

Abstract
Severe pulmonary tuberculosis (STB) is a life-threatening condition with high economic and social burden. The present study aimed to screen for distinct proteins in different stages of TB and identify biomarkers for a better understanding of TB progression and pathogenesis. Blood samples were obtained from 81 patients with STB, 80 with mild TB (MTB) and 50 healthy controls. Differentially expressed proteins were identified using liquid chromatography-tandem mass spectrometry-based label-free quantitative proteomic analysis. Functional and pathway enrichment analyses were performed for the identified proteins. The expression of potential biomarkers was further validated by western blot analysis and enzyme-linked immunosorbent assays. The accuracy, sensitivity and specificity for selected protein biomarkers in diagnosing STB were also evaluated. A total of 1,011 proteins were identified in all three groups, and 153 differentially expressed proteins were identified in patients with STB. These proteins were involved in ‘cellular process’, ‘response to stimulus’, ‘apoptotic process’, ‘immune system process’ and ‘select metabolic process’. Significant differences in protein expression were detected in α-1-acid glycoprotein 2 (ORM2), interleukin-36α (IL-36α), S100 calcium binding protein A9 (S100-A9), superoxide dismutase (SOD)1 in the STB group, compared with the MTB and control groups. The combination of plasma ORM2, IL-36α, S100A9 and SOD1 levels achieved 90.00% sensitivity and 92.16% specificity to discriminate between patients with STB and MTB, and 89.66% sensitivity and 98.9% specificity to discriminate between patients with STB and healthy controls. ORM2, S100A9, IL-36α and SOD1 were associated with the development of TB, and have the potential to distinguish between different stages of TB. Differential protein expression during disease progression may improve the current understanding of STB pathogenesis.
Keywords: severe pulmonary tuberculosis, α-1-acid glycoprotein 2, S100 calcium binding protein A9, interleukin-36α, superoxide dismutase 1, proteomics
Introduction
There were an estimated 10.4 million new cases of tuberculosis (TB) occurring in 2015, with TB still one of the top 10 causes of death worldwide (1). Disease progression is one of the most predominant causes of TB mortality and the mortality rate of patients with severe pulmonary TB is 69–80% (2). Pulmonary TB is characterized by granuloma formation, caseation and ultimately pulmonary cavitation, reflecting a complex interplay that exists between distinctive components of the innate and acquired immune responses, as well as with the pathogen itself (3).
The human body releases TB-associated proteins into the bloodstream through several signaling pathways when infected by Mycobacterium tuberculosis, and varying concentrations of proteins can be released under different severity states of the disease (4). A previous study reported that expression levels of certain proteins, such as peroxiredoxin 1 and myosin heavy chain 9, in serum samples obtained from patients with active or latent TB infections were different from those in healthy people (5). The efficiency for identification of novel biomarkers from clinical samples has been greatly improved due to the development of quantitative proteomic approaches (6). Li et al (7) demonstrated that the detection of upregulated sex hormone-binding globulin (SHBG) expression may be used to diagnose pulmonary TB with 78.74% accuracy, 75.6% sensitivity and 91.5% specificity, indicating that SHBG may be a novel potential serum biomarker for pulmonary TB. Therefore, different disease severity states may be detected by different protein biomarkers. However, the pathogenesis of TB progression remains unclear and only a limited number of biomarkers for severe TB have been reported.
In order to better characterize specific biomarkers for different severity states of TB, the present study used proteomic technology to screen distinct proteins in different stages of TB. Functional and pathway enrichment analyses were performed for distinct proteins, which may better the understanding of the pathogenesis of TB progression. Furthermore, differentially expressed proteins in serum were validated by western blot analysis and enzyme-linked immunosorbent assays (ELISAs). In addition, the accuracy, sensitivity and specificity for selected protein biomarkers in diagnosing severe TB was evaluated.
Patients and methods
Patients
Subjects were prospectively enrolled and recruited by the staff at the Beijing Chest Hospital affiliated to Capital Medical University (Beijing, China) between March and October 2015. The present study was conducted according to the principles of the Declaration of Helsinki and approved by the Ethical Committee of the Beijing Chest Hospital affiliated to Capital Medical University (Beijing, China). All participants were ≥18 years old and provided written informed consent. Individuals receiving immunomodulatory therapy, with extrapulmonary TB, autoimmune disease, chronic disease, malignancies or HIV infection were excluded.
