

Abstract
Purpose
The promise of precision oncology is that identification of genomic alterations will direct the rational use of molecularly targeted therapy. This approach is particularly applicable to neoplasms that are resistant to standard cytotoxic chemotherapy, like T-cell leukemias and lymphomas. In this study, we tested the feasibility of targeted next-generation sequencing in profiles of diverse T-cell neoplasms and focused on the therapeutic utility of targeting activated JAK1 and JAK3 in an index case.
Patients and Methods
Using Foundation One and Foundation One Heme assays, we performed genomic profiling on 91 consecutive T-cell neoplasms for alterations in 405 genes. The samples were sequenced to high uniform coverage with an Illumina HiSeq and averaged a coverage depth of greater than 500× for DNA and more than 8M total pairs for RNA. An index case of T-cell prolymphocytic leukemia (T-PLL), which was analyzed by targeted next-generation sequencing, is presented. T-PLL cells were analyzed by RNA-seq, in vitro drug testing, mass cytometry, and phospho-flow.
Results
One third of the samples had genomic aberrations in the JAK-STAT pathway, most often composed of JAK1 and JAK3 gain-of-function mutations. We present an index case of a patient with T-PLL with a clonal JAK1 V658F mutation that responded to ruxolitinib therapy. After relapse developed, an expanded clone that harbored mutant JAK3 M511I and downregulation of the phosphatase, CD45, was identified. We demonstrate that the JAK missense mutations were activating, caused pathway hyperactivation, and conferred cytokine hypersensitivity.
Conclusion
These results underscore the utility of profiling occurrences of resistance to standard regimens and support JAK enzymes as rational therapeutic targets for T-cell leukemias and lymphomas.
INTRODUCTION
T-cell neoplasms are known for their clinically aggressive behavior and for their high risk of relapse and resistance to conventional cytotoxic regimens. Adult patients with precursor neoplasms, such as acute T-cell lymphoblastic leukemia (T-ALL), or with mature neoplasms, such as T-cell non-Hodgkin lymphoma (T-NHL), have a 5-year survival rate of 20% to 30% even after intensive multiagent chemotherapy.1-6 There are rare exceptions to these dismal outcomes, such as children and adolescents who have T-ALL or anaplastic large-cell lymphoma (ALCL) with unique gene rearrangements (ie, ALK positivity or DUSP22 positivity) in whom 5-year survival rates are greater than 70% to 80% with similar chemotherapy regimens.5,7,8 However, relapsed disease is challenging to cure. Clearly, novel therapeutic approaches are needed, and the development of commercially available next-generation sequencing has raised the possibility that genomically directed therapy may be applied to T-cell leukemias and lymphomas. Genomic profiling has been performed on several histopathologic subtypes of T-cell leukemias and lymphomas to better characterize the molecular genetics.9-13 Interestingly, recent genomic profiling has discovered frequent aberrations within the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway in both precursor (T-ALL) and mature (T-NHL) T-cell neoplasms, which suggests that JAK kinase inhibition may be important therapeutically.14
JAKs are encoded by four paralogous genes, JAK1, JAK2, JAK3, and TYK2. These tyrosine kinases are recruited to cytokine receptors, where they transduce signals by phosphorylation of key substrates—most important, STAT proteins that bind DNA and regulate gene expression. JAK1 mutations have been found in 10% of childhood T-ALLs.15 Our laboratory and others have found JAK3 mutations in cutaneous T-cell lymphoma (CTCL), adult T-cell leukemia/lymphoma (ATLL), T-cell prolymphocytic leukemia (T-PLL), and natural killer/T-cell lymphoma (NKTL).16-20 Analyses of human leukemia lines and mouse models show that JAK mutations typically are activating and cause constitutive signal transduction, which may be blocked by tyrosine kinase inhibitors. Two such ATP-competitive inhibitors have been approved by the US Food and Drug Administration (FDA) for human use. Ruxolitinb is approved for use in myeloproliferative neoplasms, and tofacitinib is approved for rheumatoid arthritis.21,22
In this study, we deployed a commercially available hybrid-capture/next-generation sequencing platform to characterize major recurrent oncogene and tumor suppressor aberrations in 91 T-cell neoplasms. This targeted approach found that 33% of samples had JAK-STAT abnormalities, which included missense mutations in JAK1 and JAK3, rearrangements in JAK2 and JAK3, and missense mutations and amplifications of STAT3 and STAT5. We analyzed an index case of T-PLL, a deadly mature T-cell neoplasm with both JAK1 and JAK3 gain-of-function missense mutations.23 This patient with T-PLL had experienced progression during multiple lines of chemotherapy but experienced disease response with ruxolitinib, a JAK1/2 inhibitor. The patient eventually experienced relapse as a result of clonal expansion of T-PLL cells with gain of function of JAK3 and downregulation of CD45. To our knowledge, this study is the first to demonstrate an in vivo response to ruxolitinib in a T-cell neoplasm, which underscores the importance of the interleukin-2 receptor gamma chain IL2RG/JAK1/JAK3 cytokine pathway in the pathogenesis of T-cell neoplasms and supports inhibition of JAK enzymes as therapy.
PATIENTS AND METHODS
Patient Samples, Processing, Sequencing
Patient peripheral blood or bone marrow was banked after informed consent under a protocol approved by the Vanderbilt institutional review board. Workflows have been described as the commercially available Foundation One and Foundation One Heme assays. DNA and RNA samples were extracted from fresh liquid specimens (blood or bone marrow aspirate). Adaptor-ligated libraries were created from DNA and cDNA as described.24 Libraries were sequenced on Illumina HiSeq2500 (Illumina, San Diego, CA) to > 500× coverage depth for DNA and > 8M total pairs for RNA. DNA and RNA sequence data were processed with a customized analysis pipeline designed to accurately detect multiple classes of genomic alterations—specifically, base substitutions, indels, focal gene amplifications, homozygous gene deletions, gene fusions, and genomic rearrangements (Data Supplement).24
Gene Expression
The RNA sequencing workflow has been described previously. It also is detailed in the Data Supplement.25
Antibodies and Staining
See the Data Supplement for lists of antibodies used in this study. Cell staining also is described in Data Supplement.
Mass Cytometry and Phospho-Flow
Samples were stained and prepared for the mass cytometer as previously described. A description also is in the Data Supplement.26
Phosphatase Enzyme Assay and Inhibitor Studies
Frozen pre- and post-treatment T-PLL cells and Jurkat cells were lysed in hypotonic buffer and prepared with cytosol, as described.27 Ruxolitinib and tofacitinib were purchased from Selleck Chemicals (Houston, TX), and working solutions were prepared in dimethyl sufoxide (DMSO). T-PLL cells were plated in RPMI/10% fetal calf serum and treated with DMSO alone or with various concentrations of drug. Cell viability was quantified with a CyQuant assay (Invitrogen, Carlsbad, CA), as described.17,27 Statistical analyses were performed with GraphPad Prism (La Jolla, CA).
RESULTS
Genomic Profiling of T-Cell Leukemias and Lymphomas
We analyzed 91 occurrences of diverse T-cell leukemias and lymphomas for alterations in 405 cancer-causing genes by comprehensive hybrid capture of genomic DNA followed by next-generation sequencing (Data Supplement). On the basis of the WHO classification scheme, the cohort was composed of 25 precursor T-cell neoplasms (ie, T-ALL, acute lymphoblastic leukemias) and 66 mature T-cell neoplasms (ie, angioimmunoblastic lymphoma [AITL; n = 8], ALCL [n = 7], CTCL [n = 14], peripheral T-cell lymphoma [PTCL; n = 16], T-cell large granular leukemia [T-LGL; n = 11], T-PLL [n = 7], and NK/TL [n = 3]). The cohort showed a marked male predominance for both immature and mature T-cell neoplasms (4:1 and 1.75:1, respectively; Data Supplement) consistent with the epidemiology of these cancers.28,29 Samples were sequenced at a mean exon depth of ×489 by Illumina HiSeq. As shown in Figure 1 and the Data Supplement, the most common gene alterations was CDKN2A/B, followed by TET2, NOTCH1, JAK3, TP53, STAT3, NRAS, DNMT3A, JAK1, RHOA, MLL2, and others.
