

Abstract
Missed abortion (MA) refers to a pregnancy in which there is fetal demise without outside intervention, and additionally no uterine activity that may expel the product of conception (POC) prior to 20 weeks of gestation. Chromosomal abnormalities are the primary cause of MA and single gene defects in the POC may additionally be associated with MA; however, few studies have been conducted on the identification of mutations by whole-exome sequencing. In the present study, 19 unrelated MA POCs were collected and whole-exome sequencing was performed on the POC. Bioinformatics analysis was performed on sequence variants from a list of 286 selected candidate genes that were associated with early embryonic lethality and MA. A total of 36 sequence variants in 32 genes potentially associated with MA were identified in 15 out of 19 patients. Gene Ontology analysis suggested that these genes were enriched in biological processes in early embryonic development, including ‘chordate embryonic development’, ‘cell proliferation’ and ‘forebrain development’. Further strict in silico bioinformatics analysis predicted that the LIM domain-binding protein 1 (c.662C>T; p.S221L) variant was a highly pathogenic variant. In conclusion, the results of the present study provide researchers and clinicians with a better understanding of the etiology and molecular mechanism of human embryonic lethality and MA.
Keywords: missed abortion, whole-exome sequencing, products of conception, mutation, LIM domain-binding protein 1, embryonic lethality, miscarriage
Introduction
The term ‘missed abortion (MA)’, a type of miscarriage, refers to a pregnancy in which there is fetal demise without outside intervention, and also no uterine activity that may expel the product of conception (POC) prior to 20 weeks of gestation (1). Multiple epidemiological factors, including parental or embryonic chromosomal abnormalities, infection, immunological factors, hereditary thrombophilia, uterine abnormalities, endocrinological disorders, and nutritional and environmental factors have been associated with miscarriage (2,3). Cytogenetic analysis of the retained POC is thought to be the most effective test for identifying the cause of MA (4). Chromosomal abnormalities are the primary cause of MA, with errors in chromosome number, copy number variations and abnormalities resulting in structural defects accounting for 60–80% of MAs (4–8). However, the cause of 20–40% of MAs remains unknown, despite current detection methods.
High-throughput sequencing technology is currently widely used for identifying genetic alterations associated with MAs of unknown causes. Single gene defects in the POC may additionally be associated with MA; however, few studies have used high-throughput sequencing methods to study genetic defects in the POC.
In the present study, 19 unrelated MA POCs were collected and whole-exome sequencing (WES) was performed on the POC. Bioinformatics analysis was performed on sequence variants from a list of 286 selected candidate genes that are associated with early embryonic lethality and MA. A total of 36 sequence variants in 32 genes potentially associated with MA were identified in 15 out of 19 POCs. Gene Ontology (GO) analysis suggested that these genes were enriched in biological processes in early embryonic development, including ‘chordate embryonic development’, ‘cell proliferation’ and ‘forebrain development’. The novel genes and genetic alterations may increase knowledge of MA pathogenesis and aid future genetic counseling for MA.
Materials and methods
Subjects
A total of 19 women (between 23 and 42 years of age) participated in the present study, who experienced MA between 5–12 weeks of gestation and who were treated in the Department of Obstetrics and Gynecology of the Chinese People's Liberation Army (PLA) General Hospital (Beijing, China) between March 2017 and June 2017. Patient details are given in Table I. The inclusion criteria were as follows: The participants with spontaneous abortions in early pregnancy with unexplained etiology prior to the 12th week of gestational age and lack of any successful pregnancy in the previous history were included in the study. The exclusion criteria were as follows: The patients with a history of risk factors, including chronic infections, thrombosis, autoimmune diseases, endocrinological disorders or genital malformation were not included in the present study.
Table I.
