

 In this review, we have focused on the recent progress in metal complexes that are able to bind to β-amyloid (Aβ) species.
In this review, we have focused on the recent progress in metal complexes that are able to bind to β-amyloid (Aβ) species.
Abstract
In this review, we have focused on the recent progress in metal complexes that are able to bind to β-amyloid (Aβ) species. We have discussed various radioactive complexes of 99mTc, 68Ga, 64Cu, 89Zr, and 111In, which were designed as Aβ imaging agents for positron emission tomography (PET) and single photon emission computed tomography (SPECT) imaging, non-radioactive Re and Ru complexes as Aβ sensors using luminescence methods, and Gd3+ complexes as contrast agents for magnetic resonance imaging (MRI).
1. Introduction
Alzheimer's disease (AD) is a neurodegenerative brain disease that leads to synaptic reduction and cognitive decline. It is the most common cause of dementia and may eventually lead to death among elderly patients.1 Cerebral amyloid angiopathy (CAA) is a disorder that has been recognized as an important cause of lobar intracerebral hemorrhage (ICH), microbleeds (MBs), ischemic stroke, and dementia.2,3 It has been estimated that CAA occurs in up to 80% of patients with AD; approximately 25% of these patients experienced severe CAA.4–6 According to the amyloid cascade hypothesis, the mismetabolism of the amyloid precursor protein (APP) is the initial event in the pathogenesis of AD, which subsequently leads to the aggregation of the β-amyloid (Aβ) protein, specifically Aβ1–40/42.7,8 Histopathological studies have shown extensive Aβ deposition in the cerebral cortex in the post-mortem analysis of patients with AD, while Aβ deposition was mainly observed in the walls of the leptomeningeal and cortical arterioles of patients with CAA.9,10 The in vivo imaging of the deposition of Aβ plaques in the brain has been accepted as a useful tool in the study of the pathophysiology of neurodegenerative diseases associated with misfolded Aβ species.11 Therefore, it is of great importance to precisely detect Aβ in the preclinical diagnosis of AD and CAA with the use of noninvasive imaging techniques.12,13 To achieve this, various imaging modalities, including nuclear imaging techniques, such as positron emission tomography (PET) and single photon emission computed tomography (SPECT), magnetic resonance imaging (MRI), and optical imaging techniques, for example near-infrared (NIR) imaging methods, have been employed.14,15 In the past decades, many radiolabeled tracers aimed at the detection of Aβ in live patients with AD have been developed and evaluated. 2-(4-[11C]Methylaminophenyl)-6-hydroxybenzothiazole ([11C]PIB, 1)16–18 is the most commonly used imaging agent for PET imaging. [18F]-Flutemetamol ([18F]GE-067, 2),19,20 [18F]florbetapir ([18F]AV-45, 3),21–23 and [18F]florbetaben ([18F]BAY-94-9172, 4)24,25 were successively approved by the FDA (Fig. 1). However, noninvasive methods for the selective and accurate detection and quantification of the Aβ burden of CAA have not yet been established. The study of CAA imaging agents is still in an early stage, with only a few reported so far; examples include the 18F-polypegylated styrylpyridines (5–7) reported by Kung et al.26 and the fluorescent resorufin analogs (8–10) reported by Han et al. (Fig. 1).27
Fig. 1. Chemical structures of Aβ imaging probes designed for AD and CAA.

Metal complexes, which contain radionuclides, such as 99mTc, 68Ga, 64Cu, 89Zr, and 111In, can be utilized as PET or SPECT Aβ imaging probes. Non-radioactive metal complexes, such as phosphorescent rhenium and ruthenium complexes, which have good photophysical properties, can be used as non-conventional probes in the fluorescence imaging of amyloid formation, while gadolinium complexes can be used as MRI imaging agents for Aβ deposits. In 2014, a detailed review of metal complexes designed to bind to Aβ for the diagnosis and treatment of AD was conducted by Donnelly et al.28 However, in the intervening three years, many meaningful metal complexes have been developed for Aβ detection. Thus, this review will discuss the metal complexes designed and evaluated for the detection of Aβ deposits in AD or CAA that were developed between 2014 and 2017.
2. Radioactive metal complexes as Aβ imaging probes
2.1. 99mTc-labeled complexes for SPECT imaging
Despite the rapid increase in the number of hospitals equipped with the requisite infrastructure for PET, SPECT is still the most commonly used nuclear imaging technique. Currently, the 99mTc isotope is the most widely used radionuclide in medical diagnostic procedures using SPECT, owing to its good radioactive characteristics, such as the emission of readily detectable gamma rays (140 keV), an appropriate half-life (6.01 h), and easy production by 99mTc/99Mo generators. Thus, the development of new 99mTc-labeled imaging agents will provide simple, convenient, and widespread SPECT-based methods for the detection and quantitative analysis of Aβ plaques. All isotopes of technetium are radioactive. Rhenium is a congener of technetium in group VII and has a similar ionic radius to technetium. Thus, rhenium is always used to form analogs of 99mTc complexes for comprehensive characterization and preliminary in vitro assessments.29 Many 99mTc-labeled Aβ imaging agents have been previously reported.14,30 In the following section, we have described the significant progress towards 99mTc complexes for Aβ plaque imaging published from 2014 onwards. In order to incorporate the transition metal 99mTc into a small organic molecule, a chelating structure is essential. Two common cores, [99mTcO]3+ and [99mTc(CO)3]+, are described in this review.
2.1.1. [99mTcO]3+ complexes with tetradentate chelators
The [99mTcO]3+ core forms square pyramidal oxo-technetium complexes with multiple tetradentate chelators, such as N2S2, N2O2, N3S, and N3O bifunctional chelators (Fig. 2).31,32
Fig. 2. Chemical structures of bifunctional chelators used for 99mTc-labeled Aβ imaging probes.

The tetradentate N2S2 chelators, which include bis-aminoethanethiol (BAT) and monoamine-monoamidedithiol (MAMA), can easily form relatively small, stable, and neutral complexes with the [99mTcO]3+ core. Based on the conjugate approach, various [99mTcO]3+ complexes, such as Congo red, chrysamine G,33,34 phenylbenzothiazole,35–37 phenylbenzoxazole,38 phenylbenzofuran,39,40 flavone,41 aurone,41 chalcone,42 curcumin,43 naphthalene,44 dibenzylideneacetone45 and stilbene46 coupled with MAMA and BAT bifunctional chelators have been reported.
Cui et al. reported two novel uncharged 99mTc/Re-labeled 2-phenylbenzoxazole derivatives conjugated with BAT and MAMA chelators via a pentyloxy spacer (11 and 12, Fig. 3).38 Both compounds displayed a high binding affinity for Aβ1–42 (11 and 12, Ki = 11.1 and 14.3 nM, respectively). According to the biological evaluation, the TcOBAT complex (11, Table 1) has advantages over the TcOMAMA complex (12, Table 1) as an Aβ targeting probe, with a higher initial brain uptake (0.81% ID g–1vs. 0.43% ID g–1 at 2 min) and a faster wash-out rate (0.25% ID g–1vs. 0.30% ID g–1 at 60 min) from the brain of normal mice. Additionally, four new 2-arylbenzoxazole derivatives conjugated with the BAT chelator via a short propoxy spacer were reported ([99mTc]13–16, Fig. 3, Table 1).47 All rhenium analogs (13–16) clearly stained Aβ plaques in brain sections from transgenic (Tg) mice and patients with AD. The in vitro competition binding assay revealed that the N,N-dimethylated complexes exhibited a higher affinity to Aβ1–42 aggregates (15 and 16, Ki = 15.9 and 37.2 nM, respectively) than the N-monomethylated complexes (13 and 14, Ki = 128.2 and 393.2 nM, respectively). The affinity of the phenyl derivatives (13 and 15) was higher than that of the pyridyl analogs (14 and 16). In addition, the phenyl derivatives [99mTc]13 and [99mTc]15 displayed higher initial brain uptake (1.10 and 1.55% ID g–1 at 2 min) than the pyridyl derivatives [99mTc]14 and [99mTc]16 (0.96 and 1.24% ID g–1 at 2 min). Among these 99mTc-labeled 2-arylbenzoxazole derivatives, [99mTc]15 displayed higher initial brain uptake (1.55% ID g–1 at 2 min), but a comparable brain2min/brain60min ratio (3.38). However, the blood–brain barrier (BBB) penetrability of [99mTc]15 is still lower than ideal and requires further modifications. In addition, two stereoisomers were generated when the [99mTcO]3+ core coordinated with MAMA or BAT chelators. The X-ray crystallography of the rhenium complex 13 revealed that the syn isomer was the major product.
Fig. 3. Chemical structures of 99mTc/Re-labeled 2-arylbenzoxazoles and 2-arylbenzothiazoles conjugated with BAT and MAMA chelators.