Fig 1.
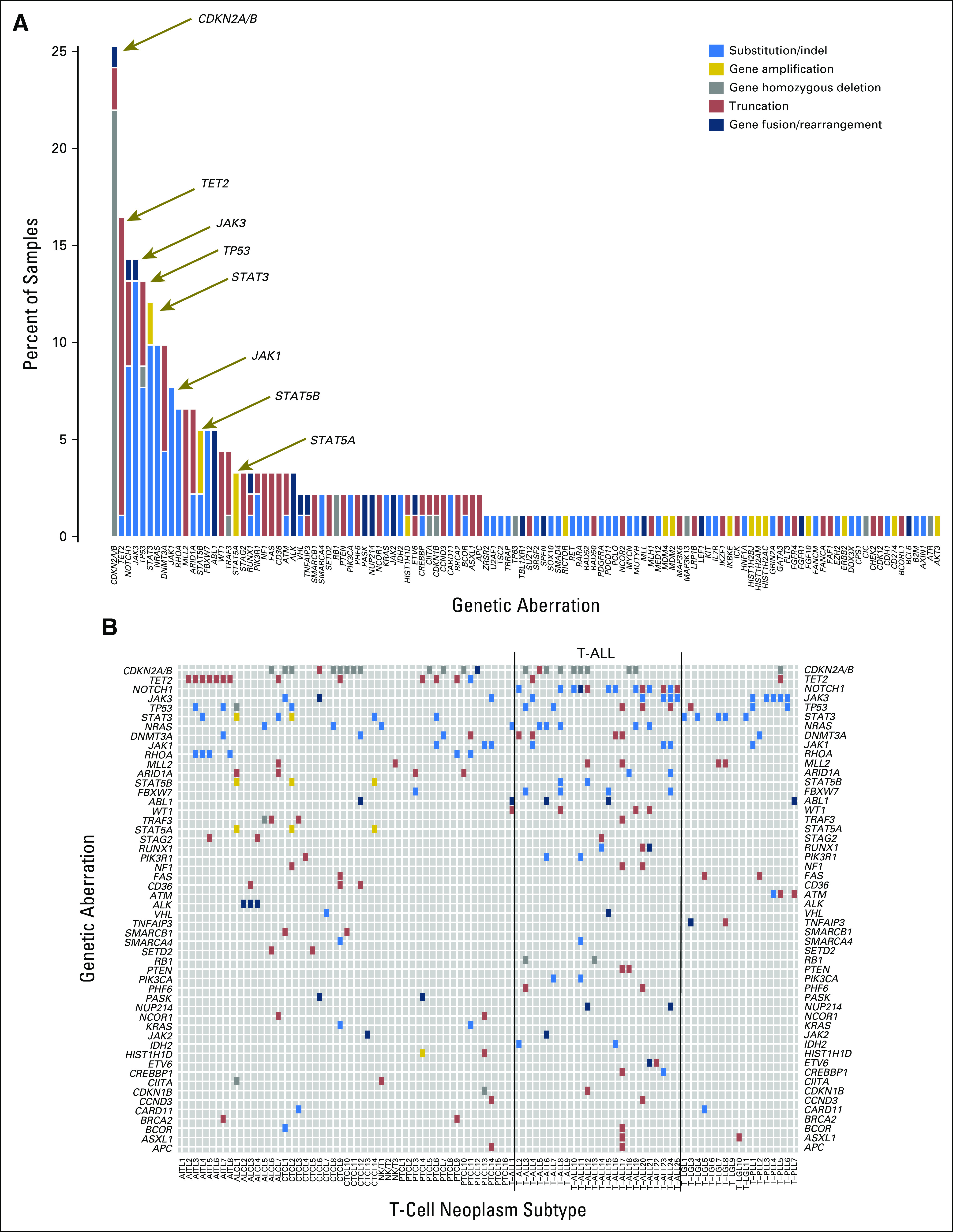
Targeted next-generation sequencing of T-cell neoplasms. (A) Bar graph shows the frequency of genetic aberrations (color coded) in 91 samples of T-cell leukemias or lymphomas. The y-axis shows the percentage of total samples. (B) Tile plot shows recurrent genetic aberrations in all 91 samples. Lines bracket immature T-cell neoplasms (T-cell acute lymphoblastic leukemias [T-ALLs]). The x-axis shows the T-cell neoplasm subtype of each individual sample: angioimmunoblastic T-cell lymphoma (AITL); anaplastic large-cell lymphoma (ALCL); cutaneous T-cell lymphoma (CTCL); natural killer/T-cell (NKT) lymphoma; peripheral T-cell lymphoma (PTCL); T-ALL; T-cell large granular leukemia (T-LGL); and T-cell prolymphocytic leukemia (T-PLL). The y-axis shows genes with boxes colored blue for substitution/insertions or deletions (indels), gold for gene amplifications, gray for homozygous deletions, dark red for truncations, and dark blue for gene fusions or rearrangements (as noted in A).
The frequencies of gene alterations were compared between immature (T-ALLs) and mature T-cell neoplasms: NOTCH1 (48% v 3%, P < .001), FBXW7 (16% v 1.5%, P = .0191), and NRAS (24% v 4.5%, P = .0117) mutations were more frequent among T-ALL than mature T-cell neoplasms. STAT3 mutations (0% v 18%, P = .032) were more common in mature T-cell neoplasms (Table 1); 31.8% (29 of 91 samples) had alterations in the JAK-STAT pathway. JAK3, which involved 13% (12 of 91 samples) of samples, was the most commonly mutated kinase, followed by JAK1, which was mutated in 7.7% (seven of 91 samples). There were two occurrences of JAK2 rearrangement (n = 1 each in CTCL and T-ALL) and five occurrences of ABL1 rearrangements (n = 1 CTCL, n = 1 T-PLL, and n = 4 T-ALL). JAK3 mutations were present with a similar frequency in T-ALL and in mature T-cell neoplasms (Fisher's exact test, P = .19) but were highly frequent in certain mature T-cell neoplasms, such as T-PLL (five [71%] of seven samples). Interestingly, all five occurrences of T-PLL had the M511I JAK3 mutation; all of the JAK3 and JAK1 mutations were in the Catalogue of Somatic Mutations in Cancer (COSMIC; http://cancer.sanger.ac.uk/cosmic) database, frequently within the pseudokinase domains of the proteins (Fig 2).30 JAK3 missense mutations have been identified previously in CTCL and ATL.16 Focal STAT3, STAT5A, and STAT5B amplifications were identified in two occurrences (n = 1 each in CTCL and ALCL), in addition to missense mutations within the SH2 domains of STAT3 and STAT5B, such as STAT3 D661Y/V in T-LGL (Fig 2). STAT5 and STAT3 gene alterations were mutually exclusive with JAK1 or JAK3 mutations. PTCL6 had mutations in STAT3 and JAK1 at 7% allele frequencies, respectively, and likely were separate clones. JAK1 and JAK3 mutations were observed in the same tumor in five occurrences, but concordant mutations in the same cell could not be confirmed (Data Supplement). JAK3 mutations were concordant with mutations in TP53 in four samples; NOTCH1 mutations, in four samples; and CDKN2A deletions, in three samples (Data Supplement).
Fig 2.

Frequent missense mutations in JAK1, JAK3, STAT5B, and STAT3. Schematic shows protein domain structures of JAK3, JAK1, STAT5B, and STAT3. Black lines under the schematics show exon numbers above and amino acid numbers below; JAK3 exons are not shown because of differential splicing. Missense mutations are denoted by blue circles, with codon changes as shown. α, alpha helical domain, a DNA binding domain; FERM, the conserved domain named for its founding members (band 4.2, ezrin, radixin, and moesin); JH1, JAK homology domain 1, the functional kinase domain; JH2, JAK homology domain 2 (also known as the pseudokinase domain); SH2, Src homology domain 2; STAT-int, STAT interaction domain.