Clinical and sequencing features of the patients and the embryos.
| Embryo ID | Patient age, years | Gestational age, weeks | Sequencing depth, x | Karyotype analysis of each embryo |
|---|---|---|---|---|
| QW001 | 30 | 6 | 170.95 | 46, XY |
| QW002 | 28 | 12 | 114.06 | 46, XX |
| QW003 | 28 | 7 | 96.61 | 46, XY |
| QW004 | 29 | 9 | 101.93 | 46, XX |
| QW005 | 30 | 7 | 92.99 | 46, XX |
| QW006 | 28 | 10 | 104.26 | 46, XY |
| QW007 | 30 | 8 | 106.04 | 46, XY |
| QW008 | 23 | 6 | 32.32 | 46, XX |
| QW009 | 42 | 14 | 131.72 | 46, XX |
| QW010 | 27 | 10 | 114.73 | 46, XX |
| QW011 | 28 | 8 | 124.83 | 46, XX |
| QW012 | 28 | 9 | 127.92 | 46, XY |
| QW013 | 28 | 8 | 116.69 | 46, XX |
| QW014 | 42 | 6 | 126.82 | 46, XY |
| QW015 | 32 | 5 | 90.60 | 46, XX |
| QW016 | 29 | 10 | 112.91 | 46, XY |
| QW017 | 31 | 7 | 87.46 | 46, XY |
| QW018 | 35 | 11 | 93.35 | 46, XY |
| QW019 | 24 | 8 | 93.06 | 46, XY |
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Chinese PLA General Hospital's research committee and with the 1964 Helsinki declaration and its later amendments, or comparable ethical standards. The present study was approved by the ethical committee of Chinese People's Liberation Army (PLA) General Hospital. Written informed consent was obtained from each participant.
The present study was conducted on 19 preserved chorionic villus samples with the normal chromosome number as determined by next-generation sequencing (NGS). WES was performed on chorionic villus genomic DNA from miscarriage samples of unknown cause.
WES analysis
Each exome was captured using Roche Nimblegen SeqCap EZ Exome v3.0 kit (Roche Applied Science, Madison, WI, USA) according to the manufacturer's protocol. Subsequently, the enriched exomes were sequenced using the Illumina HiSeq ×10 platform (Ilumina, Inc., San Diego, CA, USA). Reads were mapped against the human reference genome hg38 (https://genome.ucsc.edu/index.html) using Burrows-Wheeler Aligner (http://bio-bwa.sourceforge.net/). The single nucleotide variants (SNV) were called by SAMTools (version 0.1.19, http://samtools.sourceforge.net/) and the Genome Analysis Toolkit (GATK, version 4.0,4.0, Broad Institute, http://software.broadinstitute.org/gatk/), and ANNOVAR (http://annovar.openbioinformatics.org/en/latest/) was used for SNV annotation and filtering. Variants fulfilling the following criteria were retained: i) Missense, nonsense, frame-shift, or splice site variants; ii) absent in the dbSNP (http://www.ncbi.nlm.nih.gov/snp/), 1000 Genomes (http://browser.1000genomes.org/index.html), ESP6500 (http://evs.gs.washington.edu/EVS/), Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/) and the Genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org/) databases. Four online functional prediction tools including Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), MutationTaster (http://mutationtaster.org/) and FATHMM-MKL (http://fathmm.biocompute.org.uk/fathmmMKL.htm), were used to predict the variant effect on protein function. Constraint Metrics Z score for missense variation (9), Loss Intolerance (pLI) (10) and Haploinsufficiency Score (11) were used for evaluating the haploinsufficiency effect of each gene. DECIPHER database (https://decipher.sanger.ac.uk/) was used for identifying the previous published copy number variations and single nucleotide variants and the associated disease phenotypes. DAVID Bioinformatics Resources 6.7 (https://david-d.ncifcrf.gov/) was used for conducting the GO analysis.
Sanger sequencing validation
Sanger sequencing was used to validate the WES results and verify whether the potentially disease-causing variants identified were true variants or sequencing artifacts. Sanger sequencing for the LDB1 variant in the POC QW013 was performed using gene-specific primers as follows; the forward primer was 5′-AGGAGTGTCACAATGCTCAGATGAT-3′ and the reverse primer was 5′-GTAAACGGAGACTCAGATGGGAGAG-3′. Cycling parameters were an initial denaturation at 94°C for 5 min followed by 35 cycles of denaturation at 94°C for 20 sec, annealing at 60°C for 30 sec and extension at 72°C for 1 min, followed by a final extension at 72°C for 5 min. TransStart FastPfu DNA polymerase (TransGen Biotech Co., Ltd., Beijing, China) was used in the PCR reaction.