Table 1. Binding affinities and brain pharmacokinetics of [99mTcO]3+ complexes with tetradentate chelators.
| No. | Aβ1–42Ki (nM) | Aβ1–40Ki (nM) | Brain uptake (% ID g–1) |
Ref. | |
| 2 min | 60 min | ||||
| 11 | 11.1 | — | 0.81 | 0.25 | 38 |
| 12 | 14.3 | — | 0.43 | 0.30 | 38 |
| 13 | 128.2 | — | 1.10 | 0.31 | 47 |
| 14 | 393.2 | — | 0.96 | 0.15 | 47 |
| 15 | 15.9 | — | 1.55 | 0.40 | 47 |
| 16 | 37.2 | — | 1.24 | 0.14 | 47 |
| 17 | 56.6 | — | — | — | 48 |
| 18 | 16.0 | — | 0.69 | 0.46 | 48 |
| 19 | 33.5 | — | 0.46 | 0.40 | 48 |
| 20 | 8.4 | — | 0.59 | 0.43 | 48 |
| 21 | 2303.0 | — | — | — | 48 |
| 22 | 8.8 | — | 2.11 | 0.62 | 48 |
| 23 | 104.9 | — | — | — | 48 |
| 24 | 29.4 | — | 0.92 | 0.63 | 48 |
| 25 | 105.6 | — | — | — | 48 |
| 26 | 134.1 | — | — | — | 48 |
| 27 | 17.6 | — | 0.47 | 0.19 | 48 |
| 28 | 24.4 | — | 0.60 | 0.29 | 48 |
| 29 | 2356.2 | — | — | — | 48 |
| 30 | 93.6 | — | — | — | 48 |
| 31 | 440.9 | — | — | — | 48 |
| 32 | 75.4 | — | — | — | 48 |
| 33A | 0.72 a | 0.38 a | 0.37 | 0.08 | 52, 53 |
| 33B | 0.38 a | 0.45 a | — | — | 52, 53 |
| 34A | 16.40 a | 4.59 a | 0.28 | 0.11 | 52, 53 |
| 34B | 2.55 a | 3.37 a | — | — | 52, 53 |
| 35A | 0.26 a | 0.24 a | — | — | 52, 53 |
| 35B | 0.47 a | 0.99 a | 0.36 | 0.08 | 52, 53 |
| 36A | 2.80 a | 1.58 a | — | — | 52, 53 |
| 36B | 5.78 a | 4.96 a | 0.37 | 0.05 | 52, 53 |
| 38 | 0.56 | — | 0.35 | 0.11 | 54 |
| 39 | 855 b | — | — | — | 55 |
aExpressed as IC50 values (μM) in the presence of PIB.
bMeasured using fluorescence competition assay against thioflavin-T (ThT).
To further improve the brain pharmacokinetics of 99mTc-labeled probes for Aβ plaques, Cui et al. reported the systematic study of a series of 2-arylbenzothiazole derivatives conjugated with the BAT chelator via different lengths of alkyl linkers (n = 3–6) (17–32, Fig. 3 and Table 1).48 Different aminomethyl groups and lengths of alkyl linkers were employed to balance the binding affinity and BBB permeability. Seven rhenium complexes with relatively longer alkyl chains (18–20, 22, 24, 27, and 28) exhibited a strong binding affinity toward Aβ1–42 aggregates (Ki < 50 nM). These compounds were selected for 99mTc-labeling and further evaluation. All of the 99mTc-labeled complexes displayed good affinity and specificity to Aβ plaques in Tg mouse brain tissue in the in vitro autoradiography studies. Among them, [99mTc]22 showed both a high binding affinity (Ki = 8.8 nM) and favorable brain pharmacokinetics in normal mice (2.11% ID g–1 at 2 min and 0.62% ID g–1 at 60 min). Ex vivo autoradiography revealed that [99mTc]22 could penetrate the BBB and intensely label Aβ plaques in the brain of Tg mice. Thereafter, [99mTc]22 was selected as the first 99mTc-labeled Aβ imaging probe for SPECT/CT imaging studies in nonhuman primates. The semiquantitative data and SPECT images revealed that [99mTc]22 could penetrate the brain of rhesus monkeys to an average degree of one percent of the injected dose. All these results indicated that [99mTc]22 could serve as a potential SPECT imaging probe for Aβ plaques in AD.
However, ligands containing N2S2 chelators are air-sensitive and easily oxidized, which makes them difficult to form in a radiopharmaceutical kit. The thiol-free hydroxamamide (Ham) chelator is known to generate 99mTc-labeled complexes with high stability and high radiochemical yield under mild conditions (room temperature and neutral medium).49–51 In addition, Ham is a bidentate chelator, which may enhance the binding affinity of the bidentate complexes to Aβ plaques.
Recently, Ono et al. designed and synthesized five bidentate 99mTc–Ham complexes conjugated with the classical Aβ binding motifs of stilbene and 2-phenylbenzothiazole ([99mTc]33–36, Fig. 4 and Table 1) and dimethylaminobenzene ([99mTc]37, Fig. 4).52 According to the results for the reaction mixture analyzed by radio-HPLC, two radioactive isomers were produced in the 99mTc complexation reaction with Ham. They defined the shorter retention time isomers as either A-form ([99mTc]33A, [99mTc]34A, [99mTc]35A, [99mTc]36A, and [99mTc]37A) or B-form ([99mTc]33B, [99mTc]34B, [99mTc]35B, [99mTc]36B, and [99mTc]37B). The in vitro inhibition assay indicated that the bivalent 99mTc–Ham complexes ([99mTc]34A, [99mTc]34B, [99mTc]36A, and [99mTc]36B) displayed much higher binding affinities for amyloid aggregates than the monovalent complexes ([99mTc]33A, [99mTc]33B, [99mTc]35A, and [99mTc]35B). The high binding affinity of these 99mTc–Ham complexes was further confirmed by in vitro autoradiography studies using brain sections from Tg2576 mice. In addition, significant differences between the two isomers were observed in their binding ability; [99mTc]37 with two dimethylaminobenzene substituents did not bind to amyloid aggregates. In biodistribution studies, [99mTc]34A, which had the highest binding affinity, displayed poor brain uptake (0.28% ID g–1 at 2 min) in normal mice, which indicated that these 99mTc–Ham complexes were unable to penetrate the BBB and were thus unsuitable for application to AD diagnosis. However, they may be used as imaging agents for the diagnosis of CAA.
Fig. 4. Chemical structures of 99mTc–Ham complexes with bivalent amyloid ligands.

Thereafter, these 99mTc–Ham complexes were evaluated as CAA-specific imaging probes.53 Considering that Aβ1–40 is more abundant in the cerebrovascular amyloid of CAA, an in vitro binding study was performed using Aβ1–40 aggregates. Similar to the results using Aβ1–42 aggregates, dimer complexes displayed stronger binding than monomer complexes. The high binding ability was further verified by in vitro autoradiography studies using human CAA brain sections from two different patients; both [99mTc]34A and [99mTc]36B displayed intensive labeling of Aβ deposits. Finally, the specific binding of [99mTc]34A and [99mTc]36B to the cerebrovascular amyloid was verified by ex vivo autoradiography studies using Tg2576 mice; the complexes displayed low brain entry and selective binding to vascular Aβ deposits in the model mice, but not in the wild-type (WT) mice. These results indicated that the bivalent 99mTc–Ham complexes, [99mTc]34A and [99mTc]36B, could specifically detect CAA in vivo. However, two geometric isomers of these 99mTc–Ham complexes (A-form and B-form) were observed in the 99mTc-labeling reaction, which might complicate their clinical application.
To avoid the formation of isomers, an N-methyl-substituted Ham chelator conjugated with 2-phenylbenzothiazole was designed and evaluated.54 A single 99mTc–Ham complex, [99mTc]38, was obtained with notable stability. Although [99mTc]38 displayed lower binding affinity for Aβ aggregates (IC50 = 0.56 μM) than the parent 99mTc–Ham complex ([99mTc]38B, IC50 = 5.78 μM), specific labeling of the Aβ deposits was still observed in the in vitro autoradiography studies of brain sections from Tg2576 mice and patients with CAA. Similar to other 99mTc–Ham complexes, [99mTc]38 displayed very low brain uptake (0.35% ID g–1 at 2 min), which was favorable for CAA-specific imaging.
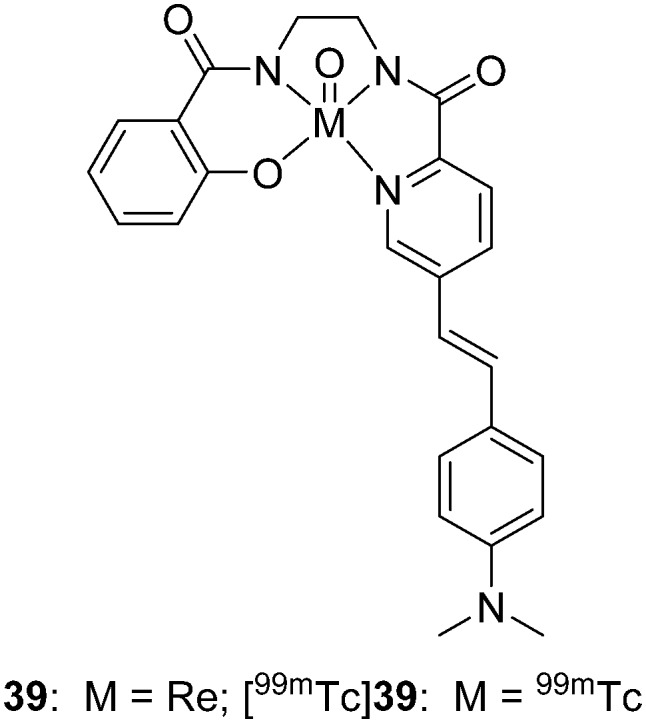
Donnelly et al. designed new non-thiol-based semirigid tetradentate hybrid N3O chelating agents to form neutral lipophilic complexes with oxotechnetium(v) and oxorhenium(v).55 Without the thiol group and tertiary amines, two advantages, antioxidation and the preclusion of the formation of syn and anti diastereomers, were achieved by this N3O chelator. The 99mTc/Re complex (39 and [99mTc]39, Fig. 5, Table 1) with a styrylpyridyl Aβ binding group was prepared. However, the binding affinity of 39 for Aβ1–42 fibrils was relatively low (Ki = 855 nM), which was unfavorable as an amyloid imaging agent. However, further biological evaluation, including autoradiography and in vivo brain penetration tests, of [99mTc]39 was not reported.
Fig. 5. Chemical structure of 99mTc/Re-labeled styrylpyridyl conjugated with the N3O chelator.
2.1.2. [99mTc(CO)3]+ complexes with various chelators
In recent years, [99mTc(CO)3]+ has been a popular core for the development of organometallic radiopharmaceuticals.56 The three water molecules in the [99mTc(CO)3(H2O)3]+ intermediate could be easily replaced by tridentate and cyclopentadiene chelators to generate charged or neutral complexes with high yield and stability.57,58
Tridentate chelators, containing pyridines, amines, and carboxylate donors conjugated to [M(CO)3]+, have rapid kinetics and good stability in aqueous solution, which allow the convenience of a kit formulation. The most commonly used tridentate chelators are picolylamine monoacetic acid (PAMA), bis(pyridin-2-ylmethyl)amine (DPA), and iminodiacetic acid (IDA), which generate 99mTc/Re(CO)3 complexes with neutral, positive, and negative charges, respectively (Fig. 2).59
Satpati et al. reported a neutral 2-phenylbenzothiazole-based 99mTc(CO)3 complex ([99mTc]40, Fig. 6) using the tridentate chelator PAMA with high radiochemical yield (95%). The total binding of [99mTc]40 to Aβ1–42 aggregates was calculated to be 4.6 ± 0.3%, and the binding was inhibited (26.7 ± 0.6%) by the addition of excessive ThT. In the in vivo studies, [99mTc]40 displayed low brain uptake (0.25 ± 0.04% ID g–1 at 2 min, Table 2), which was unfavorable as an imaging agent for the diagnosis of AD.60
Fig. 6. Chemical structures of [M(CO)3]+ (M = 99mTc or Re) complexes with tridentate chelators.