The patients were diagnosed with TB according to combined clinical criteria from the World Health Organization (8). Included patients with pulmonary TB had a positive sputum smear or culture result. The present study included 211 specimens and 161 samples, which were obtained from 161 patients with TB. Patients with pulmonary TB were further stratified according to disease severity, into a severe pulmonary TB (STB) group and a mild pulmonary TB (MTB) group. Patients were considered to be STB if three or more lung lobe alterations were observed in a chest computed tomography (CT) scan, with respiratory failure or hypoxemia (9). Patients were classified as MTB if they had less than three lung lobe alterations observed in a chest CT and normal blood oxygen. In the present study, the 161 patients were divided into 81 STB and 80 MTB cases.
Healthy subjects were recruited from a population who had no exposure to Mycobacterium tuberculosis, a negative tuberculin skin test and T-SPOT TB test result, a normal chest CT and no clinical evidence of any other diseases. A total of 50 age-matched non-Mycobacterium tuberculosis infected healthy control samples (NC) were selected. There was no significant difference in age, sex ratio, body mass index or smoking habits among patients in the STB, MTB and NC groups (all P>0.05; Table I).
Table I.
Characteristics of patients with TB and NC group.
| Patient characteristics | Severe TB | Mild TB | NC | P-value (STB vs. MTB) | P-value (STB vs. NC) |
|---|---|---|---|---|---|
| Diagnosis (n) | 81 | 80 | 50 | ||
| Age, years | 55.31±18.24 | 50.43±17.27 | 49.83±18.09 | 0.08 | 0.10 |
| Male (n) | 54 | 52 | 32 | ||
| Female (n) | 27 | 28 | 18 | 0.82 | 0.75 |
| BMI | 21.07±3.12 | 23.17±3.03 | 22.49±2.98 | 0.05 | 0.06 |
| Smoking history, n (%) | 31 (38.27) | 28 (35.00) | 19 (38.00) | 0.67 | 0.97 |
Age and BMI are presented as the mean ± standard deviation. TB, tuberculosis; BMI, body mass index; n, number of subjects; NC, negative control; STB, severe pulmonary TB; MTB, mild TB.
Plasma sampling and protein extraction
All blood samples from each group were collected from venous catheters into tubes containing EDTA. Blood samples were centrifuged at 1,400 × g for 5 min at 4°C and plasma was stored at −80°C.
Albumin and immunoglobulin G (IgG) were removed using an immunodepletion column according to the manufacturer's instructions (Agilent Technologies, Inc., Santa Clara, CA, USA). Briefly, plasma samples (600 µl) were transferred into a 1.5 ml screw cap tube and centrifuged at 10,000 × g for 30 min at 4°C. Following this, 100 µl lysis buffer (7 M urea and 2 M thiourea; pH 7.4) was added into each sample. Proteins were subsequently diluted in 50 mM NH4HCO3 solution to a final concentration of 0.5 mg/ml. Finally, trypsin (Promega Corporation, Madison, WI, USA) was added at an enzyme-to-substrate ratio of 1:50 at 37°C for 12 h. Digested supernatant fractions were stored at −80°C without further treatment until liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.
Label-free LC-MS/MS analysis
Digested peptide mixtures were pressure-loaded onto a fused silica capillary column packed with 3 µm dionex C18 reversed phase material (Phenomenex, Inc., Tianjin, China). The reverse phase sections (pore size, 100Å) were 15 cm in length and the column was washed with buffer A (water, 0.1% formic acid) and buffer B (acetonitrile, 0.1% formic acid). Following desalting, a C18 capture tip (5 mm; 300 µm) was placed in line with an Agilent 1,100 quaternary high performance liquid chromatography system (Agilent Technologies, Inc.) and analyzed using a 12-step separation. Tryptic peptides (100 µg) were eluted with a linear gradient from 2–100% buffer B over 70 min at a flow rate of 0.5 µl/min. As peptides were eluted from the micro-capillary column, they were electrosprayed directly into a mass spectrometer with (micrOTOF-Q II; Bruker Scientific Technology Co., Ltd., Beijing, China) with the application of a distal 180°C source temperature of N2 and a nebulizer pressure of 50 psi. The mass spectrometer was operated in the MS/MS (auto) mode, and the ionization mode used was positive.
Quantitative shotgun proteomics data analysis
Data-independent ion selection was monitored to select the most abundant two ions from an MS scan for analysis. Dynamic exclusion was continued for 5 min, and performed for 70 min in total. Mass spectra were processed with DataAnalysis version 4.1 (Bruker Scientific Technology Co., Ltd.) and the Mascot generic format files were searched against the NCBI human database for tryptic peptides with up to one miscleavage using Mascot software version 2.1 (Matrix Science, Ltd., London, UK).