Exceptional Response of a JAK1-Mutant T-PLL
We evaluated a 62-year-old woman in our clinic with relapsed T-PLL. She presented with constitutional symptoms, splenomegaly, and leukemic blood counts that had increased to greater than 150,000/µL and progressed through alemtuzumab; cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP); romidepsin; and pralatrexate. The T-PLL cells were CD2+CD3+CD4+CD7+CD8−CD56−CD57− (Fig 3A), had clonal T-cell receptor rearrangement (data not shown), and had infiltration of bone marrow (Figs 3B through 3E). The T-PLL cells (90% of peripheral-blood mononuclear cells) were analyzed by hybrid capture followed by next-generation sequencing. As shown in Figure 3F, the T-PLL cells had a JAK1 V658F mutation at 40% allele frequency and a JAK3 M511I mutation at 5%. Other notable mutations were in ATM and TP53 at allele frequencies of 92% and 78%, respectively, which suggests loss of heterozygosity (Data Supplement). Both JAK1 and JAK3 mutations had been described in hematologic malignancies and have proven to be oncogenic in various assays.20,30-32 The clonal JAK1 mutation could be targeted by ruxolitinib, a kinase inhibitor with activity against JAK1/2.21 The patient was agreeable to this off-label therapy and received 20 mg twice daily; her peripheral count (90% T-PLL) declined from 142,000/μl to 85,000/μl within 5 days. By day 7 day, her spleen, which was palpable at 4 cm below the costal margin before therapy, was no longer palpable. She experienced no adverse effects from ruxolitinib, and her leukocyte count stabilized to approximately 60,000/µL for more than 110 days. During the 100 days before ruxolitinib therapy, the patient required 10 apheresis units of platelets and 7 units of packed red blood cells; conversely, during ruxolitinib therapy, she received 3 apheresis units of platelets and 2 units of packed red blood cells. Unfortunately, by day 116, the leukemic blood count increased to 116,000/µL, and a bone marrow biopsy confirmed relapsed disease (Fig 3G). The patient developed worsening thrombocytopenia that did not respond to cytotoxic chemotherapy, and she died as a result of the disease. Acquired resistance to ruxolitinib and the pan-JAK inhibitor, tofacitinib, was cell intrinsic, because it was observed ex vivo. Pretreatment T-PLL cells were more sensitive to ruxolitinib (50% inhibitory concentration [IC50], 3.85 × 10−8 M) and tofacitinib (IC50, 8.64 × 10−8 M) compared with post-treatment cells (post-ruxolitinib IC50, 4.21 × 10−7 M; post-tofacitinib 50IC50, 2.86 × 10−6 M; P < .001; Fig 3H).
Fig 3.
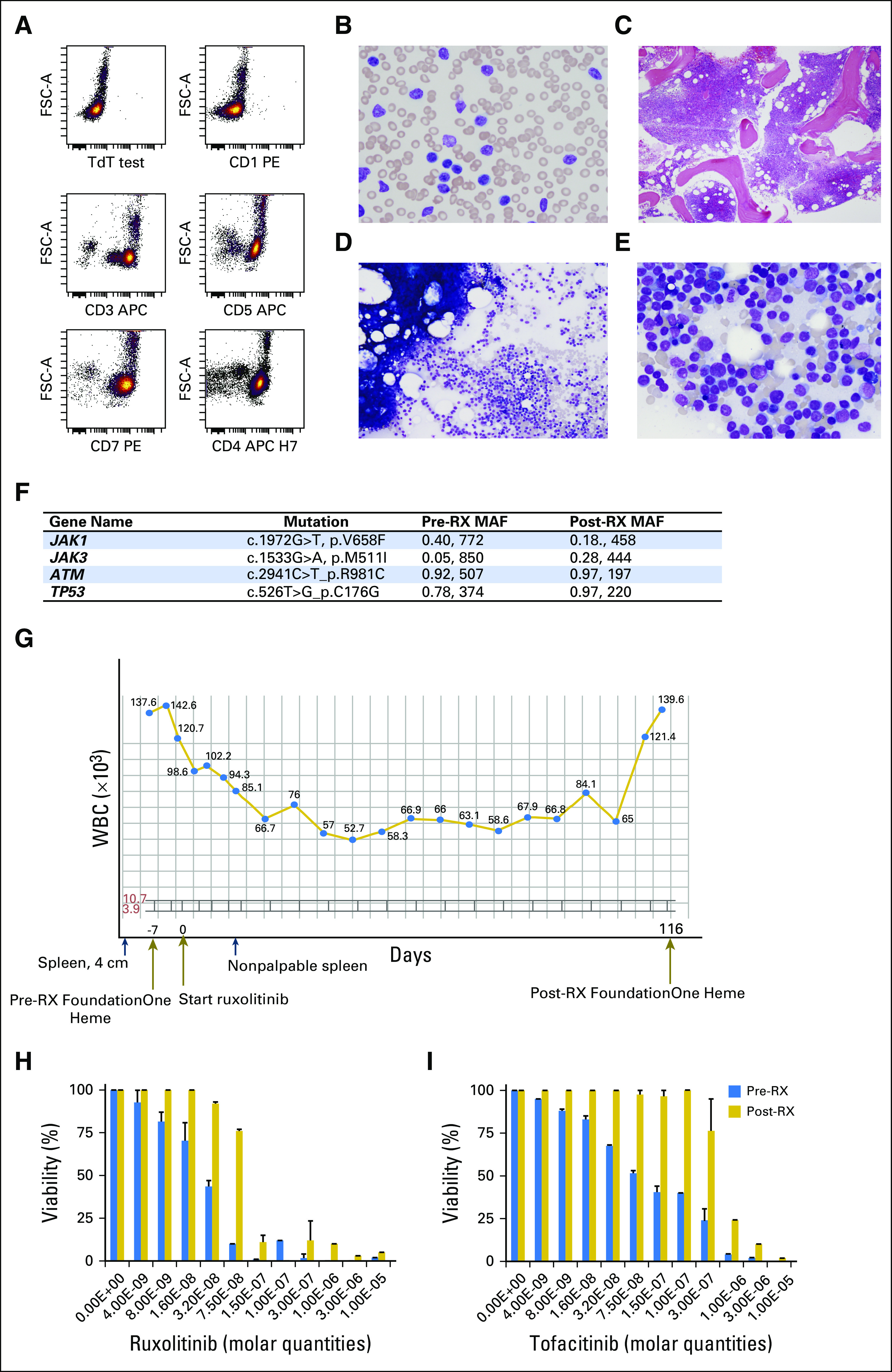
A case of T-cell prolymphocytic leukemia (T-PLL) that responded to specific JAK1 inhibitor, ruxolitinib. (A) Dot plots show the immunophenotype of the T-PLL cells at presentation as analyzed by flow cytometry; T-PLL cells were interpreted as TdT− CD1− CD3+ CD5+ CD7+ CD4+. Photomicrographs of (B) peripheral-blood smear and (C) bone marrow biopsy that shows marked hypercellularity. Bone marrow aspirates (D) with homogeneous cells and (E) at high power, which shows neoplastic cells with fine chromatin and prominent nucleoli. (F) Foundation One panel analysis of T-PLL cells before (pre-RX) and after (post-RX) ruxolitinib treatment. Major gene mutations are shown with their major allele frequencies and the number of reads. For example, JAK1 V658F was present in the pre-RX sample at 40% supported by 772 reads. (G) Plot of the total leukocytes for the patient; y-axis shows cells number (×103); gray brackets show the normal range of peripheral leukocyte number. The x-axis shows time in days with some clinical features highlighted. Dark blue arrows, time points of physical exams that show a palpable spleen 4 cm below costal margin before ruxolitinib therapy; spleen mass was not palpable after treatment. Dark gold arrows, time of first Foundation One analysis, the start of ruxolitinib therapy, and the Foundation One Heme panel analysis at relapse. (H) Bar graphs of T-PLL cells treated in vitro with varying concentrations of ruxolitinib (left panel) or tofacitinib (right panel) in molar (M) quantities; the y-axis shows cell viability. Blue bars, pre-RX samples; gold bars, post-RX/relapse samples. Error bars are standard error of the mean for three independent experiments.