Table 2. Binding affinities and brain pharmacokinetics of [CpM(CO)3] complexes.
| No. | Aβ1–42Ki (nM) | Brain uptake (% ID g–1) |
Ref. | |
| 2 min | 60 min | |||
| 40 | — | 0.25 | 0.11 | 60 |
| 42 | — | 0.25 a | 0.21 a , c | 61 |
| — | 0.24 b | 0.19 b , c | 61 | |
| 45 | 162 | — | — | 62 |
| 46 | 37 | 0.18 | 0.08 | 62 |
| 47 | 366 | — | — | 62 |
| 48 | 78 | 0.24 | 0.10 | 62 |
| 49 | 106.0 | — | — | 63 |
| 50 | 45.3 | 0.80 | 0.69 | 63 |
| 51 | 46.0 | — | — | 63 |
| 52 | 61.9 | 0.61 | 0.24 | 63 |
| 53 | 85.1 | — | — | 63 |
| 54 | 50.8 | 0.88 | 0.15 | 63 |
| 55 | 56.6 | — | — | 63 |
| 56 | 42.2, 37.0, d 64.5 e | 1.21 | 0.06 | 63 |
| 57 | 142.6 | 0.50 | 0.18 | 64 |
| 58 | 75.8 | 0.36 | 0.19 | 64 |
| 59 | 64.1 | 0.26 | 0.11 | 64 |
| 60 | 24.0 | 0.37 | 0.14 | 64 |
| 61 | 18.8 | 1.06 | 0.39 | 65 |
| 62 | 22.8 | — | — | 65 |
| 63 | 12.4 | 0.70 | 0.17 | 65 |
| 64 | 22.3 | — | — | 65 |
| 65 | 204.1 | — | — | 65 |
| 66 | 42.0 | 0.69 | 0.31 | 65 |
| 67 | 130.6 | — | — | 65 |
| 68 | 15.1 | 0.54 | 0.10 | 65 |
aThe brain uptake was measured in WT mice.
bThe brain uptake was measured in 10 month-old APP/PS1 Tg mice.
cExpressed as brain uptake determined at 30 min post-injection.
dExpressed as IC50 values (nM) for Aβ1–40 aggregates.
eExpressed as IC50 values (nM).
Donnelly et al. synthesized two pyridylamine-carboxylate tridentate ligands and two dipyridylamine ligands conjugated with stilbene-like Aβ binding motifs via a short alkyl linker.61 In the 99mTc-labeling reaction, the carboxylate-containing ligands displayed a higher radiochemical yield (>98%) than the dipyridylamine ligands (40–50%). The binding affinity of the rhenium complexes to the Aβ plaques was measured by fluorescence staining in human brain tissue. Complexes 41 and 43 resulted in almost no staining of the Aβ plaques in brain tissues from patients with AD, whereas complexes 42 and 44, with a dimethylamino group, were more promising. The neutral complex [99mTc]42 was much more stable than the cationic complex [99mTc]44, and remained stable in human serum at 37 °C for over 3 h. Therefore, [99mTc]42 was selected for further evaluation; the in vivo biodistribution studies in Tg mice and WT mice indicated that [99mTc]42 hardly crossed the BBB (0.25% ID g–1 for WT and 0.24% ID g–1 for Tg at 2 min post-injection) and no statistically significant difference between WT and Tg mice was observed at either 2 min or 30 min post-injection. The micro-SPECT studies of [99mTc]42 in Tg mice further confirmed its low brain uptake.
As described above, probes with high affinity to amyloid plaques and low BBB penetration can be used as CAA-specific imaging agents. The decreased brain uptake may help to distinguish Aβ deposits between the cerebral vessels and brain parenchyma. For decreased brain uptake, the charged 99mTc/Re(CO)3 complexes with the tridentate chelators, DPA and IDA, are a good choice.
Cui et al. reported the synthesis and characterization of four 99mTc/Re-labeled 2-phenylbenzothiazole and stilbene complexes with a relatively large DPA chelator through a conjugate approach (Fig. 6).62 The positively charged complexes were considered to have poor BBB penetrability and could specifically target Aβ plaques located on the cerebral vessel walls. Complexes with a five-carbon linker exhibited higher binding affinities (46 and 48, Ki = 37 and 78 nM, respectively, Table 2) than those with a three-carbon linker (45 and 47, Ki = 162 and 366 nM, respectively, Table 2), which suggested that the appropriate length of the carbon linker reduced the steric hindrance imposed by the bulky metal chelate core on the binding motifs. Under fluorescence staining, the rhenium complexes 46 and 48 intensely labeled the Aβ plaques associated with the blood vessels as well as in the parenchyma of brain tissue from patients with AD. As expected, reduced brain uptake was observed for [99mTc]46 and [99mTc]48 (0.18 and 0.24% ID g–1 at 2 min, respectively). However, further in vitro and ex vivo autoradiography studies were not reported.
Then, the same group developed a series of 2-phenylbenzothiazole derivatives conjugated with the chelator IDA via alkyl linkers with different lengths (n = 3–6) as negatively charged probes to detect cerebrovascular Aβ deposition.63 The rhenium surrogates 49–56 strongly stained Aβ deposits in Tg mice and AD patients, and also displayed high affinities to Aβ aggregates (Ki = 42.2–106.0 nM, Table 2). In particular, 56, with the longest carbon linker (n = 6), displayed the highest affinity toward Aβ1–42 aggregates (Ki = 42.2 nM). In addition, slightly higher binding affinities of 56 and [99mTc]56 toward aggregates of Aβ1–40 than those toward Aβ1–42 were observed in both the inhibition assay (37.0 nM vs. 64.5 nM) and the direct binding assay (10.15% vs. 6.48%). In vitro autoradiography of brain sections from Tg mice, patients with AD, and patients with CAA confirmed that [99mTc]56 possessed sufficient affinity for Aβ plaques. More interestingly, [99mTc]56 selectively labeled Aβ deposits in blood vessels, but not Aβ plaques in the parenchyma of brain tissue from AD patients, which is highly favorable for CAA-specific imaging (Fig. 7). This was the first report of a 99mTc-labeled tracer that displayed positive autoradiography results and selectively detected Aβ within the blood vessels in brain sections of patients with AD. In the biodistribution studies, the 99mTc(CO)3 complexes displayed relatively low initial brain uptake (0.61–1.21% ID g–1 at 2 min post-injection). The ex vivo autoradiography studies in Tg and WT mice further confirmed the potential of [99mTc]56 to selectively recognize Aβ deposits located in cerebral arterioles.
Fig. 7. (A) In vitro autoradiography of [99mTc]56 and [125I]IMPY on contiguous brain sections of a patient with AD. Aβ deposits within the blood vessels were indicated by triangles and Aβ plaques in the cerebral parenchyma were indicated by arrows. (B) Fluorescence staining using thioflavin-S (ThS) on the same brain sections. Adapted from ref. 63.

Cyclopentadienyl tricarbonyl complexes ([CpM(CO)3], M = Re, 99mTc) possess many attractive properties such as small size, low molecular weight, moderate lipophilicity, and high stability,58,66 which are highly favorable for BBB penetration and may be applied as Aβ imaging agents for the diagnosis of AD. Cui et al. synthesized and evaluated a series of 2-arylbenzothiazole derivatives conjugated with [CpM(CO)3] (M = 99mTc, Re) via amide and ester linkers that contained carbon chains with different lengths (n = 3, 5) (Fig. 8, Table 2).64,65
Fig. 8. Chemical structures of 2-arylbenzothiazole derivatives conjugated with the [CpM(CO)3] core.

The rhenium complexes 57–68 displayed moderate to high affinity for Aβ1–42 aggregates (Ki = 12.4–204.1 nM); in general, the tertiary N,N-dimethylamino substituents and relatively longer carbon linkers were beneficial for the retention of high Aβ affinity. In the in vitro fluorescence staining studies, these rhenium complexes displayed intense staining of Aβ deposits in both the cerebral cortex and the cortical vasculature of brain sections from Tg mice and patients with AD or CAA. Moreover, neurofibrillary tangles were also stained by these rhenium complexes. In the biodistribution studies, all the 99mTc complexes displayed poor initial brain uptake (0.26–1.06% ID g–1), which impeded their application as Aβ imaging agents for the diagnosis of AD. However, some of them were selected and evaluated as CAA imaging agents. In the in vitro autoradiography studies on brain sections from AD patients, [99mTc]68 selectively bound to the cerebrovascular Aβ deposits, but not the Aβ plaques, in the parenchyma, which is highly favorable for CAA imaging agents (Fig. 9).
Fig. 9. In vitro autoradiography of [99mTc]68 binding in contiguous brain sections from a patient with AD (68 years old, female, frontal lobe) (A and C) and the same section stained with ThS (GFP filter) (B). Higher magnification images of A and the corresponding higher magnification images stained with ThS (D). Adapted from ref. 64.

To broaden the scope of potential 99mTc(CO)3-based diagnostic imaging agents for AD, Barnard et al. synthesized a series of Re+ tricarbonyl analogs of novel acyclic and macrocyclic tridentate NHC (N-heterocyclic carbene) ligands coupled to stilbene- or benzothiazole-based amyloid binding groups (Fig. 10).67 Two linkage isomers will be formed when the acyclic ligands are used (69a, b and 70a, b), while the macrocyclic ligands produce single complexes (71 and 72). The ThT fluorescence assay of 69a, 70a, 71, and 72 to synthetic Aβ1–42 fibrils indicated that the they could bind competitively with ThT to Aβ1–42 fibrils or that they inhibited the formation of Aβ1–42 fibrils. Then, the binding capacity to Aβ plaques of complexes 70a and 71 was evaluated in human AD brain tissues. Although complex 70a showed no binding to Aβ plaques, complex 71 displayed co-localization with immunostaining, with a low background. In addition, complex 71 did not show non-specific binding to age-matched controls, according to the epifluorescence images. Thus, the NHC ligands offer possibilities for medicinal inorganic and radiopharmaceutical applications.
Fig. 10. Chemical structures of Re(CO)3–NHC complexes.

2.2. 68Ga-labeled complexes for PET imaging
Among the metal-based radionuclides for PET, gallium-68 (t1/2 = 1.13 h, β+ = 89%) is a prospective radioisotope for PET imaging. The commercial availability of 68Ga from a 68Ge/68Ga generator without an on-site cyclotron permits simple and easy preparation. Moreover, the radiolabeling procedure for 68Ga is quick and simple, requiring a single vial kit solution, and the coordination properties and features of the trivalent gallium are well known and can be easily applied to the development of radiotracers. Although 99mTc is the most widely used metal-based radionuclide for SPECT imaging, in recent years, attention has been increasingly focused on innovative biomolecules labeled with 68Ga for PET imaging.68,69
The most commonly designed chelator for 68Ga3+ is hexadentate, which is favorable for bifunctional conjugation through covalent linkage. When considering the thermodynamic stability of the 68Ga3+ complex, the best candidates are linear or macrocyclic polyamines modified with negatively charged pendant arms, such as 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA), 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA), diethylenetriaminepentaacetic acid (DTPA), and N,N′-bis[2-hydroxy-5-(carboxyethyl)benzyl]ethylenediamine-N,N′-diacetic acid (HBED) (Fig. 11).70
Fig. 11. Chemical structures of commonly designed chelators for 68Ga3+.