Results
WES analysis of MA embryos
A total of 19 patients (between 23 and 42 years of age) with MA participated in the present study (Table I). A total of 19 MA POCs (chorionic villus from 5–12 gestational weeks) were examined. The karyotypes of all POC were normal (Table I). WES was performed for each POC. The sequencing depth of each WES is listed in Table I. Therefore, WES-detected sequence variants identified in each embryo were focused upon. The present study aimed to identify direct sequence variants causing MA, which should not exist in live human beings. Therefore, polymorphisms with a minor allele frequency absent in the dbSNP, 1000 Genomes, ESP6500, ExAC and gnomAD databases were retained. Subsequently, all variants were further filtered, according to the list of 286 selected candidate genes that were associated with early embryonic lethality and MA. A total of 36 sequence variants in 32 genes potentially associated with MA were identified in 15 out of 19 patients (data not shown). All variants were in the heterozygous state.
In silico analysis of the variants
GO analysis suggested that these 32 genes were enriched in biological processes in early embryonic development, including ‘chordate embryonic development’, ‘cell proliferation’ and ‘forebrain development’ (Fig. 1). In silico analysis predicted that 12 of 36 variants were considered to be pathogenic alleles by four online prediction tools, including Polyphen-2, SIFT, Mutation Taster and FATHMM-MKL (Table II). As all the variants were heterozygous, the present study aimed to determine whether the heterozygous state of the variants influenced disease tolerance. The variation intolerance scores were analyzed using three scoring systems, including the Constraint Metrics Z score for missense variation (9), Loss Intolerance (pLI) (10) and Haploinsufficiency Score (11). Out of the 12 genes, LIM domain binding 1 gene (LDB1) was the only gene that was predicted as intolerant to variation by the three scoring systems (Table II). Sequence variant c.3064C>T; p.P1022S (Table II) in another gene, death induced obliterator-1 (DIDO1), was additionally a potential candidate gene causing MA. This variant was predicted to be a pathogenic allele by Polyphen-2, MutationTaster and FATHMM-MKL. The variant in DIDO1 was considered loss-of-function-intolerant, as predicted by the Constraint Metrics Z score for missense variation and pLI (Table II). Therefore, it was hypothesized that the variant in LDB1 (c.662C>T; p.S221L) and DIDO1 (c.3064C>T; p.P1022S) was likely to be associated with embryo lethality and MA.
Figure 1.
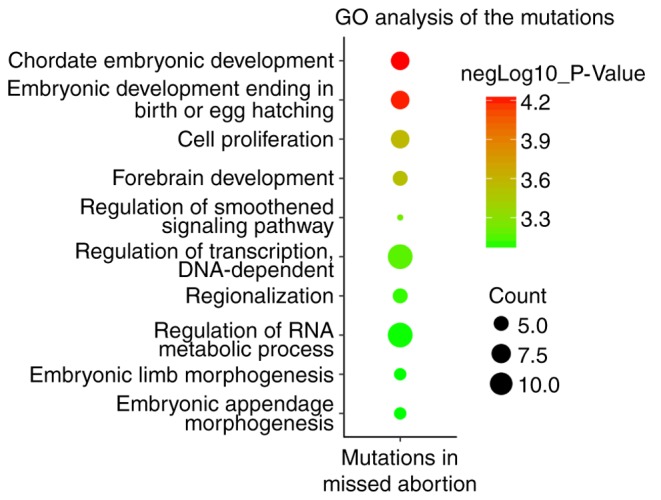
GO analysis of the 36 genes. GO analysis was performed using the DAVID Bioinformatics Resources 6.7 (https://david-d.ncifcrf.gov/). The Log10 (P-value) was calculated. Red indicates higher significance and enrichment, while green indicates relatively lower enrichment. The size of each circle indicates the gene numbers enriched in each biological process. GO, gene ontology.
Table II.