Genetic and Immunophenotypic Analysis of Resistance to Ruxolitinib Therapy
At relapse, the JAK3 M511I allele frequency had increased from 5% to 28%, whereas the allele frequency of JAK1 V658F had decreased from 40% to 18% in the T-PLL cells (Fig 3F). TP53, ATM, and other mutational frequencies did not change (Data Supplement). The pre- and post-ruxolitinib T-PLL cells were analyzed by RNA sequencing, in which the steady state abundance of mutant mRNAs approximated the allele frequencies (Data Supplement). The pre- and post-ruxolitinib/relapse T-PLL cells were analyzed for 28 cell surface antigens by mass cytometry. The multidimensional staining pattern was analyzed by viSNE, an algorithm that maps cells on to a two-dimensional plot.33 Peripheral-blood mononuclear cells from healthy donors were stratified into distinct cell populations that corresponded to specific lineages: CD4+/CD8+ T cells, natural killer cells, macrophages, and B cells (Fig 4A). The T-PLL cells from the patient case were clustered into a unique island composed of 95.3% total peripheral cells and few nonmalignant cell types (Fig 4A). The sample after relapse occurred showed two distinct leukemic populations (Figs 4A and 4B): one which resembled the pre-ruxolitinib sample (46.1%), and a new island that emerged with drug resistance (50.6%). CD45 protein expression was the major feature that discriminated the two relapsed cell populations (Figs 4C through 4D). The CD45+ population resembled the pretreatment cells except for reduced expression of CD127 (interleukin 7 receptor [IL-7R]). The CD45lo/− population showed increased CD27 (TNFRSF7), CD44 (H-CAM), CCR4, and CCR7 and reduced CD43 (leukosialin) compared with pre- or post-treatment CD45+ cells (Figs 4C through 4D).
Fig 4.
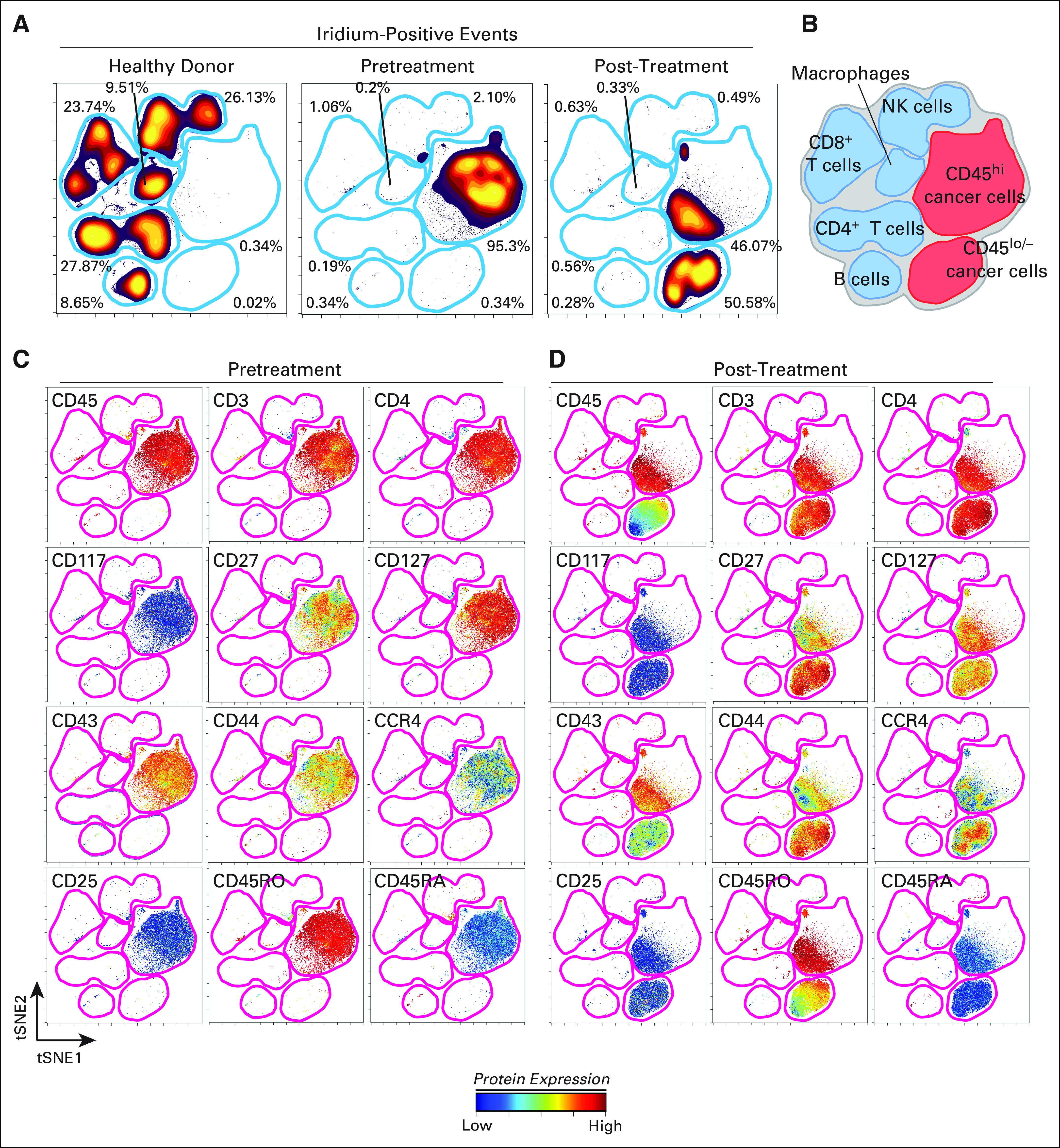
CyTOF analysis shows marked change in immunophenotype of T-cell prolymphocytic leukemia (T-PLL) cells after relapse from ruxolitinib. Peripheral-blood samples from a healthy donor and the patient with T-PLL, before ruxolitinib (pretreatment) and at relapse (post-treatment) were analyzed by mass cytometry after staining for 28 cell surface markers. (A) viSNE analysis performed on the data to allow grouping of peripheral leukocytes into distinct islands that correspond to (B) cell lineages; iridium (DNA intercalator)-positive (Ir+) events show those cells that were intact. (C) The level of expression of 12 cell surface markers in the distinct islands. (D) The same 12 markers in the post-RX sample. High-dimensional, single-cell immunophenotype was measured with a 28-marker mass cytometry panel. (A) The viSNE map shows cell density for all Ir+ (nucleic DNA intercalator) cells from a healthy donor and in pretreatment and post-treatment/relapse cells. (B) Cellular identity of each island was identified by using expression of all 28 measured proteins. Protein expression is shown on a common viSNE map for (C) pretreatment T-PLL and (D) post-treatment/relapse T-PLL. Color (for C and D) indicates the intensity of the labeled protein on a rainbow heat (ArcSinh) scales.
CD45 is a receptor tyrosine phosphatase encoded by the PTPRC gene that negatively regulates JAK-STAT and T-cell receptor signaling.34,35 CD45 downregulation coincident with clinical relapse on ruxolitinib implied that it may be a mechanism for ruxolitinib resistance. The JAK1 mutation was not detectable by Sanger sequencing, but the JAK3 mutation was clonal and present in CD45hi, CD45intermediate, and CD45− cells (Figs 5A and 5B). CD45RO and CD45RB were the mRNA isoforms expressed by the T-PLL cells (via RNA sequencing and flow cytometry; Data Supplement). Whole-transcriptome analysis on pre- and post-treatment samples found that PTPRC mRNA abundance was reduced significantly in the relapsed sample by 1.95-fold (P = 3.18E−74; Fig 5D), which approximates the 50% reduction in protein levels observed by flow cytometry. JAK1, JAK2, and JAK3 mRNAs also were downregulated significantly (Fig 5D). JAK1 and JAK3 were probed by Western blot analysis of whole-cell lysates and showed lower protein abundance in the relapsed sample compared with lysates before ruxolitinib treatment (Fig 5C).
Fig 5.