In an attempt to develop efficient and fully automated radiosynthesis of 68Ga-labeled probes for Aβ plaques, Cressier et al. reported three uncharged 68Ga3+-labeled complexes ([68Ga]73, [68Ga]74, and [68Ga]75, Fig. 12) differing in the length of the spacer between the metal-complexing DOTA macrocycle and the peptide-recognizing PIB moiety.71 However, according to the biological data, these 68Ga3+-labeled complexes were not potential Aβ imaging probes, as they showed poor brain uptake ([68Ga]73 or [68Ga]74, 0.9–1.1% ID cc–1 at 2 min in both animal models; [68Ga]75, ∼1.3% ID cc–1 at 2 min in Tg mice, ∼0.5% ID cc–1 at 2 min in control mice) and low binding affinity (73 and 75, Kd = 170 and 67 μM, respectively) for Aβ deposits. In addition, these complexes failed to detect amyloid deposits through an autoradiographic approach with human brain tissues. Future investigations are needed to enhance the binding affinity and brain uptake of these 68Ga radiotracers.
Fig. 12. Chemical structures of nat/68Ga-labeled Aβ imaging agents.

Ono et al. reported a 68Ga-labeled 2-phenylbenzofuran complex conjugated with the DOTA chelator via a propoxy linker as an Aβ imaging probe ([nat/68Ga]76, Fig. 12 and Table 3).72 The complex [natGa]76 displayed high affinity for Aβ1–42 aggregates (Ki = 10.8 nM), which was comparable with other 99mTc/Re-labeled 2-arylbenzofuran complexes. As expected, clear staining of Aβ plaques was observed in brain sections from Tg2576 mice. However, [68Ga]76 showed poor initial brain uptake (0.45% ID g–1 at 2 min) in biodistribution experiments in normal mice, which may result from the low lipophilicity (log P = –0.38) and high molecular weight (MW = 706). Therefore, further improvement of the brain uptake appears to be essential for these DOTA-based 68Ga complexes.
Table 3. Binding affinities and brain pharmacokinetics of 68Ga, 64Cu, and 89Zr labeled small molecules and mAbs.
| No. | Aβ1–42Ki (nM) | Brain uptake (% ID g–1) |
Ref. | |
| 2 min | 60 min | |||
| 73 | 170 a | 0.9–1.1 b – d | — | 71 |
| 74 | — | 0.9–1.1 b – d | — | 71 |
| 75 | 67 a | 0.5, b , c 1.3 b , d | — | 71 |
| 76 | 10.8 | 0.45 | 0.27 | 72 |
| 77 | 4.2, 3.5 e | 1.24 | 0.20 | 75 |
| 78 | 30.6 f | 0.12 | 0.08 | 76 |
| 79 | 13.4 f | 0.17 | 0.04 | 76 |
| 80 | 18.1 f | 0.31 | 0.11 | 76 |
| 81 | 6.7 f | 0.21 | 0.11 | 76 |
| 82 | 10.7 f | 0.22 | 0.07 | 76 |
| 83 | 185 f | 0.11 | 0.03 | 76 |
| 88 | 33.7 | 0.33 | 0.18 | 81 |
| 89 | 243.5 | 0.36 | 0.21 | 81 |
| 90 | 0.85 g | — | 0.50 | 82 |
aExpressed as the Kd value (μM) for Aβ1–40 aggregates.
bExpressed as % ID cc–1.
cThe brain uptake was measured in control mice.
dThe brain uptake was measured in 18 month-old APP/PS1 Tg mice.
eExpressed as the Kd value (nM).
fDetermined using human AD brain homogenates.
gExpressed as the Kd value (nM) for Aβ1–40 aggregates.
Because of the short half-life of 68Ga and the demand for protein and biomolecule labeling, rapid complexation kinetics and mild conditions are favored. However, 68Ga complexation using the DOTA chelator requires a longer reaction time and a temperature of 100 °C, and usually results in lower radiochemical yields. To address these shortcomings, other chelators including DTPA and HBED were introduced for highly efficient radiolabeling with 68Ga at room temperature.73,74
An example using a DTPA chelator was reported by Mishra et al., which is the first time that 68Ga complexes have been proposed as Aβ imaging probes ([68Ga]77, Fig. 12 and Table 3).75 The chalcone moiety was selected as the Aβ binding motif, and a homodimeric ligand was designed from the conjugation of two chalcone units with DTPA via amide linkage. [68Ga]77 was obtained under mild conditions with 85.4% efficiency and 9.5–10 MBq nmol–1 specific activity. In the in vitro binding studies, [nat/68Ga]77 displayed a high binding affinity for Aβ1–42 aggregates (Kd = 3.46 nM and Ki = 4.18 nM), which was comparable to the iodinated chalcone derivative. Owing to the incorporation of two chalcone units, the log P value determined for [68Ga]77 was 1.58, which was in the optimal range for BBB penetration. Moderate initial brain uptake (1.24% ID g–1 at 2 min) and rapid wash-out (0.20% ID g–1 at 60 min) were observed for [68Ga]77 in the in vivo biodistribution experiments, which were further validated by dynamic PET studies in normal mice. Similar to 99mTc-labeled Aβ imaging probes for the diagnosis of AD, the brain uptakes of these 68Ga complexes were still unsatisfactory.
Considering that the 68Ga complexes hardly penetrated the BBB, but may bind to Aβ aggregates on the blood vessel walls, recently, Kung et al. reported a series of novel 68Ga complexes with bivalent polypegylated styrylpyridine conjugated to HBED for the detection of CAA with PET.76 The complexes, [68Ga]78–83 (Fig. 12), were obtained with high radiochemical yields (93–98%) at room temperature and were stable in both PBS and human plasma. Compared with the monovalent complex ([nat/68Ga]83, Ki = 185 nM), the bivalent complexes, [nat/68Ga]78–82, showed improved binding affinity to Aβ plaques in post-mortem AD brain homogenates (Ki = 6.7–30.6 nM, Table 3). In vitro autoradiography of post-mortem AD brain sections confirmed their high and specific binding of Aβ plaques. In accordance with other radiometal-labeled complexes, [68Ga]78–83 showed very low initial brain uptakes (<0.31% ID g–1), which was favorable for CAA-specific imaging. The preliminary results indicated that [68Ga]78–83 displayed potential to map Aβ plaques in the blood vessels of CAA patients. However, neither in vitro autoradiography of these 68Ga complexes using post-mortem CAA brain sections nor ex vivo autoradiography using Tg mice was reported and thus the specific and selective binding to CAA was not fully confirmed.
In recent years, radiolabeled curcumin and curcuminoids were reported to have high affinity to Aβ plaques and their potential in the detection of AD was investigated.77–79 In addition, the α,β-diketone group in the middle of the structure allows curcumin to act as an OO bidentate ligand.43 Asti et al. reported three 68Ga-labeled curcumin derivatives ([nat/68Ga]84–86, Fig. 12) with a 1 : 2 metal-to-ligand molar ratio.80 All the compounds were obtained with high radiochemical yield (>95%) and showed high stability in both 0.9% NaCl solution and human serum. In preliminary binding studies, these 68Ga complexes displayed moderate-to-high affinity to Aβ1–40 aggregates and the specific binding could be inhibited by the addition of excess amounts of the corresponding curcuminoids. Although these preliminary results appear encouraging, further evaluation, including quantitative binding studies and in vivo kinetic studies of these 68Ga complexes, is needed to derive the final conclusions.
More recently, Donnelly et al. reported a new six-coordinate Ga3+ complex (87, Fig. 12) by the use of an ancillary tetradentate N2O2 Schiff base ligand and the β-diketone group of curcumin as a bidentate ligand.83 Complex 87 was found to have binding affinity for Aβ plaques in the human brain tissue of patients with AD. However, more efforts, including biological evaluation and radiolabeling, are needed for the validation of this Ga complex as an Aβ imaging agent.
2.3. 64Cu-labeled complexes for PET imaging
The positron-emitting copper-64 radionuclide (t1/2 = 12.7 h, β+ = 17.8%; β– = 38.4%) has been used in a wide range of applications in targeted radiotherapy and PET imaging.84 The half-life of 64Cu is long enough for the radiolabeling of small molecules, antibodies, proteins, and nanoparticles. In addition, its low energy positron emission, without much disruptive gamma emission, enables the output of high quality images in comparison with 18F.85 Owing to the electronic structure of Cu2+, square-planar four-coordinate, square-pyramidal or trigonal-bipyramidal five-coordinate, or octahedral six-coordinate copper complexes could be formed using various chelators with uncharged nitrogen donors and anionic oxygen or sulfur donors. A small number of 64Cu-labeled Aβ imaging probes were reported before 2014 and have been well summarized in a review paper.28,30
In 2016, Ono et al. reported two novel 64Cu-labeled 2-phenylbenzofuran derivatives conjugated with cyclen ([64Cu]88) and DOTA ([64Cu]89) as PET imaging probes for Aβ aggregates (Fig. 13 and Table 3).81 [64Cu]88 and [64Cu]89 were obtained with moderate radiochemical yields (72% and 35%) in buffer solution at 85 °C for 10 min. Compared with 88 (Ki = 33.7 nM), complex 89 (Ki = 243.5 nM), with the bulkier chelator DOTA, displayed decreased affinity to Aβ aggregates, which was consistent with the results of in vitro fluorescence staining using brain sections from Tg2576 mice, in which 88 showed better results than 89. Because of the poor brain uptake of [64Cu]88 and [64Cu]89 (0.33 and 0.36% ID g–1, respectively) in normal mice, their use as cerebral Aβ imaging probes is inadvisable. However, considering their high affinity and low brain uptake, they may be applicable to CAA or amylin imaging.
Fig. 13. Chemical structures of 64Cu-labeled 2-phenylbenzofuran derivatives conjugated with cyclen and DOTA.