In silico analysis of sequence variants demonstrated by whole-exome sequencing in the embryos from cases of missed abortion.
| Embryo ID | Gene | Reference mRNA no. | Mutation type | Variants | Aa change | Polyphen-2 | SIFT | Mutation Taster | FATHMM-MKL | M Z score | pLI | HI, % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QW001 | LIAS | NM_006859 | m | c.991T>C | p.W331R | D (0.997) | D (0) | D (1.0) | D (0.976) | 1.16 | 0.06 | 12.37 |
| QW002 | PADI6 | NM_207421 | m | c.122C>T | p.A41V | P (0.892) | N.A. | N.A. | N (0.422) | N.A. | N.A. | N.A. |
| QW004 | ATE1 | NM_007041 | m | c.929G>A | p.C310Y | P (0.855) | D (0.016) | D (1.0) | D (0.985) | −0.13 | 0.51 | 45.89 |
| QW005 | INTS1 | NM_001080453 | m | c.3934C>A | p.L1312M | B (0.183) | T (0.174) | D (0.82) | D (0.896) | 1.18 | 0.22 | 52.90 |
| PIKFYVE | NM_015040 | m | c.3683A>T | p.Q1228L | D (0.987) | T (0.245) | D (1.0) | D (0.989) | 1.58 | 0.98 | 20.31 | |
| RAC1 | NM_018890 | m | c.230G>C | p.G77A | D (0.992) | T (0.137) | D (0.997) | D (0.962) | 3.42 | 0.57 | 0.72 | |
| SCARB1 | NM_005505 | m | c.20C>A | p.A7E | D (0.997) | D (0.032) | N (1.0) | N (0.172) | 1.83 | 0.08 | 37.77 | |
| QW007 | OTX2 | NM_172337 | m | c.475C>A | p.P159T | D (0.999) | D (0.002) | D (1.0) | D (0.985) | 1.02 | 0.74 | 0.71 |
| QW008 | BPTF | NM_182641 | m | c.2882T>A | p.I961K | B (0.017) | D (0.002) | D (0.999) | D (0.912) | 4.39 | 1.00 | 36.79 |
| CREBBP | NM_004380 | m | c.3107C>A | p.T1036K | B (0.008) | T (0.527) | D (0.981) | D (0.908) | 5.58 | 1.00 | 0.62 | |
| HSF1 | NM_005526 | m | c.1462C>G | p.L488V | P (0.798) | D (0.007) | D (0.993) | D (0.938) | 0.54 | 0.59 | 48.62 | |
| NF1 | NM_000267 | m | c.1648C>A | p.L550M | D (1.0) | T (0.092) | D (0.999) | D (0.899) | 6.22 | 1.00 | 0.87 | |
| PIKFYVE | NM_015040 | m | c.3307A>G | p.K1103E | D (0.997) | T (0.222) | D (1.0) | D (0.998) | 1.58 | 0.98 | 20.31 | |
| PTCH1 | NM_000264 | m | c.3470C>A | p.A1157E | D (0.973) | T (0.071) | D (1.0) | D (0.991) | 2.86 | 1.00 | 0.48 | |
| RAPGEF2 | NM_014247 | m | c.3203T>A | p.V1068E | B (0.243) | T (0.319) | D (1.0) | D (0.993) | 3.22 | 1.00 | 21.18 | |
| QW010 | RGS14 | NM_006480 | m | c.510C>G | p.S170R | D (1.0) | D (0) | D (1.0) | D (0.916) | 1.15 | 0.02 | 61.07 |
| TRIM28 | NM_005762 | m | c.361A>C | p.K121Q | D (0.997) | T (0.172) | D (1.0) | D (0.604) | 3.16 | 1.00 | 27.80 | |
| QW011 | PTPRB | NM_001109754 | m | c.5561T>C | p.V1854A | B (0.002) | D (0.034) | N (0.97) | D (0.838) | 0.49 | 0.94 | 39.33 |
| QW012 | DIDO1 | NM_033081 | m | c.3064C>T | p.P1022S | P (0.759) | T (0.177) | D (0.929) | D (0.963) | 3.10 | 1.00 | 83.16 |
| KIF16B | NM_001199866 | m | c.3802G>T | p.V1268F | B (0.007) | D (0) | D (0.864) | N (0.096) | 0.69 | 0.00 | 44.93 | |
| ZNF568 | NM_198539 | s | c.514A>T | p.R172X | N.A. | N.A. | D (1.0) | N (0.066) | 0.27 | 0.00 | 88.58 | |
| QW013 | FAM208A | NM_015224 | m | c.1616A>G | p.H539R | D (0.991) | T (0.177) | D (1.0) | D (0.883) | 0.89 | 1.00 | 29.50 |