Bioinformatics analysis
Tandem mass spectra were searched for in the National Centre for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov) using Mascot software 2.1 (Matrix Science, Ltd.). The default parameters for the quantification software, ProfileAnalysis software version 2.0, were used throughout the analysis (Bruker Scientific Technology Co., Ltd.). The differentially expressed proteins were identified with the cut-off criteria of false discovery rate <0.01 and |log2 fold change (FC)|>1. Gene ontology (GO) annotation was carried out to categorize proteins based on biological process (BP), cellular component (CC) and molecular function (MF) using the Protein Analysis Through Evolutionary Relationships database (www.pantherdb.org) (10). Signaling pathway analysis was performed using the Search & Color Pathway tool on the Kyoto Encyclopedia of Genes and Genome (KEGG) database (www.genome.jp/kegg/pathway.html). Pathway enrichment analysis was conducted with the Database for Annotation, Visualization and Integrated Discovery (http://kobas.cbi.pku.edu.cn/anno_iden.php.) (11), with P<0.05 used as the cut-off value. Protein-protein interactions were obtained from the Search Tool for the STRING database (version 9.0; http://string-db.org/cgi/input.pl), containing known and predicted physical and functional protein-protein interactions (12).
Western blot analysis
Six different serum samples from each group were used for western blot analysis. Proteins were extracted using radioimmunoprecipitation assay lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40, 1% deoxycholic acid sodium, 0.1% SDS, 0.1M phenylmethane sulfonyl fluoride, Roche Complete protease inhibitor cocktail tablets and phosphastase inhibitor cocktail tablets). Following this, total protein was determined using a bicinchoninic acid assay, and protein samples (20 µg/lane) were subsequently separated via 8, 12 and 15% SDS-PAGE gels and transferred onto polyvinylidene fluoride membranes. Following blocking with 3% bovine serum albumin with Tris-buffered saline containing 0.1% Tween-20 (pH 8.0; Sigma-Aldrich; Merck KGaA) at 25°C for 30 min, the membranes were incubated overnight at 4°C with primary antibodies against interleukin-36α (IL-36α; cat. no. ab117925; 1:1,000), α-1-acid glycoprotein (ORM)1 (cat. no. ab134042; 1:2,000), ORM2 (cat. no. ab88869; 1:2,000), S100 calcium binding protein A9 (S100A9; cat. no. ab92057; 1:1,000), superoxide dismutase (SOD)1 (cat. no. ab51254; 1:1,000), probetacellulin precursor (BTC; cat. no. ab10417; 1:1,000), tyrosine-protein kinase Lyn isoform B (LYN; cat. no. ab32398; 1:4,000) and collagen III (cat. no. ab7778; 1:1,000; all Abcam, Cambridge, MA, USA). Membranes were washed with Tris-buffered saline (10 mmol/l Tris-HCl, 150 mmol/l NaCl and 0.1% Tween-20 containing 5% skimmed milk) prior to incubation with horseradish peroxidase-conjugated anti-rabbit IgG (cat. no. S006; 1:5,000) or anti-mouse IgG (cat. no. S002; 1:5,000; both Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h at room temperature. Transferrin (cat. no. YM3527; 1:10,000; Beyotime Institute of Biotechnology, Haimen, China) was used as an internal reference. Proteins were visualized using an electrochemiluminescence kit (Amersham Biosciences, Buckinghamshire, UK) according to the manufacturer's instructions. Proteins levels were normalized via staining with 2% Ponceau S stain for 1 min at 25°C, and transferrin (cat. no. YM3527; 1:10,000; Beyotime Institute of Biotechnology) was used as the loading control. Densitometric analysis was performed using Quant software (version 11.5; TotalLab, Ltd., Newcastle upon Tyne, UK). The experiments were performed in triplicate.
Validation of selected protein expression levels
ELISAs were performed to determinate the protein concentrations in plasma samples of STB, MTB and the NC groups. The commercial ELISA kits for four selected proteins were purchased including human IL-36α ELISA kit (cat. no. KA9520; 1:50), human ORM2 ELISA kit (cat. no. KA0480; 1:100), human protein S100-A9 ELISA kit (cat. no. KA1050; 1:50 dilution) and human SOD1 ELISA kit (cat. no. KA0680; 1:50; all Abnova, Taipei, Taiwan). The ELISA was performed according to the manufacturer's protocol. Protein levels of IL-36α, ORM2, S100-A9 and SOD1 in STB (n=72), MTB (n=71), and NC samples (n=41) were measured.
Statistical analysis
Statistical analysis was performed using SPSS version 18.0 (SPSS, Inc., Chicago, IL, USA). Qualitative variables were analyzed by the Fisher's exact test and Pearson's chi-squared test, while quantitative variables were analyzed by one-way ANOVA followed by the Student-Newman-Keuls post hoc test. P<0.05 was considered to indicate a statistically significant difference. Receiver operating characteristic curves were used to assess the diagnostic value of biomarkers.