2.4. 89Zr-labeled antibodies for PET imaging
Until now, the development of radiolabeled small molecules for the imaging of Aβ plaques has achieved great success. However, as they were all designed to bind to the β-sheet structure of Aβ fibers, the ability and specificity to detect multiple conformations of Aβ are limited.86 Immuno-PET, with radiolabeled anti-Aβ antibodies, has provided an alternative approach to overcome this issue. Anti-Aβ antibodies, which can detect various Aβ conformations, are able to provide a detailed readout of the amyloid isoform profile in individual subjects.87–89 Owing to the long biological half-life of monoclonal antibodies (mAbs), which have typical circulation times of 1–3 weeks, the radioisotopes for mAb labeling should also have a similar half-life. Indeed, several Aβ antibodies have been already labeled by radionuclides with a long half-life, such as 125I, 124I, and 64Cu.89–91 Over the past decade, zirconium-89 has been one of the most popular PET radionuclides for the labeling of mAbs and has the following advantages: appropriate half-life (t1/2 = 78.4 h) that is compatible with the circulation time of mAbs; relatively low energy positrons (average energy = 389 keV) for high resolution PET imaging and accurate quantification; the ease of production by means of small medical cyclotrons.92,93 For metal radionuclides, an appropriate chelator system must be introduced; desferrioxamine B (DFO) is the most commonly used chelator for 89Zr complexation and it is conjugated to the biomolecule before the radiolabeling reaction takes place.94–96
More recently, Fissers et al. reported a 89Zr-labeled mAb ([89Zr]90, [89Zr]Df-Bz-JRF/AβN/25, Fig. 14 and Table 3) for the immuno-PET imaging of Aβ pathology in AD.82 The bifunctional chelator p-isothiocyanatobenzyl-desferrioxamine (Df-Bz-NCS) was coupled to the amino residue of JRF/AβN/25, which is an N-terminal end-specific mAb directed against the full-length human Aβ epitope. The saturation binding assay revealed high and specific binding of [89Zr]90 to Aβ1–40 (Kd = 0.85 nM), which was further validated by in vitro autoradiography studies on brain sections from Tg mice (Fig. 15). In the biodistribution studies, [89Zr]90 demonstrated low brain uptake (0.5 ± 0.07% ID g–1 at 1 h) and slow clearance (0.36 ± 0.06% ID g–1 at 48 h). Therefore, further imaging studies in Tg mice should be performed to evaluate if the in vivo brain accumulation of [89Zr]90 was sufficient to allow accurate Aβ quantification.
Fig. 14. Chemical structures of 89Zr- and 111In-labeled mAbs and Aβ peptides.

Fig. 15. Immunostaining (A) and in vitro autoradiography (B) on brain sections of 8 month-old APPPS1-21 Tg mice (left) and age-matched WT mice (right). Red squares indicate cortical regions with known high Aβ deposition. Adapted from ref. 82.

2.5. 111In-labeled antibody and Aβ peptide for SPECT imaging
Similar to zirconium-89, indium-111 has a half-life (t1/2 = 67.3 h) that is compatible with the circulation time of mAbs; however, indium-111 decays via the emission of 171 and 245 keV gamma rays. Thus, 111In has been widely used as a SPECT radioisotope for mAbs labeling.97
Van der Weerd et al. reported an 111In-labeled fusion protein containing VHH (a llama single-domain antibody fragment used for targeting amyloid plaques in AD).98 With the aim of increasing the serum persistence, the Fc portion of the human IgG1 antibody (hinge plus CH2 and CH3 domains) was fused to the C-terminus of VHH to form VHH–Fc. Then, 111In was introduced by the conjugation of the bifunctional chelator p-SCN-Bz-DTPA to the primary amine groups on the VHH–Fc fusion protein ([111In]91, [111In]VHH–Fc–DTPA, Fig. 14). Immunofluorescence analysis confirmed that VHH–Fc–DTPA retained its affinity for Aβ deposits in brain sections from patients with AD. Despite the increased blood circulation time, [111In]91 did not efficiently reach the brain of mice, and there was no specific increase in the brains of Tg mice compared with control mice, which indicated that [111In]91 could not penetrate the BBB and bind to Aβ plaques in the brain. Further micro-SPECT imaging studies confirmed that the radioactivity of [111In]91 was not found in either Tg or WT brains.
Compared with the classical immunohistochemical techniques for mAbs against Aβ, radiolabeled Aβ analogs enable more sensitive detection of Aβ plaques. Therefore, radiolabeled Aβ1–42 and Aβ1–40 analogs could be used for amyloid imaging in living patients with AD. Pardridge et al. reported an 111In-labeled Aβ1–40 analogue that contained biotin at the amino terminus and a DTPA chelator conjugated to one of the internal lysine residues ([111In]92, [111In]DTPA–[N-biotin]–Aβ1–40, Fig. 14).99 Film autoradiography studies showed that [111In]92 could intensely bind to the Aβ plaques in brain sections of patients with AD. However, quantitative binding studies and in vivo kinetic studies of [111In]92 were not reported.
3. Non-radioactive ruthenium and rhenium complexes binding to Aβ species
Owing to the strong spin–orbit coupling, heavy-metal complexes, which have highly intense phosphorescent emission with long lifetimes, are alternative choices to short-lived conventional organic fluorescent probes for the staining of different cellular compartments, monitoring of intracellular biological functions, and targeted bioimaging in vivo.100 To date, heavy-metal complexes, including ruthenium and rhenium with d6 electron configurations, have been evaluated as Aβ specific probes to monitor the formation of Aβ oligomers/fibrils or label Aβ deposits in tissue specimens.28
Murray et al. first investigated a cationic dye ruthenium red (ammoniated ruthenium oxychloride, 93, Fig. 16) as an Aβ labeling agent.101 Under light microscopy, Aβ plaques in brain sections of Tg2576 mice were dyed red by 93; under cross-polarization, birefringence was observed when 93 bound to Aβ plaques. However, in addition to Aβ plaques, neurons in the dentate cellular layer of the hippocampus and the Purkinje layer of the cerebellum were also stained by 93, which is undesirable for Aβ-specific dyes. The binding mechanism of 93 to Aβ fibrils was electrostatic interaction. This was confirmed because divalent cations, such as calcium, could displace 93 from its binding site in the Aβ plaques and remove the red coloration. In addition, upon binding to Aβ fibrils, 93 showed birefringence and induced circular dichroic bands at 540 nm owing to induced chirality. Overall, the chirality and cation binding mechanism of 93 to Aβ fibrils may be useful in the development of novel amyloid labeling methods, as well as novel AD imaging techniques.
Fig. 16. Chemical structures of non-radioactive ruthenium and rhenium complexes.

Dipyridophenazine (DPPZ) ruthenium complexes demonstrated strong photoluminescence in the presence of Aβ fibril aggregates102 or α-synuclein aggregates,103 which made them ideal candidates for the monitoring and inhibition of the formation process of Aβ oligomers/fibrils. Based on these findings, Carlos et al. reported a novel ruthenium complex (94, Fig. 16) for multi-target treatment of AD.104 Complex 94 displayed inhibitory activity on both human AChE and BuChE without appreciable cytotoxicity. More importantly, complex 94 displayed non-covalent interactions with Aβ, and the high sensitivity of 94 to Aβ enabled real-time observation of the conformational changes of Aβ monomers to protofibrils.
Unlike the direct interaction of the ruthenium complex with Aβ species, Rajagopal et al. reported a new ruthenium complex 95 (Fig. 16) intercalated with a RNA aptamer, which could selectively bind to the Aβ monomer and soluble oligomer.105 Complex 95 displayed strong luminescence intensity in the presence of the RNA aptamer, which was apparently reduced upon the addition of Aβ monomers. Moreover, atomic force microscopy indicated that the aptamer–95 complex displayed a strong inhibitory effect on the aggregation of the Aβ peptides. Overall, this novel aptamer–Ru complex system as an Aβ sensor may contribute to the rapid diagnosis and inhibition of Aβ aggregation in clinical applications.
Non-radioactive rhenium complexes can also be used as probes for the selective and sensitive detection of Aβ fibrils. Rajagopal et al. reported two alkoxy-bridged binuclear rhenium complexes (96 and 97, Fig. 16) with two stilbene-like 4-(1-naphthylvinyl)pyridines as Aβ binding motifs.106 Luminescence titrations indicated that they exhibited strong fluorescence enhancement upon the addition of Aβ aggregates; the binding constants were calculated to be 2.2 × 105 and 2.0 × 105 M–1, respectively. It is thought that the naphthalene moiety in these rhenium complexes might strongly interact with the hydrophobic regions of Aβ via π–π stacking interactions. This speculation was confirmed by docking studies between these rhenium complexes and the Aβ peptide.
Inspired by the previously observed increase of photoluminescence for ruthenium–DPPZ complexes with Aβ fibrils, Martí et al. investigated the photoluminescence properties of a rhenium complex ([Re(CO)3(dppz)(Py)]+, 98, Fig. 16) upon interaction with fibrillar Aβ.107 Upon binding to Aβ aggregates, 98 displayed an approximately 18-fold increase in photoluminescence in the primary light switching. Additionally, a significant time-dependent enhancement of photoluminescence was observed after light irradiation (secondary light switching). Under the optimized irradiation time (7200 s), a 105-fold increase in the photoluminescence was observed for 98 in the presence of Aβ fibrils. Unlike the ruthenium–DPPZ complexes, the two light-switching events observed for 98 were unprecedented, which may allow the monitoring of Aβ aggregation in real time and the addition of another dimension to the study of these rhenium complexes.
4. Non-radioactive gadolinium complexes for MRI
MRI offers noninvasive imaging of physiological structures and functions with remarkable spatial resolution. Compared with SPECT and PET, the MRI technique offers several advantages: does not require MRI radioactive probes and thus avoids exposure to radiation; possesses better resolution (50–200 μm) for research and clinical magnets; offers anatomical information for the quantification of amyloid deposits and the precise location of the deposition areas, which are critical for the evaluation of new treatments against AD at early stages; the lower cost and wide availability of MRI around the world.108,109 However, the major limitation of MRI is its low sensitivity; MRI contrast in Aβ plaques is associated with iron accumulation, which leads to hypointense spots in T2 and 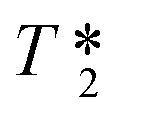 or susceptibility-weighted images.110,111 The detection of plaques that are weakly loaded with iron is more challenging. The best solution is to utilize exogenous contrast agents (CAs). So far, a modest number of MRI CAs for the diagnosis of AD have been developed, such as appropriate fluorinated small molecules,112 Aβ-targeted iron oxide nanoparticles,113 and conjugated gadolinium metal complexes.114 Owing to their high paramagnetism, relaxation enhancement, favorable in vivo dynamics, and relatively high stability, gadolinium-based MRI CAs have shown great promise.115 As the Aβ-targeted gadolinium complexes reported before 2016 are well documented in a review paper,114 herein we include just two recently reported complexes.
or susceptibility-weighted images.110,111 The detection of plaques that are weakly loaded with iron is more challenging. The best solution is to utilize exogenous contrast agents (CAs). So far, a modest number of MRI CAs for the diagnosis of AD have been developed, such as appropriate fluorinated small molecules,112 Aβ-targeted iron oxide nanoparticles,113 and conjugated gadolinium metal complexes.114 Owing to their high paramagnetism, relaxation enhancement, favorable in vivo dynamics, and relatively high stability, gadolinium-based MRI CAs have shown great promise.115 As the Aβ-targeted gadolinium complexes reported before 2016 are well documented in a review paper,114 herein we include just two recently reported complexes.
Geraldes et al. reported a negatively charged Gd3+ complex (99, Fig. 17) with a neutral ethylenediamine linker between the DOTA and the Aβ-targeting PIB moieties.116 In binding studies using a surface plasmon resonance method, 99 displayed a markedly reduced binding affinity for Aβ1–40 aggregates (Kd = 194 ± 11 μM for La analogs), which is five orders of magnitude lower than that of PIB. More interestingly, TEM images and circular dichroism spectroscopy studies indicated that 99 appeared to inhibit the formation of Aβ1–40 fibrils. Protein-based NMR also pointed to the interactions of paramagnetic 99 with Aβ1–40 in the monomer state. However, further animal studies were not reported. Owing to its low Aβ affinity and expected low brain uptake, the authors proposed that complex 99 might not be adaptable for the early MRI detection of Aβ plaques in the brain of patients with AD, but could be helpful in animal studies with BBB opening and for ex vivo MRI.
Fig. 17. Chemical structures of gadolinium complexes for MRI.