| KAT2A | NM_021078 | m | c.671C>A | p.P224H | D (1.0) | D (0) | D (1.0) | D (0.98) | 4.79 | 0.41 | 22.83 | |
| KDM1A | NM_015013 | m | c.1759A>T | p.N587Y | D (1.0) | T (0.051) | D (1.0) | D (0.991) | 5.56 | 0.99 | 5.56 | |
| LDB1 | NM_003893 | m | c.662C>T | p.S221L | D (0.985) | D (0.001) | D (1.0) | D (0.977) | 3.24 | 0.88 | 2.24 | |
| NOTCH1 | NM_017617 | m | c.2953C>G | p.P985A | D (0.969) | T (0.352) | D (1.0) | D (0.966) | 4.48 | 1.00 | 0.15 | |
| POGLUT1 | NM_152305 | m | c.832T>G | p.F278V | D (1.0) | D (0.001) | D (1.0) | D (0.937) | 1.08 | 0.00 | 20.35 | |
| QW014 | CDH5 | NM_001795 | s | c.1138C>T | p.Q380X | N.A. | N.A. | A (1.0) | D (0.736) | 0.02 | 0.15 | 32.63 |
| GAS1 | NM_002048 | m | c.610C>T | p.R204C | D (1.0) | D (0.002) | D (1.0) | D (0.772) | 4.27 | 0.62 | 29.14 | |
| QW015 | DLX3 | NM_005220 | m | c.314C>T | p.A105V | B (0.037) | T (0.119) | D (0.987) | D (0.918) | 1.87 | 0.01 | 40.57 |
| PTPRB | NM_001109754 | m | c.359T>C | p.V120A | B (0.39) | D (0.007) | N (1.0) | D (0.788) | 0.49 | 0.94 | 39.33 | |
| QW016 | INTS1 | NM_001080453 | m | c.6475C>T | p.L2159F | D (0.999) | D (0.006) | D (1.0) | D (0.963) | 1.18 | 0.22 | 52.90 |
| SALL4 | NM_020436 | m | c.733C>A | p.H245N | B (0.067) | T (0.16) | D (1.0) | D (0.865) | 1.66 | 1.00 | 38.00 | |
| QW018 | SRRT | NM_015908 | m | c.1148A>C | p.K383T | D (0.999) | T (0.249) | D (1.0) | D (0.987) | 4.61 | 0.98 | 30.36 |
| VPS26A | NM_004896 | m | c.758C>T | p.A253V | B (0.388) | D (0.019) | D (1.0) | D (0.996) | 0.94 | 0.66 | 11.04 | |
| QW019 | CREBBP | NM_004380 | m | c.2917C>A | p.P973T | P (0.952) | T (0.055) | D (1.0) | D (0.992) | 5.58 | 1.00 | 0.62 |
Polyphen-2 prediction scores range from 0 to 1 with high scores indicating probably or possibly damaging. D, probably damaging; P, possibly damaging; B, benign; N.A., not available. SIFT scores vary between 0 and 1. Variants with scores close or equal to 0 are predicted to be damaging. D, Damaging; T, tolerated; N.A., not available. Mutation Taster: The probability value is the probability of the prediction, i.e., a value close to 1 indicates a high ‘security’ of the prediction. D, probably deleterious; N, polymorphism; A, known to be deleterious. FATHMM-MKL values >0.5 are predicted to be deleterious, while those <0.5 are predicted to be neutral or benign. D, deleterious; N, neutral; M Z score, missense Z score: Positive Z scores indicate increased constraint (intolerance to variation) and therefore that the gene had fewer variants than expected. Negative Z scores are given to genes that exhibit more variants than expected. pLI: The closer pLI is to one, the more LoF-intolerant the gene appears to be. pLI >= 0.9 was considered as an extremely LoF intolerant set of genes. HI, high ranks (e.g. 0–10%) indicate a gene is more likely to exhibit HI; low ranks (e.g., 90–100%) indicate a gene is more likely to not exhibit HI. m, missense; s, stopgain; Aa, amino acid; LoF, loss of function; HI, haploinsifficiency; pLI, loss intolerance.