Results
Identification of differentially expressed proteins by label-free quantitative shotgun proteomics analysis
The mass spectra data were searched using the profile analysis search algorithm against Human RefSeq protein database (https://www.ncbi.nlm.nih.gov), which is comprised of 69,002 protein sequences. A total of 3,385 proteins were quantified across all three biological replicates in each group (n=9) using the label-free proteomics analysis method.
Based on the LC-MS/MS data, 1,011 proteins were identified across all three biological replicates in each group (n=9) with a relatively good reproducibility. Compared with the MTB samples, 299 proteins were significantly differentially expressed, including 165 upregulated and 134 downregulated proteins in STB samples. In addition, the expression levels of 512 proteins in STB samples were significantly different (284 upregulated and 228 downregulated proteins) from the NC samples. A total of 153 proteins were found to be significantly differentially expressed in STB samples, of which 82 proteins were upregulated and 71 proteins were downregulated compared with the MTB and NC samples.
Enrichment analysis and protein-protein interaction network construction
The 153 differentially expressed proteins in STB samples were used for GO database annotation in terms of BP, CC and MF (Fig. 1). In BP terms, proteins were mainly involved in ‘cellular process’, ‘responses to stimulus’, ‘apoptotic process’ and ‘immune system process’. In CC terms, the majority of the potential biomarkers were associated with the ‘organelle’, ‘cell junction’, ‘membrane’ and ‘macromolecular complex’. The enrichment analysis in MF terms revealed that ‘catalytic activity’, ‘binding’ and ‘enzyme regulatory activity’.
Figure 1.

Gene ontology annotations in terms of BP, CC and MF terms for the 153 differentially expressed proteins in plasma samples from patients with severe tuberculosis. BP, biological process; CC, cellular component; MF, molecular function.
Pathway enrichment analysis of the 153 differentially expressed proteins in the STB samples obtained 8 upregulated and 1 downregulated signaling pathway (Fig. 2). The 8 upregulated signaling pathways were ‘glycosaminoglycan biosynthesis-chondroitin sulfate/dermatan sulfate’, ‘galactose metabolism’, ‘one carbon pool by folate’, ‘axon guidance’, ‘ECM-receptor interaction’, ‘ErbB signaling pathway’, ‘butirosin and neomycin biosynthesis’ and ‘focal adhesion’. In addition, these proteins were predominantly involved in the downregulated signaling pathway ‘amyotrophic lateral sclerosis’.
Figure 2.

The enriched (A) upregulated (B) and downregulated signaling pathways for the 153 differentially expressed proteins in plasma samples from patients with severe tuberculosis. KEGG, Kyoto Encyclopedia of Genes and Genomes.
Furthermore, a network was constructed from selected protein-protein interactions (Fig. 3). This protein-protein interaction network consisted of 18 proteins, including ORM1, ORM2, S100A9, matrix metallopeptidase 16, copper chaperone for superoxide dismutase, peroxiredoxin (PRDX)1, glutathione peroxidase (GPX)2, SOD1, SOD2, PRDX2, ubiquitin C, catalase, GPX1, IL-36α, S100A8 and TIMP metallopeptidase inhibitor 2.
Figure 3.
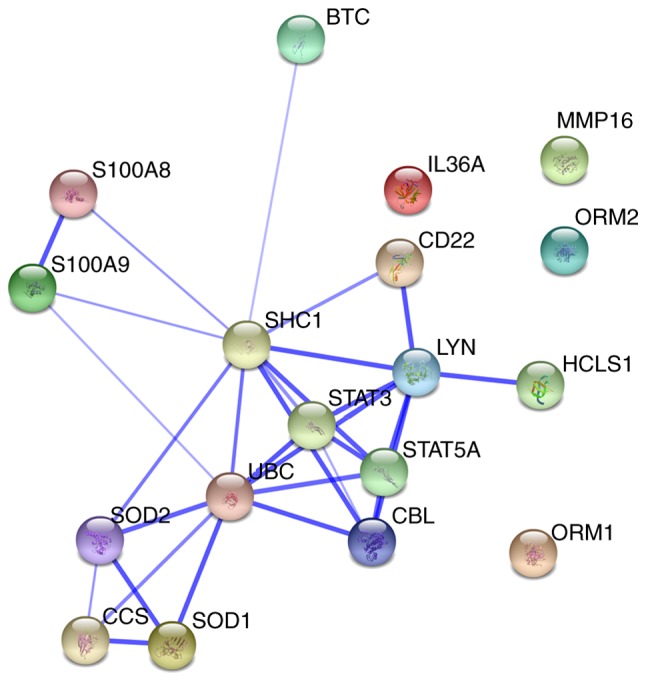
Visualization of protein-protein interaction network for dysregulated severe tuberculosis proteins. Dysregulated proteins are represented as spheres of distinct colors. Blue lines represent interactions between proteins and the thickness of the lines depicts the level of confidence associated with each interaction.