By covalently coupling curcumin and gadolinium–DOTA into a natural drug delivery platform, poly(β-l-malic acid), Holler et al. reported a nanoimaging agent (NIA) 100 (Fig. 17).117 The hydrodynamic diameter of 100 was 8.3 nm; each agent carried approximately 35 molecules of gadolinium–DOTA and curcumin. The detection of Aβ plaques by MRI in the presence of 100 displayed a significantly high contrast enhancement. Furthermore, a very clean staining pattern of Aβ plaques in mouse and human tissue, with a low background, was observed for 100. These results revealed that the nanoimaging agent 100 had a good binding ability to Aβ plaques in vitro. After conjugation with the BBB penetrating agents, this NIA might be useful for the in vivo noninvasive diagnosis of AD.
5. Conclusions
Although the etiology of AD and CAA is still not fully understood, Aβ deposition is considered to be one of the most important pathological features of these diseases. The development of Aβ-specific imaging probes for PET, SPECT, and MRI is of great importance in the early diagnosis and monitoring of the efficacy of anti-Aβ therapies for AD and CAA.
The past two decades have seen a remarkable increase in the development of 18F- and 11C-labeled Aβ probes for PET, some of which have been expanded into commercial applications. However, the development of 123I- and 99mTc-labeled Aβ probes for SPECT is trailing far behind. At present, 99mTc is still the most appropriate radioisotope for SPECT imaging. In spite of continuous efforts to develop 99mTc-labeled probes with different 99mTc cores ([99mTcO]3+ and [99mTc(CO)3]+) and chelating agents (e.g., MAMA, BAT, Ham, PAMA, IDA, and cyclopentadienyl tricarbonyl) for SPECT targeting of Aβ plaques inside the brain, none of the proposed probes could be used for the clinical diagnosis of AD. The main problem for these 99mTc-labeled probes is their low brain uptake: although they have sufficient affinity to Aβ plaques, they cannot penetrate the BBB and reach the target site. Thus, there is a need to further improve the brain uptake by structural modifications. As Aβ deposition was generated in the wall of cerebral vessels of CAA patients, there is no need to penetrate the entire BBB for imaging probes, which suggests a new application for the 99mTc-labeled probes with low brain uptake. Indeed, some probes have shown very promising results for CAA-specific imaging, as they can selectively label Aβ deposits in blood vessels, but not Aβ plaques, in the parenchyma of the brain. However, further experiments, especially human studies, will be required to confirm the utility of these 99mTc-labeled complexes as CAA imaging agents.
Other than 18F, 11C, and 99mTc, several radiometal nuclides such as 68Ga, 64Cu, 89Zr, and 111In have been incorporated into Aβ-specific small organic molecules, mAbs, and proteins via appropriate chelators (DOTA, DTPA, HBED, and DFO) as Aβ imaging agents for PET and SPECT. Similar to the 99mTc-labeled probes, they all displayed sufficient affinity for Aβ plaques, but poor BBB penetration owing to their large molecular weight and charged properties, which is unfavorable for the diagnosis of AD. However, through appropriate modifications, they may be used as imaging agents for CAA and diseases with peripheral amyloidosis.
Apart from the organic dyes, heavy-metal complexes, such as Ru and Re complexes, possessed interesting photophysical and photochemical characteristics. Some of the complexes displayed relatively long photoluminescence lifetimes and strong luminescence intensities when bound to Aβ species, which could be used as sensitive probes for the study of Aβ aggregation or labeling of Aβ deposits in tissue specimens. Compared with SPECT and PET, MRI offers an alternative approach for the detection of Aβ deposits in the brain. However, further efforts should be made to improve the Aβ binding affinity and the BBB penetrability of the reported gadolinium-based CAs.
Conflict of interest
The authors declare no competing interests.
Acknowledgments
This work was funded by the National Natural Science Foundation of China (No. 21201019 and 21571022) and the National Science and Technology Major Projects for Major New Drugs Innovation and Development (No. 2014ZX09507007-002).
References
- Roberson E. D., Mucke L. Science. 2006;314:781. doi: 10.1126/science.1132813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E. E., Greenberg S. M. Stroke. 2009;40:2601–2606. doi: 10.1161/STROKEAHA.108.536839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan A., Greenberg S. M. Ann. Neurol. 2011;70:871–880. doi: 10.1002/ana.22516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz W. D., Nelson P. T., Besser L. M., Heller K. B., Kukull W. A. Neurobiol. Aging. 2015;36:2702–2708. doi: 10.1016/j.neurobiolaging.2015.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll J. A., Yamada M., Frackowiak J., Mazur-Kolecka B., Weller R. O., Neurobiol. Aging, 2004, 25 , 589 –597 , discussion 603-584 . [DOI] [PubMed] [Google Scholar]
- Jellinger K. A. J. Neural Transm. 2002;109:813–836. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- Hardy J. J. Alzheimer's Dis. 2006;9:151–153. doi: 10.3233/jad-2006-9s317. [DOI] [PubMed] [Google Scholar]
- Hardy J., Selkoe D. J. Science. 2002;297:353. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Fryer J. D., Simmons K., Parsadanian M., Bales K. R., Paul S. M., Sullivan P. M., Holtzman D. M. J. Neurosci. 2005;25:2803–2810. doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attems J., Lintner F., Jellinger K. A. Acta Neuropathol. 2004;107:283–291. doi: 10.1007/s00401-004-0822-6. [DOI] [PubMed] [Google Scholar]
- Mathis C. A., Wang Y., Klunk W. E. Curr. Pharm. Des. 2004;10:1469–1492. doi: 10.2174/1381612043384772. [DOI] [PubMed] [Google Scholar]
- Herholz K. Lancet Neurol. 2012;11:652–653. doi: 10.1016/S1474-4422(12)70158-8. [DOI] [PubMed] [Google Scholar]
- Rowe C. C., Villemagne V. L. J. Nucl. Med. 2011;52:1733–1740. doi: 10.2967/jnumed.110.076315. [DOI] [PubMed] [Google Scholar]
- Cui M. Curr. Med. Chem. 2014;21:82–112. doi: 10.2174/09298673113209990216. [DOI] [PubMed] [Google Scholar]
- Coimbra A., Williams D. S., Hostetler E. D. Curr. Top. Med. Chem. 2006;6:629–647. doi: 10.2174/156802606776743075. [DOI] [PubMed] [Google Scholar]
- Ikonomovic M. D., Klunk W. E., Abrahamson E. E., Mathis C. A., Price J. C., Tsopelas N. D., Lopresti B. J., Ziolko S., Bi W., Paljug W. R., Debnath M. L., Hope C. E., Isanski B. A., Hamilton R. L., DeKosky S. T. Brain. 2008;131:1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk W. E., Engler H., Nordberg A., Wang Y., Blomqvist G., Holt D. P., Bergstrom M., Savitcheva I., Huang G. F., Estrada S., Ausen B., Debnath M. L., Barletta J., Price J. C., Sandell J., Lopresti B. J., Wall A., Koivisto P., Antoni G., Mathis C. A., Langstrom B. Ann. Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Mathis C. A., Wang Y., Holt D. P., Huang G. F., Debnath M. L., Klunk W. E. J. Med. Chem. 2003;46:2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- Heurling K., Leuzy A., Zimmer E. R., Lubberink M., Nordberg A. Eur. J. Nucl. Med. Mol. Imaging. 2016;43:362–373. doi: 10.1007/s00259-015-3208-1. [DOI] [PubMed] [Google Scholar]
- Koole M., Lewis D. M., Buckley C., Nelissen N., Vandenbulcke M., Brooks D. J., Vandenberghe R., Van Laere K. J. Nucl. Med. 2009;50:818–822. doi: 10.2967/jnumed.108.060756. [DOI] [PubMed] [Google Scholar]
- Wong D. F., Rosenberg P. B., Zhou Y., Kumar A., Raymont V., Ravert H. T., Dannals R. F., Nandi A., Brasic J. R., Ye W., Hilton J., Lyketsos C., Kung H. F., Joshi A. D., Skovronsky D. M., Pontecorvo M. J. J. Nucl. Med. 2010;51:913–920. doi: 10.2967/jnumed.109.069088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister-James J., Pontecorvo M. J., Clark C., Joshi A. D., Mintun M. A., Zhang W., Lim N., Zhuang Z., Golding G., Choi S. R., Benedum T. E., Kennedy P., Hefti F., Carpenter A. P., Kung H. F., Skovronsky D. M. Semin. Nucl. Med. 2011;41:300–304. doi: 10.1053/j.semnuclmed.2011.03.001. [DOI] [PubMed] [Google Scholar]