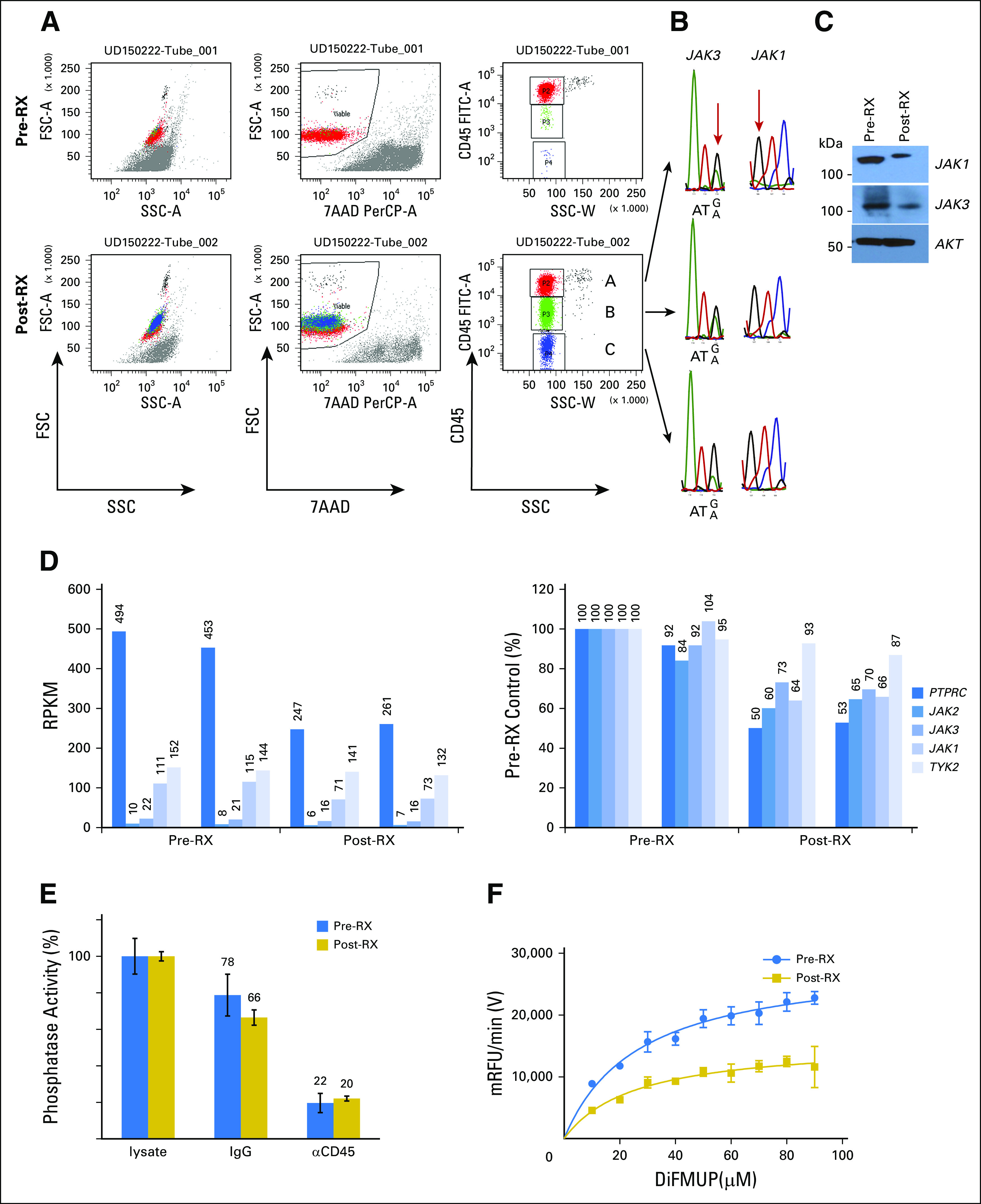
Clinical resistance correlates with downregulation of CD45 protein, mRNA, and phosphatase activity. (A-C) Flow cytometry dot plots show (A) staining patterns of T-cell prolymphocytic leukemia (T-PLL) cells before (pre-RX) and after (post-RX) ruxolitinib treatment/relapse for forward scatter (FSC) and side scatter (SSC), 7-aminoactinomycin D (7-AAD), and anti-CD45. Three distinct populations were noted on the basis of CD45 expression; (B) cells were flow sorted, extracted for genomic DNA, subjected to polymerase chain reaction for relevant exons, and sequenced. (C) Western blot analysis of pre-RX and post-RX/relapse T-PLL cells for total JAK1, JAK3, and AKT proteins. (D) RNA-seq results for PTPRC, JAK2, JAK3, JAK1, and TYK2. Vertical line separates pre-RX and post-RX/relapse samples; both were analyzed in duplicate. Left panel, RPKM for T-PLL, reads per kilobase of gene per million reads; right panel, RPKM normalized to pre-RX reads (set at 100%). (E) Bar graphs show measured in vitro phosphatase activity from cytosolic lysates prepared from T-PLL cells before (blue) and after (gold) ruxolitinib. The second set of assays was done after pull down with isotype control IgG/protein A/G. The third set of assays was done after incubation with anti-CD45/protein A/G; values were normalized to cytosolic lysates before immunodepletion (set at 100%); error bars show standard error of the mean from quadruplicates. (F) Graph shows a kinetic phosphatase assay of cytosolic lysates prepared from pre-RX and post-RX/relapse T-PLL cells. The x-axis shows varying concentrations of phosphatase substrate, DiFMUP (6, 8-difluoro-4-methylumbiliferyl phosphate), in micromolar; the y-axis, velocity of product formation in mRFU/min.
Next, the tyrosine phosphatase activity was analyzed in lysates prepared from T-PLL cells pre- and post-ruxolitinib/relapse therapy. Immunodepletion with a specific antibody against CD45 reduced total tyrosine phosphatase activity to 20% to 22% of normal, which confirmed that CD45 was the major enzyme to contribute to this enzyme activity in T-PLL cells (Fig 5E). The specific activity of tyrosine phosphatase in pre-ruxolitinib lysates was 3.40 µmol/min/mg (95% CI, 3.10 to 3.70 µmol/min/mg), which decreased 54% to 1.85 µmol/min/mg (95% CI, 1.58 to 2.12 µmol/min/mg) in the post-ruxolitinib/relapse sample (P < .001; Fig 5F). Thus, the cellular phosphatase activity reflected the decreased CD45 protein levels, which paralleled the decrease in PTPRC mRNA abundance. Interestingly, there was another T-PLL case with a JAK3 M511I mutation that had downregulated CD45 at the time of disease relapse after treatment with combination cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy (Data Supplement).
Enhanced Phosphorylations Downstream of JAK1/JAK3 at Relapse
CD45 has been shown to directly dephosphorylate JAK1 and JAK3, which suggests that its loss of function should show increased activity of JAK3 or JAK1. To analyze JAK activity, we probed the intracellular phosphorylation of key substrates in T-PLL cells pre- and post-ruxolitinib/relapse treatment by flow cytometry. The percentage of T cells that expressed basal levels of phospho-STAT1 (p-STAT1) and p-STAT5 were increased in pre-ruxolitinib cells and in post-ruxolitinib/relapse cells compared with healthy T cells (p-STAT1: 22.72% and 21.24%, respectively, v 0.79%; p-STAT5: 57.52% and 54.84%, respectively, v 2.21%), whereas no cells in any condition expressed basal phosphorylation of p-STAT6, which served as a control (Fig 6B). T-PLL cells with the lowest expression of CD45 had the highest proportion of p-STAT5–positive cells at baseline before any stimulation (Fig 6C). To assess whether T-PLL cells had increased sensitivity to cytokine stimulation in addition to increased basal signaling, we stimulated each sample with 20 ng/mL of cytokine for 15 minutes. Signaling was quantified as previously described36,37 by using the median fluorescence intensity of per-cell phosphoprotein to create a fold-change. Increased phosphorylation of STAT5 was seen after in vitro stimulation with IL-2, IL-4, IL-7, IL-21, and interferon gamma, but not with IL-9, in both pre- and post-ruxolitinib/relapse–treated samples compared with healthy T cells (Fig 6D). A comparison between pre- and post-ruxolitinib/relapse T-PLL cells showed similar p-STAT5 levels at baseline and after stimulation by cytokines (Fig 6D). For example, there was an IL-2–induced 1.49-fold change in p-STAT5 for pre-ruxolitinib samples and 1.44-fold (comparison of arcsine transformed raw values) change for post-ruxolitinib/relapse samples. To understand the effects of CD45 expression, we gated on CD45hi, CD45lo, and CD45− post-ruxolitinib/relapse T-PLL cells and analyzed p-STAT5 and p-STAT6 basally and in response to stimuli. CD45hi had the lowest p-STAT5 response to IL-2 (0.23-fold), followed by CD45lo (0.61-fold) and CD45− (1.24-fold; Fig 6D); this pattern was seen for all common gamma chain cytokines compared with control (p-STAT6; Figs 6D and 6E). In summary, T-PLL cells were hyper-responsive to common gamma chain cytokines, and CD45 expression was negatively correlated with p-STAT5 at relapse.