Analysis of the variant in LDB1
LDB1 serves important roles in the regulation of a variety of processes in early embryonic development, including heart formation (12), head and brain development (12–17), limb patterning (18), and eye development (16). In previous studies, the development of LDB1 null mutant mice was arrested at embryonic day 8.5 (E8.5) and mice succumbed at E9-E10 (12,19). Therefore, LDB1 may be a good candidate gene for human early embryonic lethality and MA. In the present study, the heterozygous c.662C>T variant in LDB1 was also validated by Sanger sequencing (Fig. 2A). DNA samples from the patient and her husband were unavailable, therefore it was not possible to analyze whether this variant was inherited or generated de novo. This variant and the flanking region were additionally highly conserved in humans and other animals including zebrafish (Fig. 2B), which reflects the highly conserved role of LDB1 in early embryonic development across different species.
Figure 2.
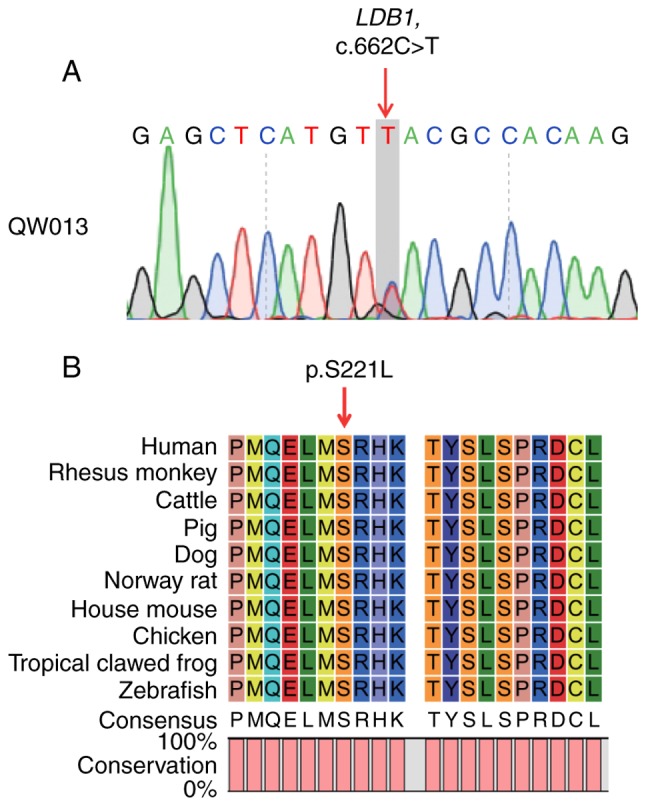
Analysis of the LDB1 variant. (A) Sanger sequencing validated the heterozygous c.662C>T variant in the LDB1 gene. The red arrow indicates the mutation site. (B) Amino acid sequence alignment of LDB1 in different species. The red arrow indicates the mutational amino acid. Serine at position 211 was 100% conserved (full red columns) in all species. LDB1, LIM domain-binding protein 1.
Discussion
In the present study, WES was performed on 19 POC and using a strict filtering strategy, 36 rare sequence variants associated with MA were identified. GO analysis suggested that these 32 genes were enriched in biological processes in early embryonic development, including ‘chordate embryonic development’, ‘cell proliferation’ and ‘forebrain development’. Further strict in silico bioinformatics analysis predicted the LDB1 (c.662C>T; p.S221L) variant to be a highly pathogenic variant.
Previous studies suggest that chromosomal abnormalities account for 60–80% of MA cases (4–8). However, to the best of the authors' knowledge, no studies have been performed on POC using NGS technology to elucidate single gene defects. Therefore, the present study attempted to dissect the genetic causes of the remaining 20–40% of MA cases. A total of 36 rare sequence variants in 32 genes potentially associated with MA were identified in 15 out of 19 patients. Of the 32 genes, seven genes [lycine acetyltransferase 2A, lycine demethylase 1A (KDM1A), spalt like transcription factor 4, heat shock transcription factor 1, integrator subunit complex 1, patched 1 and growth arrest specific 1] were significantly enriched in the biological process of ‘chordate embryonic development’, and five genes (KDM1A, notch 1, neurofibromin 1, orthodenticle homeobox 2 and Rac family small GTPase 1) were enriched in ‘forebrain development’. The embryos succumbed or arrested if each of the above genes were knocked out.