- Kung H. F., Choi S. R., Qu W., Zhang W., Skovronsky D. J. Med. Chem. 2010;53:933–941. doi: 10.1021/jm901039z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne V. L., Ong K., Mulligan R. S., Holl G., Pejoska S., Jones G., O'Keefe G., Ackerman U., Tochon-Danguy H., Chan J. G., Reininger C. B., Fels L., Putz B., Rohde B., Masters C. L., Rowe C. C. J. Nucl. Med. 2011;52:1210–1217. doi: 10.2967/jnumed.111.089730. [DOI] [PubMed] [Google Scholar]
- Rowe C. C., Ackerman U., Browne W., Mulligan R., Pike K. L., O'Keefe G., Tochon-Danguy H., Chan G., Berlangieri S. U., Jones G., Dickinson-Rowe K. L., Kung H. P., Zhang W., Kung M. P., Skovronsky D., Dyrks T., Holl G., Krause S., Friebe M., Lehman L., Lindemann S., Dinkelborg L. M., Masters C. L., Villemagne V. L. Lancet Neurol. 2008;7:129–135. doi: 10.1016/S1474-4422(08)70001-2. [DOI] [PubMed] [Google Scholar]
- Zha Z., Choi S. R., Ploessl K., Lieberman B. P., Qu W., Hefti F., Mintun M., Skovronsky D., Kung H. F. J. Med. Chem. 2011;54:8085–8098. doi: 10.1021/jm2009106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B. H., Zhou M. L., Vellimana A. K., Milner E., Kim D. H., Greenberg J. K., Chu W., Mach R. H., Zipfel G. J. Mol. Neurodegener. 2011;6:86. doi: 10.1186/1750-1326-6-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayne D. J., Lim S., Donnelly P. S. Chem. Soc. Rev. 2014;43:6701–6715. doi: 10.1039/c4cs00026a. [DOI] [PubMed] [Google Scholar]
- Jurisson S. S., Lydon J. D. Chem. Rev. 1999;99:2205–2218. doi: 10.1021/cr980435t. [DOI] [PubMed] [Google Scholar]
- Hickey J. L., Donnelly P. S. Coord. Chem. Rev. 2012;256:2367–2380. [Google Scholar]
- Liu S., Edwards D. S. Chem. Rev. 1999;99:2235–2268. doi: 10.1021/cr980436l. [DOI] [PubMed] [Google Scholar]
- Bandoli G., Tisato F., Dolmella A., Agostini S. Coord. Chem. Rev. 2006;250:561–573. [Google Scholar]
- Dezutter N. A., de Groot T. J., Busson R. H., Janssen G. A., Verbruggen A. M. J. Labelled Compd. Radiopharm. 1999;42:309–324. [Google Scholar]
- Dezutter N. A., Dom R. J., de Groot T. J., Bormans G. M., Verbruggen A. M. Eur. J. Nucl. Med. 1999;26:1392–1399. doi: 10.1007/s002590050470. [DOI] [PubMed] [Google Scholar]
- Serdons K., Verduyckt T., Cleynhens J., Bormans G., Verbruggen A. J. Labelled Compd. Radiopharm. 2008;51:357–367. [Google Scholar]
- Chen X., Yu P., Zhang L., Liu B. Bioorg. Med. Chem. Lett. 2008;18:1442–1445. doi: 10.1016/j.bmcl.2007.12.071. [DOI] [PubMed] [Google Scholar]
- Serdons K., Verduyckt T., Cleynhens J., Terwinghe C., Mortelmans L., Bormans G., Verbruggen A. Bioorg. Med. Chem. Lett. 2007;17:6086–6090. doi: 10.1016/j.bmcl.2007.09.055. [DOI] [PubMed] [Google Scholar]
- Wang X., Cui M., Yu P., Li Z., Yang Y., Jia H., Liu B. Bioorg. Med. Chem. Lett. 2012;22:4327–4331. doi: 10.1016/j.bmcl.2012.05.010. [DOI] [PubMed] [Google Scholar]
- Cheng Y., Ono M., Kimura H., Ueda M., Saji H. J. Med. Chem. 2012;55:2279–2286. doi: 10.1021/jm201513c. [DOI] [PubMed] [Google Scholar]
- Ono M., Fuchi Y., Fuchigami T., Kobashi N., Kimura H., Haratake M., Saji H., Nakayama M. ACS Med. Chem. Lett. 2010;1:443–447. doi: 10.1021/ml100140d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M., Ikeoka R., Watanabe H., Kimura H., Fuchigami T., Haratake M., Saji H., Nakayama M. Bioorg. Med. Chem. Lett. 2010;20:5743–5748. doi: 10.1016/j.bmcl.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Ono M., Ikeoka R., Watanabe H., Kimura H., Fuchigami T., Haratake M., Saji H., Nakayama M. ACS Chem. Neurosci. 2010;1:598–607. doi: 10.1021/cn100042d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagnou M., Benaki D., Triantis C., Tsotakos T., Psycharis V., Raptopoulou C. P., Pirmettis I., Papadopoulos M., Pelecanou M. Inorg. Chem. 2011;50:1295–1303. doi: 10.1021/ic102228u. [DOI] [PubMed] [Google Scholar]
- Cui M., Tang R., Li Z., Ren H., Liu B. Bioorg. Med. Chem. Lett. 2011;21:1064–1068. doi: 10.1016/j.bmcl.2010.11.096. [DOI] [PubMed] [Google Scholar]
- Yang Y., Cui M., Jin B., Wang X., Li Z., Yu P., Jia J., Fu H., Jia H., Liu B. Eur. J. Med. Chem. 2013;64:90–98. doi: 10.1016/j.ejmech.2013.03.057. [DOI] [PubMed] [Google Scholar]
- Zhang J., Zhou X., Qin X. J. Radioanal. Nucl. Chem. 2012;292:1377–1383. [Google Scholar]
- Wang X., Cui M., Jia J., Liu B. Eur. J. Med. Chem. 2015;89:331–339. doi: 10.1016/j.ejmech.2014.10.046. [DOI] [PubMed] [Google Scholar]
- Zhang X., Yu P., Yang Y., Hou Y., Peng C., Liang Z., Lu J., Chen B., Dai J., Liu B., Cui M. Bioconjugate Chem. 2016;27:2493–2504. doi: 10.1021/acs.bioconjchem.6b00444. [DOI] [PubMed] [Google Scholar]
- Thipyapong K., Uehara T., Tooyama Y., Braband H., Alberto R., Arano Y. Inorg. Chem. 2011;50:992–998. doi: 10.1021/ic101714q. [DOI] [PubMed] [Google Scholar]
- Nakayama M., Xu L. C., Koga Y., Harada K., Sugii A., Nakayama H., Tomiguchi S., Kojima A., Ohyama Y., Takahashi M., Okabayashi I. Appl. Radiat. Isot. 1997;48:571–577. [Google Scholar]
- Nakayama M., Saigo H., Koda A., Ozeki K., Harada K., Sugii A., Tomiguchi S., Kojima A., Hara M., Nakashima R., Ohyama Y., Takahashi M., Takata J., Karube Y. Appl. Radiat. Isot. 1994;45:735–740. [Google Scholar]
- Iikuni S., Ono M., Watanabe H., Matsumura K., Yoshimura M., Harada N., Kimura H., Nakayama M., Saji H. Mol. Pharmaceutics. 2014;11:1132–1139. doi: 10.1021/mp400499y. [DOI] [PubMed] [Google Scholar]
- Iikuni S., Ono M., Watanabe H., Matsumura K., Yoshimura M., Kimura H., Ishibashi-Ueda H., Okamoto Y., Ihara M., Saji H. Sci. Rep. 2016;6:25990. doi: 10.1038/srep25990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iikuni S., Ono M., Watanabe H., Yoshimura M., Ishibashi-Ueda H., Ihara M., Saji H. PLoS One. 2016;11:e0163969. doi: 10.1371/journal.pone.0163969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayne D. J., White J. M., McLean C. A., Villemagne V. L., Barnham K. J., Donnelly P. S. Inorg. Chem. 2016;55:7944–7953. doi: 10.1021/acs.inorgchem.6b00972. [DOI] [PubMed] [Google Scholar]
- Morais M., Paulo A., Gano L., Santos I., Correia J. D. G. J. Organomet. Chem. 2013;744:125–139. [Google Scholar]
- Alberto R., Schibli R., Waibel R., Abram U., Schubiger A. P. Coord. Chem. Rev. 1999;190–192:901–919. [Google Scholar]
- Alberto R., Ortner K., Wheatley N., Schibli R., Schubiger A. P. J. Am. Chem. Soc. 2001;123:3135–3136. doi: 10.1021/ja003932b. [DOI] [PubMed] [Google Scholar]
- Schibli R., La Bella R., Alberto R., Garcia-Garayoa E., Ortner K., Abram U., Schubiger P. A. Bioconjugate Chem. 2000;11:345–351. doi: 10.1021/bc990127h. [DOI] [PubMed] [Google Scholar]
- Satpati D., Korde A., Sarma H. D., Banerjee S. J. Radioanal. Nucl. Chem. 2014;302:1339–1344. [Google Scholar]
- Hayne D. J., North A. J., Fodero-Tavoletti M., White J. M., Hung L. W., Rigopoulos A., McLean C. A., Adlard P. A., Ackermann U., Tochon-Danguy H., Villemagne V. L., Barnham K. J., Donnelly P. S. Dalton Trans. 2015;44:4933–4944. doi: 10.1039/c4dt02969k. [DOI] [PubMed] [Google Scholar]
- Jia J., Cui M., Dai J., Wang X., Ding Y.-S., Jia H., Liu B. MedChemComm. 2014;5:153–158. [Google Scholar]
- Jia J., Cui M., Dai J., Liu B. Mol. Pharmaceutics. 2015;12:2937–2946. doi: 10.1021/acs.molpharmaceut.5b00209. [DOI] [PubMed] [Google Scholar]
- Jia J., Cui M., Dai J., Liu B. Dalton Trans. 2015;44:6406–6415. doi: 10.1039/c5dt00023h. [DOI] [PubMed] [Google Scholar]
- Jia J., Zhou K., Dai J., Liu B., Cui M. Eur. J. Med. Chem. 2016;124:763–772. doi: 10.1016/j.ejmech.2016.09.001. [DOI] [PubMed] [Google Scholar]
- Bernard J., Ortner K., Spingler B., Pietzsch H. J., Alberto R. Inorg. Chem. 2003;42:1014–1022. doi: 10.1021/ic0204575. [DOI] [PubMed] [Google Scholar]
- Chan C. Y., Noor A., McLean C. A., Donnelly P. S., Barnard P. J. Chem. Commun. 2017;53:2311–2314. doi: 10.1039/c6cc10066j. [DOI] [PubMed] [Google Scholar]
- Velikyan I. Theranostics. 2014;4:47–80. doi: 10.7150/thno.7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch F., Riss P. J. Curr. Top. Med. Chem. 2010;10:1633–1668. doi: 10.2174/156802610793176738. [DOI] [PubMed] [Google Scholar]
- Fani M., André J. P., Maecke H. R. Contrast Media Mol. Imaging. 2008;3:53–63. doi: 10.1002/cmmi.232. [DOI] [PubMed] [Google Scholar]
- Cressier D., Dhilly M., Cao Pham T. T., Fillesoye F., Gourand F., Maïza A., Martins A. F., Morfin J.-F., Geraldes C. F. G. C., Tóth É., Barré L. Mol. Imaging Biol. 2016;18:334–343. doi: 10.1007/s11307-015-0906-9. [DOI] [PubMed] [Google Scholar]