Fig 6.
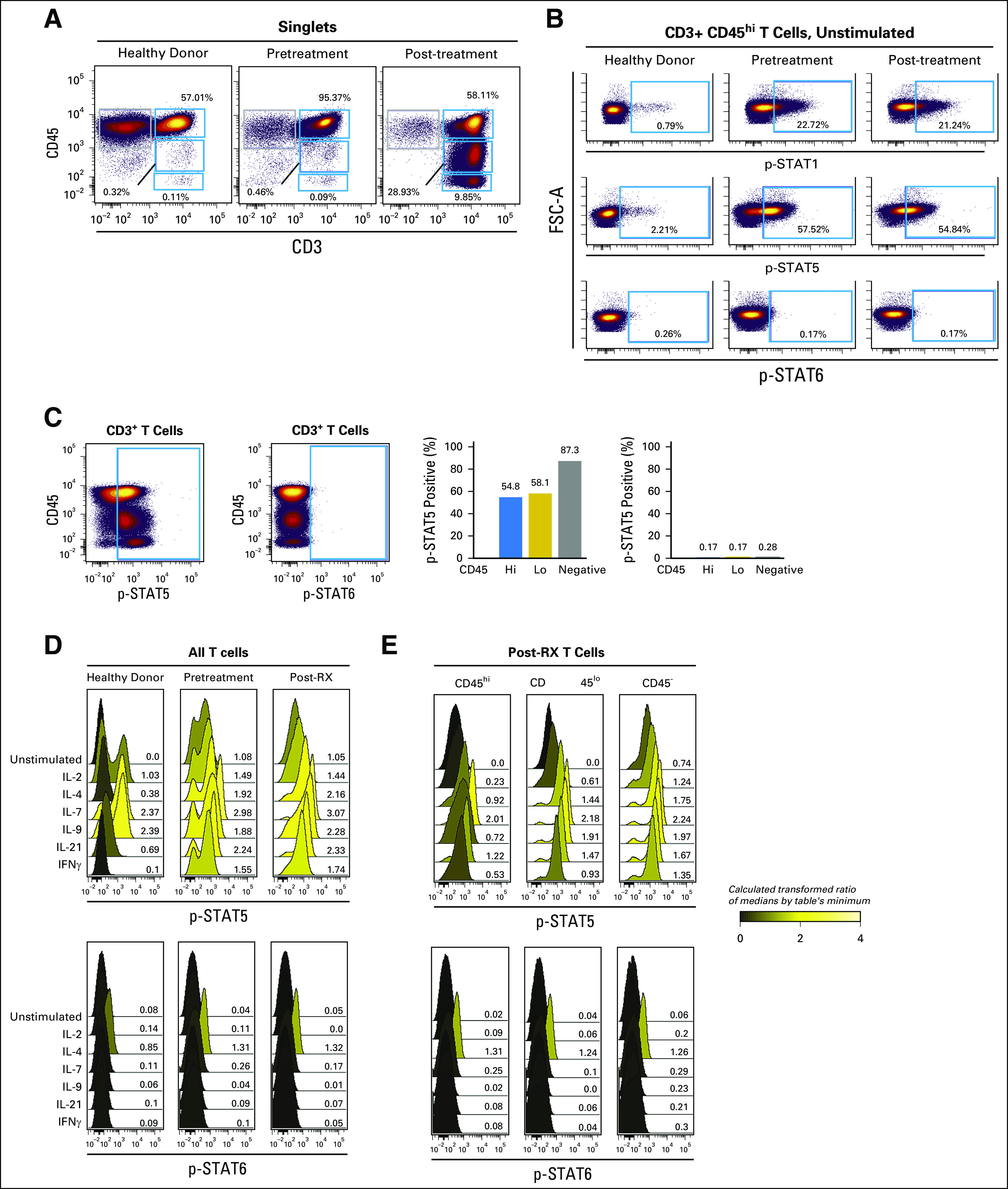
T-cell prolymphocytic leukemia (T-PLL) cells have constitutive phospho-STAT1 (p-STAT1) and p-STAT5. Intracellular signaling responses, monitored by using phospho-flow cytometry, of peripheral-blood mononuclear cells (PBMCs) from healthy donors, and from the patient with T-PLL before (pretreatment) and after (post-treatment) relapse experienced during ruxolitinib treatment. (A) Biaxial plots show expression of CD45 and CD3 on healthy donor and T-PLL samples. Blue gates indicate populations defined by expression of CD45 (hi, lo, and negative). CD3+ included peripheral T cells in health donor and all T-PLL cells. (B) Biaxial plots show basal p-STAT-1, p-STAT5, and p-STAT6 in CD3+ CD45hi cells. (C) Biaxial plot shows CD45 versus p-STAT5 and p-STAT6 at the time of relapse. Right panel, bar graph of percentage of cells with p-STAT5 or p-STAT6 for cells that express CD45hi, CD45lo, and CD45neg. (D) Graphs show p-STAT5 and p-STAT6 for health donor T cells, pretreatment T-PLL, and post-treatment T-PLL unstimulated or after 15 minutes of cytokine stimulation at 20 ng/mL.(E) Similar graphs of cytokine-stimulated p-STAT5 and p-STAT6 for post-treatment T-PLL cells with CD45hi, CD45lo, or CD45neg gating. Values are from median fluorescent intensities that were ArcSinh transformed and expressed as a fold change from the table’s minimum value. Graphs are shaded from black to yellow to reflect ratios from zero to four, as shown by the scale. Y-axes show counts, and x-axes show fluorescence.
DISCUSSION
In this study, diverse T-cell neoplasms were profiled by targeted next-generation sequencing of the exomes from approximately 400 known tumor suppressors and oncogenes. Among oncogenic mutations, JAK-STAT (33%) alterations were the most common. The cohort was composed of occurrences submitted to Foundation Medicine as a result of relapsed or resistant disease, so the observed genetic alterations may be specific to advanced-stage disease or therapeutic resistance. Nevertheless, the mutation frequencies for JAK3 (13% of occurrences) and for JAK1 (8.7%) were consistent with genomic profiling studies focused on specific disease subtypes (ie, T-ALL, ATLL, T-PLL, and CTCL15-19; less often, AITL and PTCL).38,39 The JAK mutations in this study were mutually exclusive with STAT3 and STAT5 gene alterations, as expected, because STAT3 and STAT5 proteins are downstream of IL2RG (common gamma chain)/JAK1/JAK3–restricted cytokines. Interestingly, recent data in cell lines and mouse models suggest that JAK1 enzyme activity is required for mutant JAK3 effects.40-42 However, as in the index case, the mutations were co-occurring but not present in the same cell; allele frequencies approached 50% for JAK1 and JAK3 mutations in the same tumor.