LDB1 is a critical gene involved in embryonic morphogenesis (12–17,19). LDB1 gene deficiency leads to early embryonic arrest and embryo loss between E9-E10 (12,19). Therefore, LDB1 is a good candidate gene for embryonic lethality. Additionally, LDB1 was searched for in Online Mendelian Inheritance in Man (OMIM) and PubMed; however, no study demonstrating LDB1 gene mutations associated with developmental diseases was identified. Therefore, it was hypothesized that human embryos harboring pathogenic mutations in LDB1 may lead to embryonic lethality, meaning that no child carrying the LDB1 pathogenic mutation would be born; this may explain why no LDB1 mutation was observed in OMIM. Furthermore, in the present study, the c.662C>T; p.S221L variant in LDB1 was predicted as a pathogenic allele by a number of prediction tools, which, along with the extreme rarity and conservation of the variant and flanking regions, suggested that the LDB1 mutation in the POC was associated with MA.
Ablation of DIDO1 in mice causes embryonic lethality during the gastrulation stage (20). DIDO heterozygous deletion mice demonstrate abnormalities in their spleen, bone marrow and peripheral blood (21). By checking the DECIPHER database (https://decipher.sanger.ac.uk/), two heterozygous missense variants in DIDO1 were identified, one of which may be associated with abnormalities in the head, cardiovascular system, ear, integument, nervous system and skeletal system. Therefore, the variant in the present study may also be associated with embryonic developmental abnormalities.
In conclusion, the present study identified 36 rare sequence variants in 19 POCs associated with MA. Further bioinformatics analysis predicted that the LDB1 (c.662C>T; p.S221 L) variant is a highly pathogenic variant and may be associated with embryonic lethality. Taken together, the results of the present study provide researchers and clinicians with a better understanding of the etiology and molecular mechanism of human embryonic lethality and MA.
Acknowledgements
The authors would like to thank the participants in the present study for their consent and support to publish this article.
Funding
The present study was supported by the National Natural Science Foundation of China (grant no. 81571411) and the Military Medical Innovation Project (grant no. 16JS011).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
MF and SM conducted the experiments; MF, CW, SJ and LL performed the WES data analysis and in silico analysis of sequence variants; MF, SJ, YM, SM and CW collected the POC samples; and YM and HP designed the experiments and wrote the manuscript. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Chinese PLA General Hospital's research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This research was approved by the ethical committee of Chinese People's Liberation Army (PLA) General Hospital. Written informed consent was obtained from each participant.
Patient consent for publication
Written informed consent was obtained from each participant.
Competing interests
The authors declare they have no competing interests.
References
- 1.Griebel CP, Halvorsen J, Golemon TB, Day AA. Management of spontaneous abortion. Am Fam Physician. 2005;72:1243–1250. [PubMed] [Google Scholar]
- 2.Clifford K, Rai R, Watson H, Regan L. An informative protocol for the investigation of recurrent miscarriage: Preliminary experience of 500 consecutive cases. Hum Reprod. 1994;9:1328–1332. doi: 10.1093/oxfordjournals.humrep.a138703. [DOI] [PubMed] [Google Scholar]
- 3.Hatasaka HH. Recurrent miscarriage: Epidemiologic factors, definitions, and incidence. Clin Obstet Gynecol. 1994;37:625–634. doi: 10.1097/00003081-199409000-00016. [DOI] [PubMed] [Google Scholar]
- 4.Segawa T, Kuroda T, Kato K, Kuroda M, Omi K, Miyauchi O, Watanabe Y, Okubo T, Osada H, Teramoto S. Cytogenetic analysis of the retained products of conception after missed abortion following blastocyst transfer: A retrospective, large-scale, single-centre study. Reprod Biomed Online. 2017;34:203–210. doi: 10.1016/j.rbmo.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Alberman ED, Creasy MR. Frequency of chromosomal abnormalities in miscarriages and perinatal deaths. J Med Genet. 1977;14:313–315. doi: 10.1136/jmg.14.5.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang HK, Luo FW, Geng Q, Li J, Liu QZ, Chen WB, Li F, Xie JS. Analysis of fetal chromosomal karyotype and etiology in 252 cases of early spontaneous abortion. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2011;28:575–578. doi: 10.3760/cma.j.issn.1003-9406.2011.05.024. (In Chinese) [DOI] [PubMed] [Google Scholar]