- Watanabe H., Ono M., Iikuni S., Yoshimura M., Matsumura K., Kimura H., Saji H. Bioorg. Med. Chem. Lett. 2014;24:4834–4837. doi: 10.1016/j.bmcl.2014.08.058. [DOI] [PubMed] [Google Scholar]
- Eder M., Wangler B., Knackmuss S., LeGall F., Little M., Haberkorn U., Mier W., Eisenhut M. Eur. J. Nucl. Med. Mol. Imaging. 2008;35:1878–1886. doi: 10.1007/s00259-008-0816-z. [DOI] [PubMed] [Google Scholar]
- Motekaitis R. J., Martell A. E., Welch M. J. Inorg. Chem. 1990;29:1463–1467. [Google Scholar]
- Chauhan K., Datta A., Adhikari A., Chuttani K., Kumar Singh A., Mishra A. K. Org. Biomol. Chem. 2014;12:7328–7337. doi: 10.1039/c4ob00941j. [DOI] [PubMed] [Google Scholar]
- Zha Z., Song J., Choi S. R., Wu Z., Ploessl K., Smith M., Kung H. Bioconjugate Chem. 2016;27:1314–1323. doi: 10.1021/acs.bioconjchem.6b00127. [DOI] [PubMed] [Google Scholar]
- Cui M., Ono M., Kimura H., Liu B., Saji H. J. Med. Chem. 2011;54:2225–2240. doi: 10.1021/jm101404k. [DOI] [PubMed] [Google Scholar]
- Rokka J., Snellman A., Zona C., La Ferla B., Nicotra F., Salmona M., Forloni G., Haaparanta-Solin M., Rinne J. O., Solin O. Bioorg. Med. Chem. 2014;22:2753–2762. doi: 10.1016/j.bmc.2014.03.010. [DOI] [PubMed] [Google Scholar]
- Ryu E. K., Choe Y. S., Lee K. H., Choi Y., Kim B. T. J. Med. Chem. 2006;49:6111–6119. doi: 10.1021/jm0607193. [DOI] [PubMed] [Google Scholar]
- Asti M., Ferrari E., Croci S., Atti G., Rubagotti S., Iori M., Capponi P. C., Zerbini A., Saladini M., Versari A. Inorg. Chem. 2014;53:4922–4933. doi: 10.1021/ic403113z. [DOI] [PubMed] [Google Scholar]
- Watanabe H., Kawasaki A., Sano K., Ono M., Saji H. Bioorg. Med. Chem. 2016;24:3618–3623. doi: 10.1016/j.bmc.2016.06.001. [DOI] [PubMed] [Google Scholar]
- Fissers J., Waldron A. M., De Vijlder T., Van Broeck B., Pemberton D. J., Mercken M., Van Der Veken P., Joossens J., Augustyns K., Dedeurwaerdere S., Stroobants S., Staelens S., Wyffels L. Mol. Imaging Biol. 2016;18:598–605. doi: 10.1007/s11307-016-0935-z. [DOI] [PubMed] [Google Scholar]
- Lange J. L., Hayne D. J., Roselt P., McLean C. A., White J. M., Donnelly P. S. J. Inorg. Biochem. 2016;162:274–279. doi: 10.1016/j.jinorgbio.2016.02.029. [DOI] [PubMed] [Google Scholar]
- Blower P. J., Lewis J. S., Zweit J. Nucl. Med. Biol. 1996;23:957–980. doi: 10.1016/s0969-8051(96)00130-8. [DOI] [PubMed] [Google Scholar]
- Smith S. V. J. Inorg. Biochem. 2004;98:1874–1901. doi: 10.1016/j.jinorgbio.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Nordberg A. Lancet Neurol. 2004;3:519–527. doi: 10.1016/S1474-4422(04)00853-1. [DOI] [PubMed] [Google Scholar]
- McLean D., Cooke M. J., Albay 3rd R., Glabe C., Shoichet M. S. ACS Chem. Neurosci. 2013;4:613–623. doi: 10.1021/cn300226q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glabe C. G. Trends Biochem. Sci. 2004;29:542–547. doi: 10.1016/j.tibs.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Sehlin D., Fang X. T., Cato L., Antoni G., Lannfelt L., Syvanen S. Nat. Commun. 2016;7:10759. doi: 10.1038/ncomms10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson K., Sehlin D., Syvanen S., Svedberg M. M., Philipson O., Soderberg L., Tegerstedt K., Holmquist M., Gellerfors P., Tolmachev V., Antoni G., Lannfelt L., Hall H., Nilsson L. N. J. Alzheimer's Dis. 2013;37:29–40. doi: 10.3233/JAD-130029. [DOI] [PubMed] [Google Scholar]
- McLean D., Cooke M. J., Wang Y., Green D., Fraser P. E., George-Hyslop P. S., Shoichet M. S. PLoS One. 2012;7:e51958. doi: 10.1371/journal.pone.0051958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G., Seibold U., Schirrmacher R., Wängler B., Wängler C. Molecules. 2013;18:6469–6490. doi: 10.3390/molecules18066469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severin G. W., Engle J. W., Nickles R. J., Barnhart T. E. Med. Chem. 2011;7:389–394. doi: 10.2174/157340611796799186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perk L. R., Vosjan M. J., Visser G. W., Budde M., Jurek P., Kiefer G. E., van Dongen G. A. Eur. J. Nucl. Med. Mol. Imaging. 2010;37:250–259. doi: 10.1007/s00259-009-1263-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosjan M. J., Perk L. R., Visser G. W., Budde M., Jurek P., Kiefer G. E., van Dongen G. A. Nat. Protoc. 2010;5:739–743. doi: 10.1038/nprot.2010.13. [DOI] [PubMed] [Google Scholar]
- Miller M. J. Chem. Rev. 1989;89:1563–1579. [Google Scholar]
- Chakraborty S., Liu S. Curr. Top. Med. Chem. 2010;10:1113–1134. doi: 10.2174/156802610791384243. [DOI] [PubMed] [Google Scholar]
- Rotman M., Welling M. M., van den Boogaard M. L., Moursel L. G., van der Graaf L. M., van Buchem M. A., van der Maarel S. M., van der Weerd L. Nucl. Med. Biol. 2015;42:695–702. doi: 10.1016/j.nucmedbio.2015.03.003. [DOI] [PubMed] [Google Scholar]
- Kurihara A., Pardridge W. M. Bioconjugate Chem. 2000;11:380–386. doi: 10.1021/bc9901393. [DOI] [PubMed] [Google Scholar]
- Friedman A. E., Chambron J. C., Sauvage J. P., Turro N. J., Barton J. K. J. Am. Chem. Soc. 1990;112:4960–4962. [Google Scholar]
- Cook N. P., Archer C. M., Fawver J. N., Schall H. E., Rodriguez-Rivera J., Dineley K. T., Martí A. A., Murray I. V. J. ACS Chem. Neurosci. 2013;4:379–384. doi: 10.1021/cn300219n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook N. P., Torres V., Jain D., Martí A. A. J. Am. Chem. Soc. 2011;133:11121–11123. doi: 10.1021/ja204656r. [DOI] [PubMed] [Google Scholar]
- Cook N. P., Kilpatrick K., Segatori L., Martí A. A. J. Am. Chem. Soc. 2012;134:20776–20782. doi: 10.1021/ja3100287. [DOI] [PubMed] [Google Scholar]
- Silva D. E. S., Cali M. P., Pazin W. M., Carlos-Lima E., Salles Trevisan M. T., Venâncio T., Arcisio-Miranda M., Ito A. S., Carlos R. M. J. Med. Chem. 2016;59:9215–9227. doi: 10.1021/acs.jmedchem.6b01130. [DOI] [PubMed] [Google Scholar]
- Babu E., Muthu Mareeswaran P., Sathish V., Singaravadivel S., Rajagopal S. Talanta. 2015;134:348–353. doi: 10.1016/j.talanta.2014.11.020. [DOI] [PubMed] [Google Scholar]
- Sathish V., Babu E., Ramdass A., Lu Z.-Z., Velayudham M., Thanasekaran P., Lu K.-L., Rajagopal S. Talanta. 2014;130:274–279. doi: 10.1016/j.talanta.2014.06.070. [DOI] [PubMed] [Google Scholar]
- Aliyan A., Kirby B., Pennington C., Martí A. A. J. Am. Chem. Soc. 2016;138:8686–8689. doi: 10.1021/jacs.6b04411. [DOI] [PubMed] [Google Scholar]
- Wadghiri Y. Z., Hoang D. M., Wisniewski T., Sigurdsson E. M. Methods Mol. Biol. 2012;849:435–451. doi: 10.1007/978-1-61779-551-0_30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huddleston D. E., Small S. A. Nat. Clin. Pract. Neurol. 2005;1:96–105. doi: 10.1038/ncpneuro0046. [DOI] [PubMed] [Google Scholar]
- Jack Jr. C. R., Garwood M., Wengenack T. M., Borowski B., Curran G. L., Lin J., Adriany G., Grohn O. H., Grimm R., Poduslo J. F. Magn. Reson. Med. 2004;52:1263–1271. doi: 10.1002/mrm.20266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer M., Bauer H., Mintorovitch J., Requardt M., Weinmann H. J. Invest. Radiol. 2005;40:715–724. doi: 10.1097/01.rli.0000184756.66360.d3. [DOI] [PubMed] [Google Scholar]
- Tooyama I., Yanagisawa D., Taguchi H., Kato T., Hirao K., Shirai N., Sogabe T., Ibrahim N. F., Inubushi T., Morikawa S. Ageing Res. Rev. 2016;30:85–94. doi: 10.1016/j.arr.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Beckmann N., Gerard C., Abramowski D., Cannet C., Staufenbiel M. J. Neurosci. 2011;31:1023–1031. doi: 10.1523/JNEUROSCI.4936-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salerno M., Santo Domingo Porqueras D. Coord. Chem. Rev. 2016;327–328:27–34. [Google Scholar]
- Verwilst P., Park S., Yoon B., Kim J. S. Chem. Soc. Rev. 2015;44:1791–1806. doi: 10.1039/c4cs00336e. [DOI] [PubMed] [Google Scholar]
- Martins A. F., Oliveira A. C., Morfin J.-F., Laurents D. V., Tóth É., Geraldes C. F. G. C. J. Biol. Inorg. Chem. 2016;21:83–99. doi: 10.1007/s00775-015-1316-9. [DOI] [PubMed] [Google Scholar]
- Patil R., Gangalum P. R., Wagner S., Portilla-Arias J., Ding H., Rekechenetskiy A., Konda B., Inoue S., Black K. L., Ljubimova J. Y., Holler E. Macromol. Biosci. 2015;15:1212–1217. doi: 10.1002/mabi.201500062. [DOI] [PMC free article] [PubMed] [Google Scholar]