s, 3H), 3.64–3.57 (m, 1H), 2.62 (dd, J = 4.0, 8.0 Hz, 1H), 2.47–2.39 (m, 1H), 1.66–1.25 (m, 6H), 0.97–0.86 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 192.71, 165.42, 153.34, 129.70, 113.56, 106.65, 98.12, 55.30, 53.44, 43.67, 34.95, 27.49, 22.59, 13.93; LC-MS (ESI): m/z calcd. for C14H19NO2: 233.14, found 234.17 [M + H]+.
4.1.3.23. 2-Butyl-7-fluoro-2,3-dihydroquinolin-4(1H)-one (5w)
MR: 90–93 °C; IR (KBr, cm–1): 3347 (NH), 1645 (CO); 1H NMR (300 MHz, CDCl3): δ 7.83 (t, J = 8.7 Hz, 1H), 6.46–6.39 (m, 1H), 6.30 (d, J = 10.2 Hz, 1H), 4.42 (s, br, NH, 1H), 3.67–3.58 (m, 1H), 2.70–2.63 (m, 1H), 2.51–2.41 (m, 1H), 1.64–1.58 (m, 2H), 1.38–1.30 (m, 4H), 0.95–0.91 (m, 3H); 13C NMR (75 MHz, CDCl3): δ 192.63, 167.42 (d), 153.08 (d), 130.51 (d), 115.95, 106.26 (d), 101.26 (d), 53.30, 43.53, 34.74, 27.42, 22.54, 13.92; LC-MS (ESI): m/z calcd. for C13H16FNO: 221.12, found 222.12 [M + H]+.
4.2. Biology (α-glucosidase enzyme inhibition assay)
4.2.1. Test concentration
Stock solutions (10 mM) of the standards and each test substance were prepared in 100% dimethyl sulfoxide (DMSO, Sigma Chemical Co., St. Louis, MO) and further diluted with PBS to obtain experimental concentrations of 1–0.25 mM (also 0.125 and 0.062 for the potent compounds).
4.2.2. Protocol for α-glucosidase enzyme assay
An inhibitory activity assay procedure was performed by optimizing the previously reported method (Damsud et al., 2013).14 The α-glucosidase enzyme (0.1 U ml–1) was dissolved in 1 mM phosphate buffer (pH 6.9) and was freshly prepared every time. The initial step was performed by mixing 10 μL of the test sample with 40 μL of the prepared enzyme solution and the mixture was incubated for 15–20 min at 37 °C. Then 40 μL of p-nitrophenyl-α-d-glycopyranoside (PNPG) substrate (0.1 mM) was added and the mixture was incubated for 45–60 min. The release of p-nitrophenol by quenching was determined with the addition of 100 μL of 0.1 M Na2CO3. Further, the enzymatic activity was spectrophotometrically quantified in proportion to the level of p-nitrophenol with the absorbance reading at 415 nm. A blank reading was taken without the test compound and with the enzyme and substrates. Acarbose, voglibose and miglitol were used as positive controls.
4.2.3. Data interpretation
The readings taken at 415 nm were further evaluated for their percentage inhibition of the test compounds (Table 3). The equation used was: ((Ablank – Asample)/Ablank) × 100
The IC50 values were calculated by plotting graphs with the percentage inhibition on the y-axis and log concentrations on the x-axis using GraphPad Prism v6.0. The IC50 values are reported as the best fit values after normalization of data (n = 3).
Conclusions
In conclusion, we have developed an efficient methodology by employing acid catalysts for the synthesis of aza-flavanones, evaluated their α-glucosidase inhibition activity and applied molecular docking studies. The aza-flavanone derivatives 5a, 5g, 5h, 5r, and 5w were identified as potent α-glucosidase inhibitors compared to reference standards. Particularly compound 5g displayed a 6–8 fold higher activity compared to reference standards. This indicates that this particular library may be a valuable addition to understand numerous cellular processes involving α-glucosidase inhibition. Based on the present results, these derivatives can be taken forward to assist in accelerating the drug discovery of anti-diabetics.
Conflict of interest
The authors declare no competing interest.