- 7.Muñoz M, Arigita M, Bennasar M, Soler A, Sanchez A, Borrell A. Chromosomal anomaly spectrum in early pregnancy loss in relation to presence or absence of an embryonic pole. Fertil Steril. 2010;94:2564–2568. doi: 10.1016/j.fertnstert.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 8.Philipp T, Philipp K, Reiner A, Beer F, Kalousek DK. Embryoscopic and cytogenetic analysis of 233 missed abortions: Factors involved in the pathogenesis of developmental defects of early failed pregnancies. Hum Reprod. 2003;18:1724–1732. doi: 10.1093/humrep/deg309. [DOI] [PubMed] [Google Scholar]
- 9.Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, Kosmicki JA, Rehnström K, Mallick S, Kirby A, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. 2014;46:944–950. doi: 10.1038/ng.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang N, Lee I, Marcotte EM, Hurles ME. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mukhopadhyay M, Teufel A, Yamashita T, Agulnick AD, Chen L, Downs KM, Schindler A, Grinberg A, Huang SP, Dorward D, Westphal H. Functional ablation of the mouse Ldb1 gene results in severe patterning defects during gastrulation. Development. 2003;130:495–505. doi: 10.1242/dev.00225. [DOI] [PubMed] [Google Scholar]
- 13.Kim S, Zhao Y, Lee JM, Kim WR, Gorivodsky M, Westphal H, Geum D. Ldb1 is essential for the development of isthmic organizer and midbrain dopaminergic neurons. Stem Cells Dev. 2016;25:986–994. doi: 10.1089/scd.2015.0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Y, Flandin P, Vogt D, Blood A, Hermesz E, Westphal H, Rubenstein JL. Ldb1 is essential for development of Nkx2.1 lineage derived GABAergic and cholinergic neurons in the telencephalon. Dev Biol. 2014;385:94–106. doi: 10.1016/j.ydbio.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao Y, Kwan KM, Mailloux CM, Lee WK, Grinberg A, Wurst W, Behringer RR, Westphal H. LIM-homeodomain proteins Lhx1 and Lhx5, and their cofactor Ldb1, control Purkinje cell differentiation in the developing cerebellum. Proc Natl Acad Sci USA. 2007;104:13182–13186. doi: 10.1073/pnas.0705464104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plautz CZ, Zirkle BE, Deshotel MJ, Grainger RM. Early stages of induction of anterior head ectodermal properties in Xenopus embryos are mediated by transcriptional cofactor ldb1. Dev Dyn. 2014;243:1606–1618. doi: 10.1002/dvdy.24193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costello I, Nowotschin S, Sun X, Mould AW, Hadjantonakis AK, Bikoff EK, Robertson EJ. Lhx1 functions together with Otx2, Foxa2, and Ldb1 to govern anterior mesendoderm, node, and midline development. Genes Dev. 2015;29:2108–2122. doi: 10.1101/gad.268979.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tzchori I, Day TF, Carolan PJ, Zhao Y, Wassif CA, Li L, Lewandoski M, Gorivodsky M, Love PE, Porter FD, et al. LIM homeobox transcription factors integrate signaling events that control three-dimensional limb patterning and growth. Development. 2009;136:1375–1385. doi: 10.1242/dev.026476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Lee JY, Gross J, Song SH, Dean A, Love PE. A requirement for Lim domain binding protein 1 in erythropoiesis. J Exp Med. 2010;207:2543–2550. doi: 10.1084/jem.20100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Futterer A, Raya A, Llorente M, Izpisúa-Belmonte JC, de la Pompa JL, Klatt P, Martínez-A C. Ablation of Dido3 compromises lineage commitment of stem cells in vitro and during early embryonic development. Cell Death Differ. 2012;19:132–143. doi: 10.1038/cdd.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fütterer A, Campanero MR, Leonardo E, Criado LM, Flores JM, Hernández JM, Miguel San JF, Martínez-A C. Dido gene expression alterations are implicated in the induction of hematological myeloid neoplasms. J Clin Invest. 2005;115:2351–2362. doi: 10.1172/JCI24177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.