Confirmation of differential levels of candidate protein expression by western blot
The expression levels of eight differentially expressed proteins (ORM1, SOD1, IL-36α, ORM2, S100A9, BTC, LYN and collagen III) with the highest values of |log2FC| were detected by western blot analysis. Six proteins among these eight differentially expressed proteins were demonstrated to be differentially expressed. In the plasma samples of the STB group, ORM1, IL-36α, ORM2, S100A9 and collagen III were upregulated, and SOD1 was downregulated, compared with the MTB and NC groups (Fig. 4A). This trend matched with what had been previously observed in the label-free quantitative proteomics analysis. Densitometry demonstrated that the protein expression levels of each protein were significantly different between the STB and NC groups (Fig. 4B; P<0.05) The plasma expression levels of ORM1, ORM2, IL-36α, and collagen III were significantly higher in the STB group compared with those in the MTB and NC groups (Fig. 4B).
Figure 4.

Western blot analysis of eight selected proteins in plasma samples of the STB, MTB and NC groups. (A) Protein expression was determined by western blot analysis. (B) Densitometric analysis of the eight plasma proteins in the STB group compared with the MTB and NC groups. *P<0.05 and **P<0.01. N/S, not significant; STB, severe pulmonary tuberculosis; MTB, mild pulmonary tuberculosis; NC, healthy control samples; BTC, betacellulin; SOD, superoxide dismutase; ORM, α-1-acid glycoprotein; LYN, LYN proto-oncogene, Src family tyrosine kinase; IL-36α, interleukin-36α.
Expression levels of IL-36α, ORM2, S100A9 and SOD1 by ELISA
The plasma expression of four candidate proteins including ORM2, S100A9, IL-36α and SOD1 were measured by ELISA (Fig. 5). IL-36α and collagen III were not detected due to lack of commercially-available ELISA kits and relatively lower values of |log2FC|. The expression levels of ORM2, S100A9, IL-36α and SOD1 in the STB group were significantly differently compared with the other two groups (P<0.05; Table II). The expression trend of these proteins was consistent with the results obtained from proteomics analysis.
Figure 5.

Validation of ORM2, S100A9, IL-36α and SOD1 expression in plasma. Levels of these candidate biomarkers were measured by enzyme-linked immunosorbent assay in the NC (n=41), MTB (n=71), STB (n=72) groups. n, number of subjects. Median values are depicted by the red horizontal lines. *P<0.05 and **P<0.01. ORM2, α-1-acid glycoprotein 2; S100A9, S100 calcium binding protein A9; IL-36α, interleukin-36α; SOD, superoxide dismutase; STB, severe pulmonary tuberculosis; MTB, mild pulmonary tuberculosis; NC, healthy control samples.
Table II.
The median, upper and lower quartiles of plasma concentrations of ORM2, S100A9, IL-36α and SOD1in the STB, MTB and NC groups.
| Characteristic | STB | MTB | NC | P-value (STB vs. MTB) | P-value (STB vs. NC) |
|---|---|---|---|---|---|
| ORM2 (mg/ml) | 614.49 | 514.57 | 386.75 | <0.001 | <0.001 |
| Lower quartile (mg/ml) | 481.23 | 335.96 | 268.39 | ||
| Upper quartile (mg/ml) | 805.93 | 639.64 | 488.43 | ||
| S100A9 (mg/ml) | 14.90 | 5.07 | 2.55 | 0.03 | 0.001 |
| Lower quartile (mg/ml) | 11.37 | 2.70 | 1.69 | ||
| Upper quartile (mg/ml) | 19.34 | 6.95 | 3.25 | ||
| IL-36α (ng/ml) | 92.15 | 59.55 | 21.90 | 0.02 | <0.001 |
| Lower quartile (mg/ml) | 48.42 | 40.63 | 11.54 | ||
| Upper quartile (mg/ml) | 203.25 | 105.38 | 51.03 | ||
| SOD1 (mg/ml) | 771.70 | 985.75 | 1,021.03 | <0.001 | <0.001 |
| Lower quartile (mg/ml) | 648.26 | 652.7 | 638.53 | ||
| Upper quartile (mg/ml) | 913.40 | 1,257.81 | 1,290.86 |
ORM2, S100A9, S100 calcium binding protein A9; IL-36α, interleukin-36α; SOD1, superoxide dismutase 1; STB, severe tuberculosis; MTB, mild tuberculosis; NC, healthy control samples.