Most important, the JAK1 and JAK3 mutations were functionally significant, because they induced constitutive phosphorylations of downstream STAT5 proteins. p-STAT3 was not observed in the T-PLL cells (data not shown), although it is an important downstream substrate in other T-cell neoplasms, such as ALCL.43 Thus, signaling patterns downstream of JAK1/JAK3 may be unique to disease subtype. STAT1 and STAT5 phosphorylations in T-PLL could be inhibited by the specific JAK inhibitors ruxolitinib and tofacitinib.44 In the index case, treatment with ruxolitinib induced an impressive clinical response. These compelling data argue for oncogene dependence upon the JAK-STAT pathway in T-PLL, results that may extend to other T-cell neoplasms with JAK mutations. Furthermore, this oral drug worked when intensive parenteral therapies had failed to control the disease.
This study showed two cell-intrinsic mechanisms to account for resistance to ruxolitinib: expansion of the mutant JAK3 T-PLL clone and downregulation of CD45. In in vitro studies, the IC50 of ruxolitinib for JAK1 is 3.3 nM; for JAK2, it is 2.8 nM45; and for JAK3, it is 428 nM.45 This diminished potency against JAK3 probably accounted for the expansion of the mutant JAK3 clone from 10% before ruxolitinib to 56% at relapse. The T-PLL cells showed cross-resistance to tofacitinib, which has a nanomolar IC50 for JAK3.46,47 The downregulation of CD45 protein appears to be an additional resistance mechanism at relapse. Reduced CD45 proteins reduced the total tyrosine phosphatase activity in the T-PLL cells, which correlated with increased p-STAT5. PTPRC mRNA abundance was decreased, which suggests either enhanced mRNA degradation or transcriptional repression. The expression pattern of CD45 in a clonal T-PLL population resembles position-effect variegation, an epigenetic phenomenon.48,49 Because the primary leukemia samples were consumed, we were unable to directly transduce PTPRC cDNA to test if JAK inhibitor sensitivity could be restored. Thus, a cooperative genetic interaction between PTPRC loss of function and JAK3 M511I remains speculative. We observed an additional case of T-PLL with a clonal JAK3 M511I mutation that had similarly downregulated CD45 after disease relapse during CHOP chemotherapy, which suggests that downregulation of the CD45 protein may play a role in chemotherapy resistance. Notably, Porcu et al50 discovered deletion, missense, and nonsense mutations in PTPRC in T-ALL—evidence that supports a tumor suppressor role for PTPRC in this disease. Furthermore, Porcu et al50 also showed concordant loss of function in PTPRC and gain of function in JAK1 or IL7R, which suggests that these two hits cooperate in T-ALL pathogenesis. In fact, the authors described augmented p-STAT5 when PTPRC was knocked down by small interfering RNA. Our studies on the index case are similar to these findings, because we also observed an inverse correlation between CD45 levels and p-STAT5, albeit in T-PLL. Our studies do suggest that PTPRC may be a tumor suppressor in more mature T-cell neoplasms in addition to precursor T-ALL and that its loss of function may be an important resistance mechanism to ruxolitinib. Finally, the data presented in this study support the design of larger phase I/II clinical trials to test ruxolitinib on its own or in combination with cytotoxic therapies in T-cell neoplasms. A basket design in which patients with rare T-cell subtypes may be enrolled on the basis of the presence of JAK1 or JAK3 mutations may be most informative.
ACKNOWLEDGMENT
We thank Stephen Brandt, MD, Scott Hiebert, PhD, Carlos Arteaga, MD, and Justin Balko, PhD, for helpful discussions and Kevin Weller, PhD, for his expert advice.
Footnotes
Supported in part by National Cancer Institute (NCI) Award No. R01CA207530; the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, and Biomedical Laboratory Research and Development (Grant No. I01BX001799); and by an American Society of Hematology bridge grant. Additional support was provided by NIH/NCI Grants No. R00 CA143231 (J.M.I.) and F31 CA199993 (A.G.) and by the Vanderbilt-Ingram Cancer Center (Grant No. P30 CA68485). Flow and mass cytometry experiments were performed in the Vanderbilt Flow Cytometry Shared Resource, which is supported by Vanderbilt Ingram Cancer Center (Grant No. P30 CA68485) and Vanderbilt Digestive Disease Research Center (Grant No. DK058404).
The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health.
AUTHOR CONTRIBUTIONS
Conception and design: Kai Wang, Utpal Davé
Collection and assembly of data: Allison Greenplate, Kai Wang, Rati M. Tripathi, Norma Palma, Vincent A. Miller, Nishitha M. Reddy, Lina Kozhaya, Jonathan M. Irish, Utpal Davé
Provision of study material or patients: Nishitha M. Reddy, Xueyan Chen
Administrative support: Siraj Ali, Utpal Davé
Financial support: Utpal Davé
Data analysis and interpretation: Allison Greenplate, Kai Wang, Rati M. Tripathi, Siraj M. Ali, Phil J. Stephens, Vincent A. Miller, Yu Shyr, Yan Guo, Nishitha M. Reddy, Derya Unutmaz, Xueyan Chen, Jonathan M. Irish, Utpal Davé
Manuscript writing: All authors
Final approval of manuscript: All authors
Agree to be accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Allison Greenplate
Research Funding: Incyte
Kai Wang
No relationship to disclose
Rati M. Tripathi
No relationship to disclose
Norma Palma
Employment: Agios
Employment: Foundation Medicine
Stock and Other Ownership Interests: Agios, Foundation Medicine
Siraj M. Ali
Employment: Foundation Medicine
Stock and Other Ownership Interests: Exelixis, Blueprint Medicines, Agios
Patents, Royalties, Other Intellectual Property: Patents via Foundation Medicine; patents via Seres Health on microbiome stuff in non-neoplastic disease (I)
Phil J. Stephens
Employment: Foundation Medicine
Leadership: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Vincent A. Miller
Employment: Foundation Medicine
Leadership: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Patents, Royalties, Other Intellectual Property: Receive periodic royalties related to T790M patent awarded to Memorial Sloan Kettering Cancer Center
Yu Shyr
Consulting or Advisory Role: Aduro Biotech, Janssen Research and Development, Novartis, Roche, Genentech
Yan Guo
No relationship to disclose
Nishitha M. Reddy
Consulting or Advisory Role: Celgene, Abbvie, Gilead Sciences, Bristol-Myers Squibb
Lina Kozhaya
No relationship to disclose
Derya Unutmaz
No relationship to disclose
Xueyan Chen
No relationship to disclose
Jonathan M. Irish
Research Funding: Janssen (Inst), Incyte (Inst), Pharmacyclics (Inst)
Patents, Royalties, Other Intellectual Property: Methods and Compositions for Risk Stratification, US Patents (US 7,393,656, US 7,939,278, US 8,206,939, US 8,309,316, US 8,394,599)
Other Relationship: Cytobank
Utpal Davé
No relationship to disclose
REFERENCES
- 1.Coustan-Smith E, Mullighan CG, Onciu M, et al. : Early T-cell precursor leukemia: A subtype of very high-risk acute lymphoblastic leukemia. Lancet Oncol 10:147-156, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patrick K, Wade R, Goulden N, et al. : Outcome for children and young people with early T-cell precursor acute lymphoblastic leukemia treated on a contemporary protocol, UKALL 2003. Br J Haematol 166:421-424, 2014 [DOI] [PubMed] [Google Scholar]
- 3.Yeoh EJ, Ross ME, Shurtleff SA, et al. : Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 1:133-143, 2002 [DOI] [PubMed] [Google Scholar]
- 4.Chiaretti S, Li X, Gentleman R, et al. : Gene expression profile of adult T-cell acute lymphocytic leukemia identifies distinct subsets of patients with different response to therapy and survival. Blood 103:2771-2778, 2004 [DOI] [PubMed] [Google Scholar]
- 5.Vose J, Armitage J, Weisenburger D: International peripheral T-cell and natural killer/T-cell lymphoma study: Pathology findings and clinical outcomes. J Clin Oncol 26:4124-4130, 2008 [DOI] [PubMed] [Google Scholar]