Supplementary Material
Acknowledgments
The authors wish to sincerely thank GVK Biosciences Pvt. Ltd., for the Higher Education Program, financial support and encouragement. Help from the analytical department for all spectral analysis is highly appreciated. We are thankful to Dr. Sudhir Kumar Singh for his immense support and motivation. Dr Bathini and Dileep are thankful to the Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers (Govt. of India). We are also thankful to JNTUH, Hyderabad and BITS-Pilani, Hyderabad.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c7md00162b
References
- (a) Dabhi A. S., Bhatt N. R., Shah M. J. J. Clin. Diagn. Res. 2013;7:3023–3027. doi: 10.7860/JCDR/2013/6373.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Delgado H., Lehmann T., Bobbioni-Harsch E., Ybarra J., Golay A. Diabetes Metab. 2002;28:195–200. [PubMed] [Google Scholar]
- (a) Seo W. D., Kim J. H., Kang J. E., Ryu H. W., Curtis-Long M. J., Lee H. S., Yang M. S., Park K. H. Bioorg. Med. Chem. Lett. 2005;15:5514–5516. doi: 10.1016/j.bmcl.2005.08.087. [DOI] [PubMed] [Google Scholar]; (b) Chang C. C., Ho S. L., Lee S. S. Bioorg. Med. Chem. 2015;23:3383–3396. doi: 10.1016/j.bmc.2015.04.053. [DOI] [PubMed] [Google Scholar]
- (a) Ma D., Xia C., Jiang J., Zhang J., Tang W. J. Org. Chem. 2003;68:442–451. doi: 10.1021/jo026125z. [DOI] [PubMed] [Google Scholar]; (b) Ma D., Xia C., Jiang J., Zhang J. Org. Lett. 2001;3:2189–2191. doi: 10.1021/ol016043h. [DOI] [PubMed] [Google Scholar]
- Kawaii S., Endo K., Tokiwano T., Yoshizawa Y. Anticancer Res. 2012;32:2819–2826. [PubMed] [Google Scholar]
- (a) Hradil P., Hlavac J., Soural M., Hajdúch M., Kolar M., Vecerova R. Mini-Rev. Med. Chem. 2009;9:696–708. doi: 10.2174/138955709788452720. [DOI] [PubMed] [Google Scholar]; (b) Zhang S. X., Feng J., Kuo S. C., Brossi A., Hamel E., Tropsha A., Lee K. H. J. Med. Chem. 2000;43:167–177. doi: 10.1021/jm990333a. [DOI] [PubMed] [Google Scholar]; (c) Xia Y., Yang Z. Y., Xia P., Bastow K. F., Tachibana Y., Kuo S. C., Hamel E., Hackl T., Lee K. H. J. Med. Chem. 1998;41:1155–1163. doi: 10.1021/jm9707479. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar S., Pushpavalli S. N., Chatla S., Mukhopadhyay D., Ganganna B., Vijeender K., Srihari P., Reddy C. R., Ramaiah M. J., Bhadra U. Bioorg. Med. Chem. Lett. 2012;22:645–648. doi: 10.1016/j.bmcl.2011.10.061. [DOI] [PubMed] [Google Scholar]
- Evans B., Rittle K., Bock M., DiPardo R., Freidinger R., Whitter W., Lundell G., Veber D., Anderson P. J. Med. Chem. 1988;31:2235–2246. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- Ahmed N., van Lier J. E. Tetrahedron Lett. 2006;47:2725–2729. [Google Scholar]
- Ahmed N., van Lier J. E. Tetrahedron Lett. 2007;48:13–15. [Google Scholar]
- Chelghoum M., Bahnous M., Bouraiou A., Bouacida S., Belfaitah A. Tetrahedron Lett. 2012;53:4059–4061. [Google Scholar]
- Chandrasekhar S., Vijeender K., Sridhar C. Tetrahedron Lett. 2007;48:4935–4937. [Google Scholar]
- Pandit R. P., Sharma K., Lee Y. R. Synthesis. 2015;47:3881–3890. [Google Scholar]
- Chopade P. R., Davis T. A., Prasad E., Flowers R. A. Org. Lett. 2004;6:2685–2688. doi: 10.1021/ol049129u. [DOI] [PubMed] [Google Scholar]
- Damsud T., Adisakwattana S., Phuwapraisirisan P. Phytochem. Lett. 2013;6:350–354. [Google Scholar]
- Schrödinger Release 2014-1: Maestro, version 9.7, Schrödinger, LLC, New York, NY, 2014.
- Yamamoto K., Miyake H., Kusunoki M., Osaki S. FEBS J. 2010;277:4205–4214. doi: 10.1111/j.1742-4658.2010.07810.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.