Diagnostic value of IL-36α, ORM2, S100A9 and SOD1
The sensitivity, specificity and accuracy of ORM2 (79.3%, 48.0% and 0.587), S100A9 (86.2%, 90.0% and 0.891), IL-36α (82.4%, 67.4% and 0.791), and SOD1 (79.3%, 31.9% and 0.525) in discriminating between STB and MTB were presented in Fig. 6A. Furthermore, the combination of these four proteins used for discriminating STB from MTB achieved 90.00% sensitivity, 92.16% specificity and 0.965 accuracy.
Figure 6.

Efficacy of SOD1, S100A9, IL-36α and ORM2 in discriminating STB from MTB and the NC group. ROC curves of SOD1, S100A9, IL-36α and ORM2 in discriminating (A) STB from MTB, and (B) STB from the NC group. S100A9, S100 calcium binding protein A9; IL-36α, interleukin-36α; SOD, superoxide dismutase; ORM2, α-1-acid glycoprotein; STB, severe pulmonary tuberculosis; MTB, mild pulmonary tuberculosis; NC, healthy control samples; ROC, receiver operating characteristic.
For discriminating between STB and NC, the sensitivity, specificity, and accuracy of ORM2 was 98.0%, 35.4% and 0.720; S100A9, 62.0%, 84.3% and 0.729; IL-36α, 51.9%, 77.3% and 0.555; and SOD1 36.2%, 94.1% and 0.635, respectively (Fig. 6B). In addition, the combination of these four proteins for STB discrimination from NC achieved a sensitivity of 89.66%, specificity of 98.9% and accuracy of 0.981.
Discussion
Proteomics is a high throughput analytic method and proteins identified by mass spectrometry may be useful in the diagnosis and clinical staging of disease. For example, Zhang et al (13) identified potential protein biomarkers for different stages of chronic obstructive pulmonary disease using proteomic technology. Drug resistance, lesion location, complications and patient management may aggravate TB (14), although the specific mechanisms involved in TB progression are complicated and remain unclear. In the present study, the proteomic spectra of plasma samples from patients with STB, MTB and the NC group were analyzed using a LC-MS/MS-based label-free quantitative method. The disease severity of this study was defined by lung CT and blood oxygen results. Based on the LC-MS/MS data, 1,011 proteins were identified in these three groups from all three biological replicates per group. Furthermore, 153 significantly differentially expressed proteins in STB group were identified, compared with the MTB and NC groups. The GO annotations indicated that progression of TB may be associated with ‘responses to stimuli’, ‘apoptotic process’, ‘immune system process’ and ‘select metabolic process’.
The identified differentially expressed proteins were validated by further experimentation. Western blot analysis and ELISAs further demonstrated that the expression of ORM2, S100A9, IL-36α and SOD1 in the plasma samples of the STB group were significantly different from the other two groups. These four proteins have previously been reported to be involved in several pathophysiological processes of pulmonary TB. ORM2 is a precursor of α-2-acid glycoprotein is a major acute-phase protein with an important role in cell inflammation, cell-mediated immunity suppression, as well as enhancing the growth of bacilli and promoting disease progression (15). The synthesis of ORM2 is increased during various acute-phase responses and is mainly produced by alveolar macrophages and type II pneumocytes in the early stage of pulmonary tuberculosis, and by foamy macrophages located in tuberculous areas at the advanced stage (16). In the present study, expression levels of ORM2 significantly increased in patients with STB, compared with cases of MTB or the NC group. Therefore, it was hypothesized that ORM2 may be a potential protein biomarker for TB progression. IL-36α is a member of the interleukin-1 cytokine family, that is upregulated in inflammatory disease states (17). Intratracheal instillation of IL-36α enhances IL-36α receptor mRNA expression in the lungs of C57BL/6 and IL-1αβ (−/-) mice (18). The present study demonstrated that IL-36α expression was markedly increased in patients with STB, compared with patients with MTB and the NC group. The expression of these proteins at high levels in the plasma of individuals with STB indicated that the inflammatory response may serve an important role in STB-induced injury. However, the precise effect of these proteins on the development of TB requires further exploration.
S100A9 is a major calcium-binding protein in neutrophils and monocytes that is also recognized as a damage-associated molecular-pattern molecule. Gopal et al (19) reported that S100A9 expression is upregulated in patients with TB, and may have a major pathological role in TB by mediating neutrophil accumulation and inflammation. Greater areas of lung tissue infected with mycobacterium tuberculosis, inflammation and severe hypoxia increase S100A9 expression, leading to aggravation of lung injury inflammation (20). In the present study, hypoxic conditions were more severe in STB group compared with the MTB and NC groups, and S100A9 expression was revealed to be upregulated in the STB group compared with the MTB and NC groups. This suggests that there may be an association between hypoxemia and S100A9 expression.