- 6.Weisenburger DD, Savage KJ, Harris NL, et al. : Peripheral T-cell lymphoma, not otherwise specified: A report of 340 cases from the International Peripheral T-Cell Lymphoma Project. Blood 117:3402-3408, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Parrilla Castellar ER, Jaffe ES, Said JW, et al. : ALK-negative anaplastic large-cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood 124:1473-1480, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wood BL, Winter SS, Dunsmore KP, et al. : T-lymphoblastic leukemia (T-ALL) shows excellent outcome, lack of significance of the early thymic precursor (ETP) immunophenotype, and validation of the prognostic value of end-induction minimal residual disease (MRD) in Children’s Oncology Group (COG) study AALL0434. Blood 124:1-2, 2014. 24993873 [Google Scholar]
- 9.Sakata-Yanagimoto M, Enami T, Yoshida K, et al. : Somatic RHOA mutation in angioimmunoblastic T-cell lymphoma. Nat Genet 46:171-175, 2014 [DOI] [PubMed] [Google Scholar]
- 10.McKinney M, Moffitt AB, Gaulard P, et al. : The genetic basis of hepatosplenic T-cell lymphoma. Cancer Discov 7:369-379, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi J, Goh G, Walradt T, et al. : Genomic landscape of cutaneous T-cell lymphoma. Nat Genet 47:1011-1019, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palomero T, Couronné L, Khiabanian H, et al. : Recurrent mutations in epigenetic regulators, RHOA and FYN kinase, in peripheral T-cell lymphomas. Nat Genet 46:166-170, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kataoka K, Nagata Y, Kitanaka A, et al. : Integrated molecular analysis of adult T-cell leukemia/lymphoma. Nat Genet 47:1304-1315, 2015 [DOI] [PubMed] [Google Scholar]
- 14.Chen E, Staudt LM, Green AR: Janus kinase deregulation in leukemia and lymphoma. Immunity 36:529-541, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullighan CG, Zhang J, Harvey RC, et al. : JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci USA 106:9414-9418, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGirt LY, Jia P, Baerenwald DA, et al. : Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood 126:508-519, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elliott N, Cleveland S, Grann V: FERM domain mutations induce gain of function in JAK3 in adult T-cell leukemia/lymphoma. Blood 118:3911-3921, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergmann AK, Schneppenheim S, Seifert M, et al. : Recurrent mutation of JAK3 in T-cell prolymphocytic leukemia. Genes Chromosomes Cancer 53:309-316, 2014 [DOI] [PubMed] [Google Scholar]
- 19.Kiel MJ, Velusamy T, Rolland D, et al. : Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood 124:1460-1472, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeong EG, Kim MS, Nam HK, et al. : Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin Cancer Res 14:3716-3721, 2008 [DOI] [PubMed] [Google Scholar]
- 21.Yang LP, Keating GM: Ruxolitinib: In the treatment of myelofibrosis. Drugs 72:2117-2127, 2012 [DOI] [PubMed] [Google Scholar]
- 22.Scott LJ: Tofacitinib: A review of its use in adult patients with rheumatoid arthritis. Drugs 73:857-874, 2013 [DOI] [PubMed] [Google Scholar]
- 23.Dearden C: How I treat prolymphocytic leukemia. Blood 120:538-551, 2012 [DOI] [PubMed] [Google Scholar]
- 24.Frampton GM, Fichtenholtz A, Otto GA, et al. : Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31:1023-1031, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Layer JH, Alford CE, McDonald WH, et al. : LMO2 oncoprotein stability in T-cell leukemia requires direct LDB1 binding. Mol Cell Biol 36:488-506, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leelatian N, Diggins KE, Irish JM: Characterizing phenotypes and signaling networks of single human cells by mass cytometry. Single cell protein analysis. Methods Mol Biol 1346:99-113, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodings C, Tripathi R, Cleveland SM, et al. : Enforced expression of E47 has differential effects on Lmo2-induced T-cell leukemias. Leuk Res 39:100-109, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han X, Kilfoy B, Zheng T, et al. : Lymphoma survival patterns by WHO subtype in the United States, 1973-2003. Cancer Causes Control 19:841-858, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Boucheix C, David B, Sebban C, et al. : Immunophenotype of adult acute lymphoblastic leukemia, clinical parameters, and outcome: An analysis of a prospective trial including 562 tested patients (LALA87). Blood 84:1603-1612, 1994 [PubMed] [Google Scholar]
- 30.Forbes SA, Bindal N, Bamford S, et al. : COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 39:D945-D950, 2011 suppl 1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mullighan CG, Collins-Underwood JR, Phillips LA, et al. : Rearrangement of CRLF2 in B-progenitor– and Down syndrome–associated acute lymphoblastic leukemia. Nat Genet 41:1243-1246, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamashita Y, Yuan J, Suetake I, et al. : Array-based genomic resequencing of human leukemia. Oncogene 29:3723-3731, 2010 [DOI] [PubMed] [Google Scholar]
- 33.Amir AD, Davis KL, Tadmor MD, et al. : viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 31:545-552, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irie-Sasaki J, Sasaki T, Matsumoto W, et al. : CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature 409:349-354, 2001 [DOI] [PubMed] [Google Scholar]
- 35.Koretzky GA, Picus J, Schultz T, et al. : Tyrosine phosphatase CD45 is required for T-cell antigen receptor and CD2-mediated activation of a protein tyrosine kinase and interleukin 2 production. Proc Natl Acad Sci USA 88:2037-2041, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Irish JM, Myklebust JH, Alizadeh AA, et al. : B-cell signaling networks reveal a negative prognostic human lymphoma cell subset that emerges during tumor progression. Proc Natl Acad Sci USA 107:12747-12754, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Myklebust JH, Brody J, Kohrt HE, et al. : Distinct patterns of B-cell receptor signaling in non-Hodgkin lymphomas identified by single-cell profiling. Blood 129:759-770, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vallois D, Dobay MPD, Morin RD, et al. : Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood 128:1490-1502, 2016 [DOI] [PubMed] [Google Scholar]
- 39.Boddicker RL, Razidlo GL, Dasari S, et al. : Integrated mate-pair and RNA sequencing identifies novel, targetable gene fusions in peripheral T-cell lymphoma. Blood 128:1234-1235, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haan C, Rolvering C, Raulf F, et al. : JAK1 has a dominant role over JAK3 in signal transduction through γc-containing cytokine receptors. Chem Biol 18:314-323, 2011 [DOI] [PubMed] [Google Scholar]
- 41.Degryse S, de Bock CE, Cox L, et al. : JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood 124:3092-3100, 2014 [DOI] [PubMed] [Google Scholar]
- 42.Losdyck E, Hornakova T, Springuel L, et al. : Distinct acute lymphoblastic leukemia (ALL)-associated Janus kinase 3 (JAK3) mutants exhibit different cytokine-receptor requirements and JAK inhibitor specificities. J Biol Chem 290:29022-29034, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiarle R, Simmons WJ, Cai H, et al. : Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med 11:623-629, 2005 [DOI] [PubMed] [Google Scholar]
- 44.Flanagan ME, Blumenkopf TA, Brissette WH, et al. : Discovery of CP-690,550: A potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. J Med Chem 53:8468-8484, 2010 [DOI] [PubMed] [Google Scholar]
- 45.Quintás-Cardama A, Vaddi K, Liu P, et al. : Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: Therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 115:3109-3117, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Changelian PS, Moshinsky D, Kuhn CF, et al. : The specificity of JAK3 kinase inhibitors. Blood 111:2155-2157, 2008 [DOI] [PubMed] [Google Scholar]
- 47.Karaman MW, Herrgard S, Treiber DK, et al. : A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 26:127-132, 2008 [DOI] [PubMed] [Google Scholar]
- 48.Girton JR, Johansen KM: Chromatin structure and the regulation of gene expression: The lessons of PEV in Drosophila. Adv Genet 61:1-43, 2008 [DOI] [PubMed] [Google Scholar]
- 49. Cleveland SM, Goodings C, Tripathi RM, et al. LMO2 induces T-cell leukemia with epigenetic deregulation of CD4. Exp Hematol 2014;42:581-593.e5. [DOI] [PMC free article] [PubMed]
- 50.Porcu M, Kleppe M, Gianfelici V, et al. : Mutation of the receptor tyrosine phosphatase PTPRC (CD45) in T-cell acute lymphoblastic leukemia. Blood 119:4476-4479, 2012 [DOI] [PubMed] [Google Scholar]