SOD1 is the predominant SOD isoform in the majority of cells and tissues, accounting for 70–80% of total cellular SOD activity (21). SOD1 inhibits inflammation by preventing oxidative fragmentation of hyaluronan and catalyzing the dismutation of the superoxide anion to oxygen and hydrogen peroxide. TB is associated with oxidative stress and the induction of host antioxidants to counteract this response. Golubović et al (22) reported that serum SOD expression was significantly lower in patients with TB compared with healthy individuals, and imbalance in this coordinated system lead to increased oxidative stress. In addition, it has been indicated that the concentration of SOD decreases in patients with pulmonary failure (23). Furthermore, SOD proteins considerably reduced the production of NO and oxygen radicals and ameliorated impairment in cell immunological function during early infection (24). The present study demonstrated that SOD1 levels were significantly lower in patients with STB, compared with the MTB and NC groups. Therefore, SOD1 may be a protective factor against pulmonary TB. This protein may serve a key role in maintaining the balance between oxidation and antioxidation, thereby inhibiting the inflammatory damage process (25). The current study also demonstrated that a combination of ORM2, S100A9, IL-36α and SOD1 distinguished between patients with STB and MTB (sensitivity, 90.00%; specificity, 92.16%), as well as between patients with STB and the NC group (sensitivity, 89.66%; specificity, 98.9%). These results revealed that these four differentially expressed proteins may be useful in discriminating between the different severities of TB.
In conclusion, 1,011 proteins were identified in STB, MTB and NC groups based on LC-MS/MS-based label-free quantitative analysis. A total of 153 differentially expressed proteins were identified in the plasma samples obtained from patients with STB. Proteins including ORM2, S100A9, IL-36α and SOD1 were determined to be associated with the development of TB, and may be useful as biomarkers to ascertain pulmonary TB clinical severity. Therefore, differential protein expression during disease progression may improve the understanding of STB pathogenesis. The present study provided meaningful data and novel ideas to determine the severity of TB.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the Beijing Natural Science Foundation (grant no. 7164245), the Beijing Nova Program (grant no. Z161100004916080), the National Science and Technology Major Project of China (grant no. 2015ZX10004801-003), the Collaborative Innovation Center of Infectious Diseases (grant no. PXM2016_014226_000052), the ‘Beijing Municipal Administration of Hospitals’ Youth Program (grant no. QML20151501) and Tongzhou District Science and Technology Committee (grant no. KJ2017CX076).
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
QiuL, ZZ and QiL conceived and designed the study. QiuL, LP, FH and BL performed western blotting analyses. HJ, AX and QiL collected samples, and performed data analysis and interpretation. All authors wrote and approved the final version of the manuscript to be published.
Ethics approval and consent to participate
The study was conducted according to the principles of the Declaration of Helsinki and was approved by the Ethical Committee of the Beijing Chest Hospital affiliated to Capital Medical University (Beijing, China). All participants provided written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Global tuberculosis report 2016. World Health Organization; Geneva: 2016. pp. 1–201. [Google Scholar]
- 2.Waitt CJ, Squire SB. A systematic review of risk factors for death in adults during and after tuberculosis treatment. Int J Tuberc Lung Dis. 2011;15:871–885. doi: 10.5588/ijtld.10.0352. [DOI] [PubMed] [Google Scholar]
- 3.Barry CE, III, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, Schnappinger D, Wilkinson RJ, Young D. The spectrum of latent tuberculosis: Rethinking the biology and intervention strategies. Nat Rev Microbiol. 2009;7:845–855. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Altena R, Duggirala S, Gröschel MI, van der Werf TS. Immunology in tuberculosis: Challenges in monitoring of disease activity and identifying correlates of protection. Curr Pharm Des. 2011;17:2853–2862. doi: 10.2174/138161211797470228. [DOI] [PubMed] [Google Scholar]
- 5.Zhang X, Liu F, Li Q, Jia H, Pan L, Xing A, Xu S, Zhang Z. A proteomics approach to the identification of plasma biomarkers for latent tuberculosis infection. Diagn Microbiol Infect Dis. 2014;79:432–437. doi: 10.1016/j.diagmicrobio.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Serada S, Naka T. Screening for novel serum biomarker for monitoring disease activity in rheumatoid arthritis using iTRAQ technology-based quantitative proteomic approach. Methods Mol Biol. 2014;1142:99–110. doi: 10.1007/978-1-4939-0404-4_12. [DOI] [PubMed] [Google Scholar]
- 7.Li C, He X, Li H, Zhou Y, Zang N, Hu S, Zheng Y, He M. Discovery and verification of serum differential expression proteins for pulmonary tuberculosis. Tuberculosis (Edinb) 2015;95:547–554. doi: 10.1016/j.tube.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 8.World Health Organization, corp-author. International Union Against Tuberculosis and Lung Disease; Royal Netherlands Tuberculosis Association: Revised international definitions in tuberculosis control. Int J Tuberc Lung Dis. 2001;5:213–215. [PubMed] [Google Scholar]
- 9.Lichtenstein DA, Mezière GA. Relevance of lung ultrasound in the diagnosis of acute respiratory failure: The BLUE protocol. Chest. 2008;134:117–125. doi: 10.1378/chest.07-2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45(D1):D183–D189. doi: 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 12.Satoh K, Maniwa T, Oda T, Matsumoto K. Proteomic profiling for the identification of serum diagnostic biomarkers for abdominal and thoracic aortic aneurysms. Proteome Sci. 2013;11:27. doi: 10.1186/1477-5956-11-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X, Zhang J, Li Q, Wu X, Wang T, Wang Y. SELDI-TOF-MS in chronic obstructive pulmonary disease. Chin Sci Bull. 2013;58:pp634–640. doi: 10.1007/s11434-012-5460-0. [DOI] [Google Scholar]
- 14.Hongmei Chen, Xiaoguang WU, Liping MA. The clinical analysis of the hospitalized elderly patients died from tuberculosis. Chin J Antituberculosis. 2014;36:55–58. (In Chinese) [Google Scholar]
- 15.Fournier T, Medjoubi-N N, Porquet D. Alpha-1-acid glycoprotein. Biochim Biophys Acta. 2000;1482:157–171. doi: 10.1016/S0167-4838(00)00153-9. [DOI] [PubMed] [Google Scholar]
- 16.Cordero Martìnez E, Gonzàlez MM, Aguilar LD, Orozco EH, Pando Hernàndez R. Alpha-1-acid glycoprotein, its local production and immunopathological participation in experimental pulmonary tuberculosis. Tuberculosis (Edinb) 2008;88:203–211. doi: 10.1016/j.tube.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 17.Ichii O, Kimura J, Okamura T, Horino T, Nakamura T, Sasaki H, Elewa YHA, Kon Y. IL-36α regulates tubulointerstitial inflammation in the mouse kidney. Front Immunol. 2017;8:1346. doi: 10.3389/fimmu.2017.01346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramadas RA, Ewart SL, Iwakura Y, Medoff BD, LeVine AM. IL-36α exerts pro-inflammatory effects in the lungs of mice. PLoS One. 2012;7:e45784. doi: 10.1371/journal.pone.0045784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, Junecko Fallert BA, Reinhart TA, Kolls J, Báez-Saldaña R, et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med. 2013;188:1137–1146. doi: 10.1164/rccm.201304-0803OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuipers MT, Vogl T, Aslami H, Jongsma G, van den Berg E, Vlaar AP, Roelofs JJ, Juffermans NP, Schultz MJ, van der Poll T, et al. High levels of S100A8/A9 proteins aggravate ventilator-induced lung injury via TLR4 signaling. PLoS One. 2013;8:e68694. doi: 10.1371/journal.pone.0068694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strålin P, Karlsson K, Johansson BO, Marklund SL. The interstitium of the human arterial wall contains very large amounts of extracellular superoxide dismutase. Arterioscler Thromb Vasc Biol. 1995;15:2032–2036. doi: 10.1161/01.ATV.15.11.2032. [DOI] [PubMed] [Google Scholar]
- 22.Golubović S, Stanković I, Ristić L, Cosić V, Dordević I, Radović M. Antioxidant enzymes and lipid peroxidation products in patients with pulmonary tuberculosis. Med Pregl. 2010;63:450–453. doi: 10.2298/MPNS1008450G. (In Serbian) [DOI] [PubMed] [Google Scholar]
- 23.Bouhafs RK, Jarstrand C. Phagocyte-induced lipid peroxidation of lung surfactant. Pediatr Pulmonol. 1999;27:322–327. doi: 10.1002/(SICI)1099-0496(199905)27:5<322::AID-PPUL5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 24.Liao D, Fan Q, Bao L. The role of superoxide dismutase in the survival of Mycobacterium tuberculosis in macrophages. Jpn J Infect Dis. 2013;66:480–488. doi: 10.7883/yoken.66.480. [DOI] [PubMed] [Google Scholar]
- 25.Gao F, Koenitzer JR, Tobolewski JM, Jiang D, Liang J, Noble PW, Oury TD. Extracellular superoxide dismutase inhibits inflammation by preventing oxidative fragmentation of hyaluronan. J Biol Chem. 2008;283:6058–6066. doi: 10.1074/jbc.M709273200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.